

Sedoheptulose Kinase SHPK Expression in Glioblastoma: Emerging Role of the Nonoxidative Pentose Phosphate Pathway in Tumor Proliferation

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Results
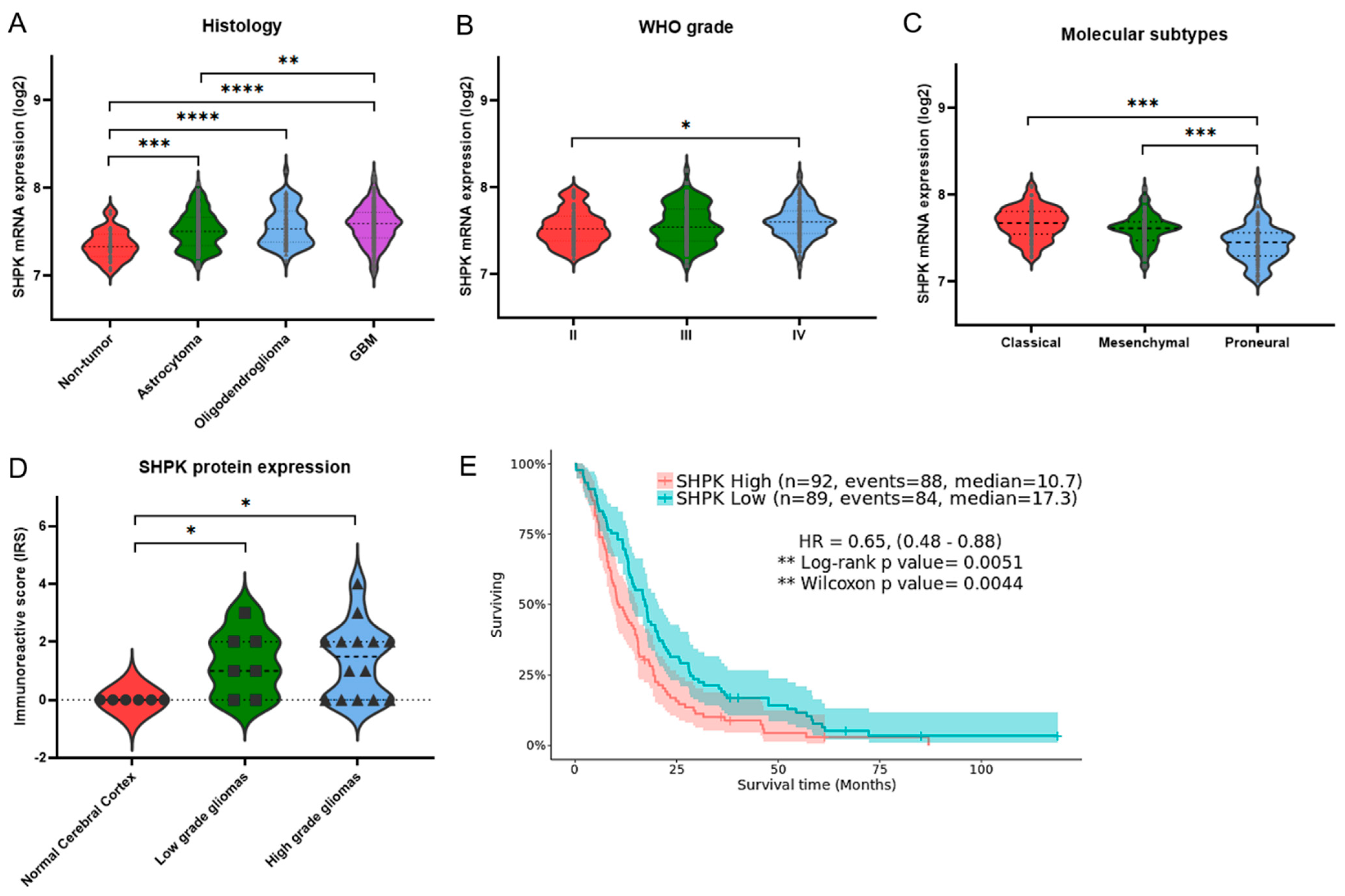
2.1. Correlation between SHPK Expression and Clinical Characteristics of Glioma Patients
2.2. SHPK-Related Biological Process
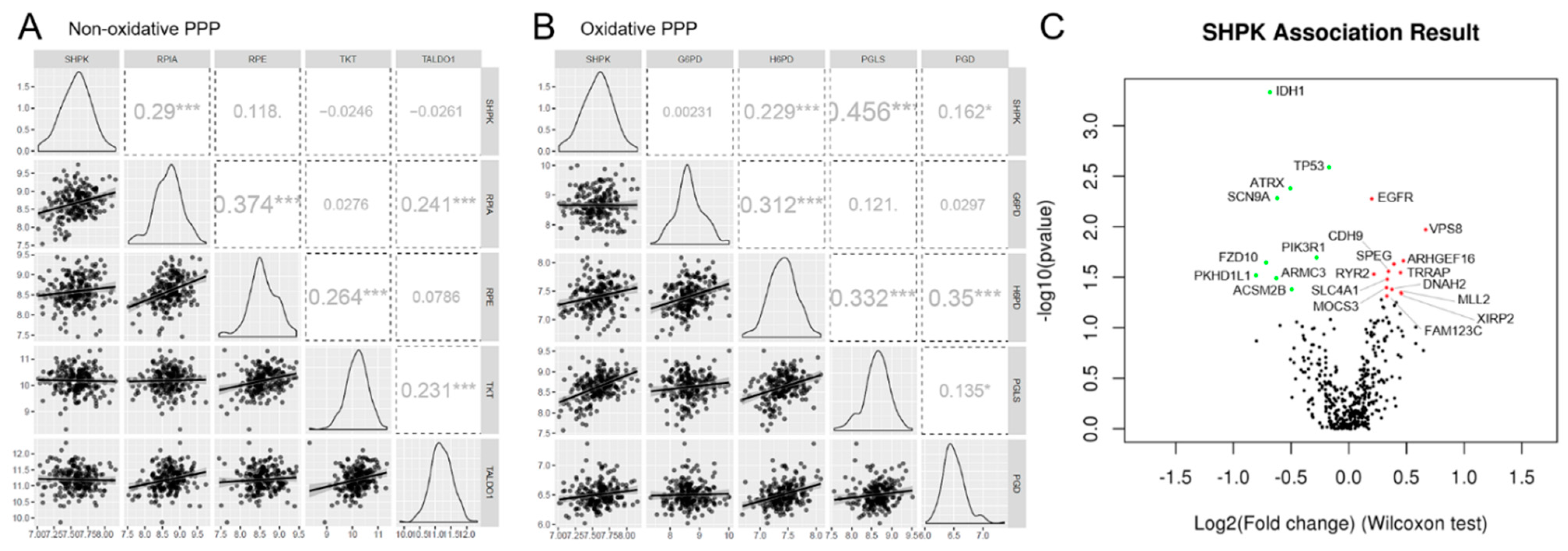
2.3. SHPK Correlation with Other PPP Enzymes
2.4. SHPK Expression and Mutational Status Association
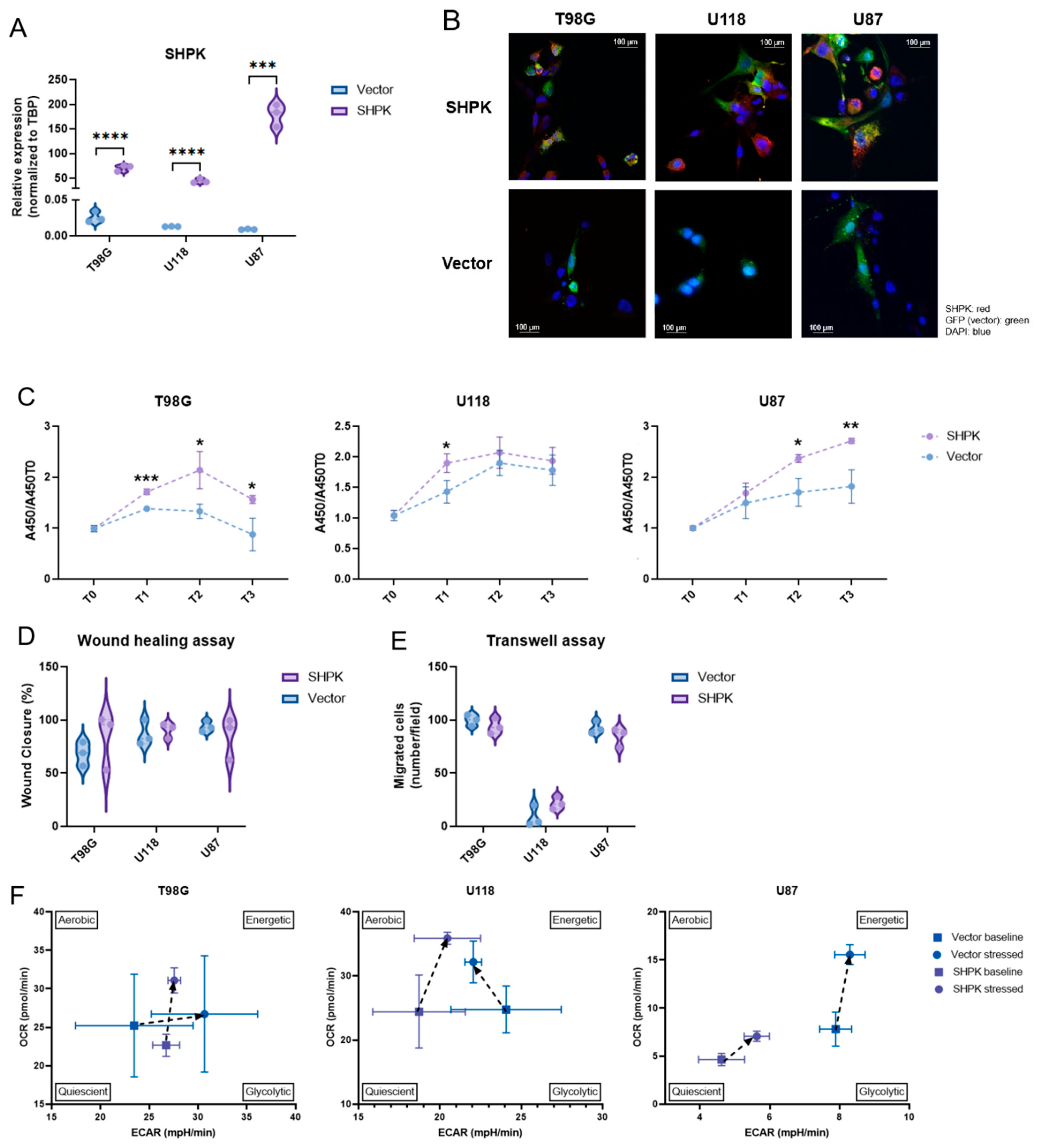
2.5. SHPK Overexpression and Cell Functional Studies
3. Discussion
4. Materials and Methods
4.1. Glioma Samples and Normal Controls
4.2. Immunohistochemistry of Histological Sections
4.3. Cell Lines and Transfection
4.4. SHPK mRNA Expression of Cell Lines
4.5. Immunofluorescence
4.6. Cell Viability Assay
4.7. Wound Healing Assay
4.8. Transwell Assay
4.9. Clonogenic Survival Assay
4.10. Cell Energy Phenotype Test
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. S5), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Lara-Velazquez, M.; Al-Kharboosh, R.; Jeanneret, S.; Vazquez-Ramos, C.; Mahato, D.; Tavanaiepour, D.; Rahmathulla, G.; Quinones-Hinojosa, A. Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, I.E.; Preul, M.C. Historical Perspective on Surgery and Survival with Glioblastoma: How Far Have We Come? In World Neurosurgery; Elsevier: Amsterdam, The Netherlands, 2021; Volume 149, pp. 148–168. [Google Scholar] [CrossRef]
- Verburg, N.; de Witt Hamer, P.C. State-of-the-art imaging for glioma surgery. Neurosurg. Rev. 2020, 44, 1331–1343. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Oliver, L.; Lalier, L.; Salaud, C.; Heymann, D.; Cartron, P.F.; Vallette, F.M. Drug resistance in glioblastoma: Are persisters the key to therapy? Cancer Drug Resist. 2020, 3, 287–301. [Google Scholar] [CrossRef]
- Gianfaldoni, S.; Gianfaldoni, R.; Wollina, U.; Lotti, J.; Tchernev, G.; Lotti, T. An Overview on Radiotherapy: From Its History to Its Current Applications in Dermatology. Open Access Maced. J. Med Sci. 2017, 5, 521–525. [Google Scholar] [CrossRef]
- Corso, C.D.; Bindra, R.S.; Mehta, M.P. The role of radiation in treating glioblastoma: Here to stay. J. Neuro-Oncol. 2017, 44, 479–485. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Di Ianni, N.; Musio, S.; Pellegatta, S. Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int. J. Mol. Sci. 2021, 22, 4460. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncol. 2015, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, J.L.; Jalasutram, A.; Asuthkar, S.; Sahagun, J.; Park, S.; Ravindra, A.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Beyond glucose: Alternative sources of energy in glioblastoma. Theranostics 2021, 11, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors (Review). Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro-Oncol. 2016, 18, 1219–1229. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Yang, C.-A.; Huang, H.-Y.; Lin, C.-L.; Chang, J.-G. G6PD as a predictive marker for glioma risk, prognosis and chemosensitivity. J. Neuro-Oncol. 2018, 139, 661–670. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Madhavan, S.; Zenklusen, J.-C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping Personalized Medicine Become a Reality through Integrative Translational Research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Assal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Seker-Polat, F.; Degirmenci, N.P.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. [Google Scholar] [CrossRef]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles. Int. J. Mol. Sci. 2017, 18, 2774. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.-G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, N.; El-Baba, C.; Araji, K.; El-Khoury, R.; Usta, J.; Darwiche, N. The Pentose Phosphate Pathway in Cancer: Regulation and Therapeutic Opportunities. Chemotherapy 2021, 66, 179–191. [Google Scholar] [CrossRef]
- Kathagen-Buhmann, A.; Maire, C.L.; Weller, J.; Schulte, A.; Matschke, J.; Holz, M.; Ligon, K.L.; Glatzel, M.; Westphal, M.; Lamszus, K. The secreted glycolytic enzyme GPI/AMF stimulates glioblastoma cell migration and invasion in an autocrine fashion but can have anti-proliferative effects. Neuro-Oncol. 2018, 20, 1594–1605. [Google Scholar] [CrossRef]
- Stanke, K.M.; Wilson, C.; Kidambi, S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates with Lower Survival. Front. Mol. Biosci. 2021, 8, 1281. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Ehlers, J.; Schötterl, S.; Naumann, U. Molecular Mechanisms of Glioma Cell Motility. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 73–93. [Google Scholar] [CrossRef]
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021, 38, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Batsios, G.; Taglang, C.; Cao, P.; Gillespie, A.M.; Najac, C.; Subramani, E.; Wilson, D.M.; Flavell, R.R.; Larson, P.E.Z.; Ronen, S.M.; et al. Imaging 6-Phosphogluconolactonase Activity in Brain Tumors In Vivo Using Hyperpolarized δ-[1-13C] gluconolactone. Front. Oncol. 2021, 11, 1194. [Google Scholar] [CrossRef]
- Xu, X.; Wang, L.; Zang, Q.; Li, S.; Li, L.; Wang, Z.; He, J.; Qiang, B.; Han, W.; Zhang, R.; et al. Rewiring of purine metabolism in response to acidosis stress in glioma stem cells. Cell Death Dis. 2021, 12, 227. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Cossu, V.; Bonanomi, M.; Bauckneht, M.; Ravera, S.; Righi, N.; Miceli, A.; Morbelli, S.; Orengo, A.M.; Piccioli, P.; Bruno, S.; et al. Two high-rate pentose-phosphate pathways in cancer cells. Sci. Rep. 2020, 10, 22111. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Xiang, C.; Zhong, F.; Liu, Y.; Dong, Q.; Li, K.; Shi, W.; Ding, C.; Qin, L.; He, F. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J. Exp. Clin. Cancer Res. 2019, 38, 154. [Google Scholar] [CrossRef]
- Dos Santos, G.C.; Saldanha-Gama, R.; De Brito, N.M.; Renovato-Martins, M.; Barja-Fidalgo, C. Metabolomics in cancer and cancer-associated inflammatory cells. J. Cancer Metastasis Treat. 2021, 7, 1. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.; Bernard, A.; Hau, A.; et al. Altered metabolic landscape in IDH -mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef]
- Boren, J.; Montoya, A.R.; De Atauri, P.; Comin-Anduix, B.; Cortes, A.; Centelles, J.J.; Frederiks, W.M.; Van Noorden, C.J.; Cascante, M. Metabolic control analysis aimed at the ribose synthesis pathways of tumor cells: A new strategy for antitumor drug development. Mol. Biol. Rep. 2002, 29, 7–12. [Google Scholar] [CrossRef]
- Ramos-Montoya, A.; Lee, W.-N.P.; Bassilian, S.; Lim, S.; Trebukhina, R.V.; Kazhyna, M.V.; Ciudad, C.J.; Noé, V.; Centelles, J.J.; Cascante, M. Pentose phosphate cycle oxidative and nonoxidative balance: A new vulnerable target for overcoming drug resistance in cancer. Int. J. Cancer 2006, 119, 2733–2741. [Google Scholar] [CrossRef]
- Frederiks, W.M.; Vizan, P.; Bosch, K.S.; Vreeling-Sindelárová, H.; Boren, J.; Cascante, M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. Int. J. Exp. Pathol. 2008, 89, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Pandith, A.A.; Qasim, I.; Baba, S.M.; Koul, A.; Zahoor, W.; Afroze, D.; Lateef, A.; Manzoor, U.; A Bhat, I.; Sanadhya, D.; et al. Favorable role of IDH1/2 mutations aided with MGMT promoter gene methylation in the outcome of patients with malignant glioma. Futur. Sci. OA 2020, 7, FSO663. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Yao, Y.; Xu, H.-Z.; Qin, Z.-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients with Glioblastomas. Medicine 2016, 95, e2583. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Alzahrani, A.S. Isocitrate Dehydrogenase IDH1 and IDH2 Mutations in Human Cancer: Prognostic Implications for Gliomas. Br. J. Biomed. Sci. 2022, 79, 10208. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Xu, L.; Zhang, J.; Cao, H. Analysis of the EGFR Amplification and CDKN2A Deletion Regulated Transcriptomic Signatures Reveals the Prognostic Significance of SPATS2L in Patients with Glioma. Front. Oncol. 2021, 11, 713. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| abs(LogFC) < 1 | Reactome | ID | p-Value | APOD | POPDC3 | CNTN2 | NKAIN2 | RUNDC3B | KCNQ5 | RUNDC3A | RYR2 | MYRIP | NDN | FAM155A | FAIM2 | SPOCK1 | CHGA | PKP4 | SNCA | RAB3A | CHGB | JPH4 | TMEFF1 | NPY1R | PAK3 | DOCK3 | NAP1L3 | FA2H | XKR4 |
| Transmission across Chemical Synapses | 112315 | 8.79 × 10−13 | |||||||||||||||||||||||||||
| Neuronal System | 112316 | 1.15 × 10−12 | |||||||||||||||||||||||||||
| Serotonin Neurotransmitter Release Cycle | 181429 | 3.97 × 10−8 | |||||||||||||||||||||||||||
| Dopamine Neurotransmitter Release Cycle | 212676 | 3.35 × 10−7 | |||||||||||||||||||||||||||
| Neurotransmitter release cycle | 112310 | 7.56 × 10−6 | |||||||||||||||||||||||||||
| Neurotransmitter receptors and postsynaptic signal transmission | 112314 | 1.47 × 10−5 | |||||||||||||||||||||||||||
| Glutamate Neurotransmitter Release Cycle | 210500 | 2.18 × 10−5 | |||||||||||||||||||||||||||
| Acetylcholine Neurotransmitter Release Cycle | 264642 | 1.10 × 10−4 | |||||||||||||||||||||||||||
| Norepinephrine Neurotransmitter Release Cycle | 181430 | 1.54 × 10−4 | |||||||||||||||||||||||||||
| GABA receptor activation | 977443 | 5.65 × 10−4 | |||||||||||||||||||||||||||
| Protein–protein interactions at synapses | 6794362 | 6.42 × 10−3 | |||||||||||||||||||||||||||
| GABA synthesis, release, reuptake and degradation | 888590 | 8.24 × 10−3 | |||||||||||||||||||||||||||
| Long-term potentiation | 9620244 | 1.58 × 10−2 | |||||||||||||||||||||||||||
| Neurotoxicity of clostridium toxins | 168799 | 3.70 × 10−2 | |||||||||||||||||||||||||||
| abs(LogFC) > 1 | Reactome | ID | p-Value | PTX3 | COL1A2 | IGFBP2 | ADM | CRISPLD1 | VEGFA | COL3A1 | IGFBP3 | CDCA7L | |||||||||||||||||
| Integrin cell surface interactions | 216083 | 8.21 × 10−6 | |||||||||||||||||||||||||||
| Nonintegrin membrane-ECM interactions | 3000171 | 2.45 × 10−5 | |||||||||||||||||||||||||||
| ECM proteoglycans | 3000178 | 1.16 × 10−4 | |||||||||||||||||||||||||||
| Crosslinking of collagen fibrils | 2243919 | 1.84 × 10−4 | |||||||||||||||||||||||||||
| Scavenging by Class A Receptors | 3000480 | 2.32 × 10−4 | |||||||||||||||||||||||||||
| Collagen formation | 1474290 | 3.23 × 10−4 | |||||||||||||||||||||||||||
| Extracellular matrix organization | 1474244 | 4.40 × 10−4 | |||||||||||||||||||||||||||
| Syndecan interactions | 3000170 | 8.80 × 10−4 | |||||||||||||||||||||||||||
| Assembly of collagen fibrils and other multimeric structures | 2022090 | 9.94 × 10−4 | |||||||||||||||||||||||||||
| Collagen biosynthesis and modifying enzymes | 1650814 | 1.72 × 10−3 | |||||||||||||||||||||||||||
| Collagen chain trimerization | 8948216 | 7.61 × 10−3 | |||||||||||||||||||||||||||
| Anchoring fibril formation | 2214320 | 7.90 × 10−3 | |||||||||||||||||||||||||||
| Collagen degradation | 1442490 | 3.37 × 10−2 |
| Gene | Log2FC (Median) | p-Value | FDR (BH) | Event_SD | Event_TD |
|---|---|---|---|---|---|
| PKHD1L1 | −0.8033 | 0.0304 | 0.8846 | 141 | 3 |
| FZD10 | −0.7171 | 0.0227 | 0.8846 | 141 | 3 |
| IDH1 | −0.6885 | 0.0005 | 0.2030 | 141 | 8 |
| ARMC3 | −0.6270 | 0.0327 | 0.8846 | 141 | 3 |
| SCN9A | −0.6201 | 0.0052 | 0.4584 | 141 | 5 |
| ATRX | −0.5071 | 0.0042 | 0.4584 | 141 | 8 |
| ACSM2B | −0.4953 | 0.0418 | 0.8846 | 141 | 3 |
| PIK3R1 | −0.2773 | 0.0201 | 0.8846 | 141 | 12 |
| TP53 | −0.1720 | 0.0026 | 0.4584 | 141 | 45 |
| EGFR | 0.1987 | 0.0053 | 0.4584 | 141 | 45 |
| RYR2 | 0.2172 | 0.0296 | 0.8846 | 141 | 12 |
| MOCS3 | 0.3278 | 0.0403 | 0.8846 | 141 | 3 |
| FAM123C | 0.3311 | 0.0490 | 0.8846 | 141 | 4 |
| SLC4A1 | 0.3325 | 0.0332 | 0.8846 | 141 | 4 |
| CDH9 | 0.3434 | 0.0277 | 0.8846 | 141 | 5 |
| DNAH2 | 0.3726 | 0.0419 | 0.8846 | 141 | 5 |
| SPEG | 0.3916 | 0.0234 | 0.8846 | 141 | 4 |
| TRRAP | 0.4470 | 0.0284 | 0.8846 | 141 | 4 |
| MLL2 | 0.4534 | 0.0449 | 0.8846 | 141 | 4 |
| XIRP2 | 0.4578 | 0.0463 | 0.8846 | 141 | 3 |
| ARHGEF16 | 0.4721 | 0.0218 | 0.8846 | 141 | 3 |
| VPS8 | 0.6664 | 0.0108 | 0.7768 | 141 | 3 |
| A (Percentage of Positive Cells) | B (Intensity of Staining) | IRS Score (A × B) |
|---|---|---|
| 0 = no positive cells | 0 = no color reaction | 0 = negative |
| 1 = <25% of positive cells | 1 = weak reaction | 1–2 = mild |
| 2 = 25–75% of positve cells | 2 = moderate reaction | 3–6 = moderate |
| 3 = >75% of positive cells | 3 = intense reaction | 7–9 = strong |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceschi, S.; Lessi, F.; Morelli, M.; Menicagli, M.; Pasqualetti, F.; Aretini, P.; Mazzanti, C.M. Sedoheptulose Kinase SHPK Expression in Glioblastoma: Emerging Role of the Nonoxidative Pentose Phosphate Pathway in Tumor Proliferation. Int. J. Mol. Sci. 2022, 23, 5978. https://doi.org/10.3390/ijms23115978
Franceschi S, Lessi F, Morelli M, Menicagli M, Pasqualetti F, Aretini P, Mazzanti CM. Sedoheptulose Kinase SHPK Expression in Glioblastoma: Emerging Role of the Nonoxidative Pentose Phosphate Pathway in Tumor Proliferation. International Journal of Molecular Sciences. 2022; 23(11):5978. https://doi.org/10.3390/ijms23115978
Chicago/Turabian StyleFranceschi, Sara, Francesca Lessi, Mariangela Morelli, Michele Menicagli, Francesco Pasqualetti, Paolo Aretini, and Chiara Maria Mazzanti. 2022. "Sedoheptulose Kinase SHPK Expression in Glioblastoma: Emerging Role of the Nonoxidative Pentose Phosphate Pathway in Tumor Proliferation" International Journal of Molecular Sciences 23, no. 11: 5978. https://doi.org/10.3390/ijms23115978
APA StyleFranceschi, S., Lessi, F., Morelli, M., Menicagli, M., Pasqualetti, F., Aretini, P., & Mazzanti, C. M. (2022). Sedoheptulose Kinase SHPK Expression in Glioblastoma: Emerging Role of the Nonoxidative Pentose Phosphate Pathway in Tumor Proliferation. International Journal of Molecular Sciences, 23(11), 5978. https://doi.org/10.3390/ijms23115978