
Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model

 , ,
, ,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
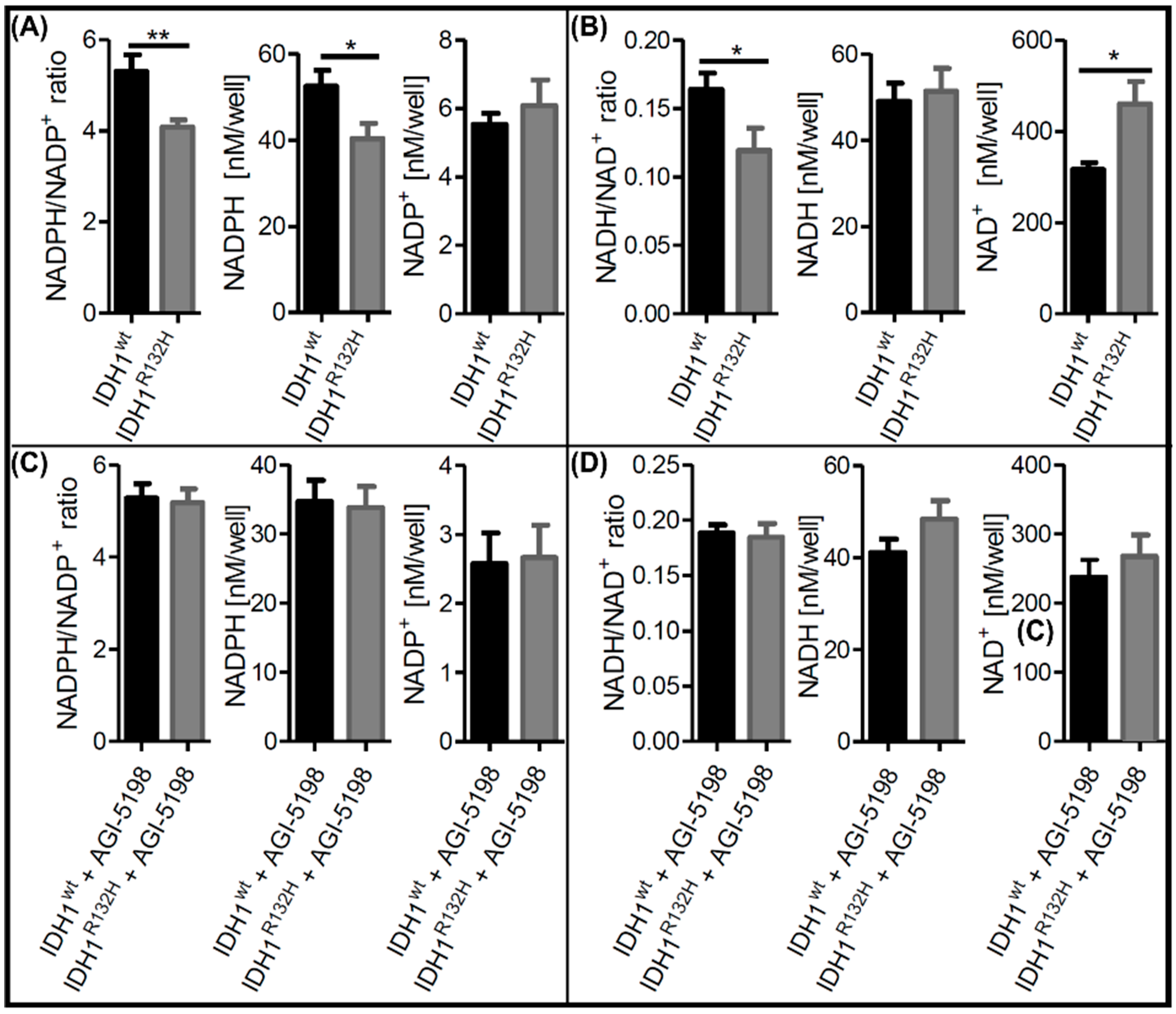
2.1. Alterations in NAD+ and NADP+ Metabolism in IDH1R132H Cells Could Be Directly Attributed to the Neoenzymatic Function of IDH1R132H
2.2. IDH1R132H Altered NAMPT Expression
2.3. Knockdown of NAMPT and other NAD+ Synthesis Enzymes Selectively Reduced the Viability of IDH1R132H Cells
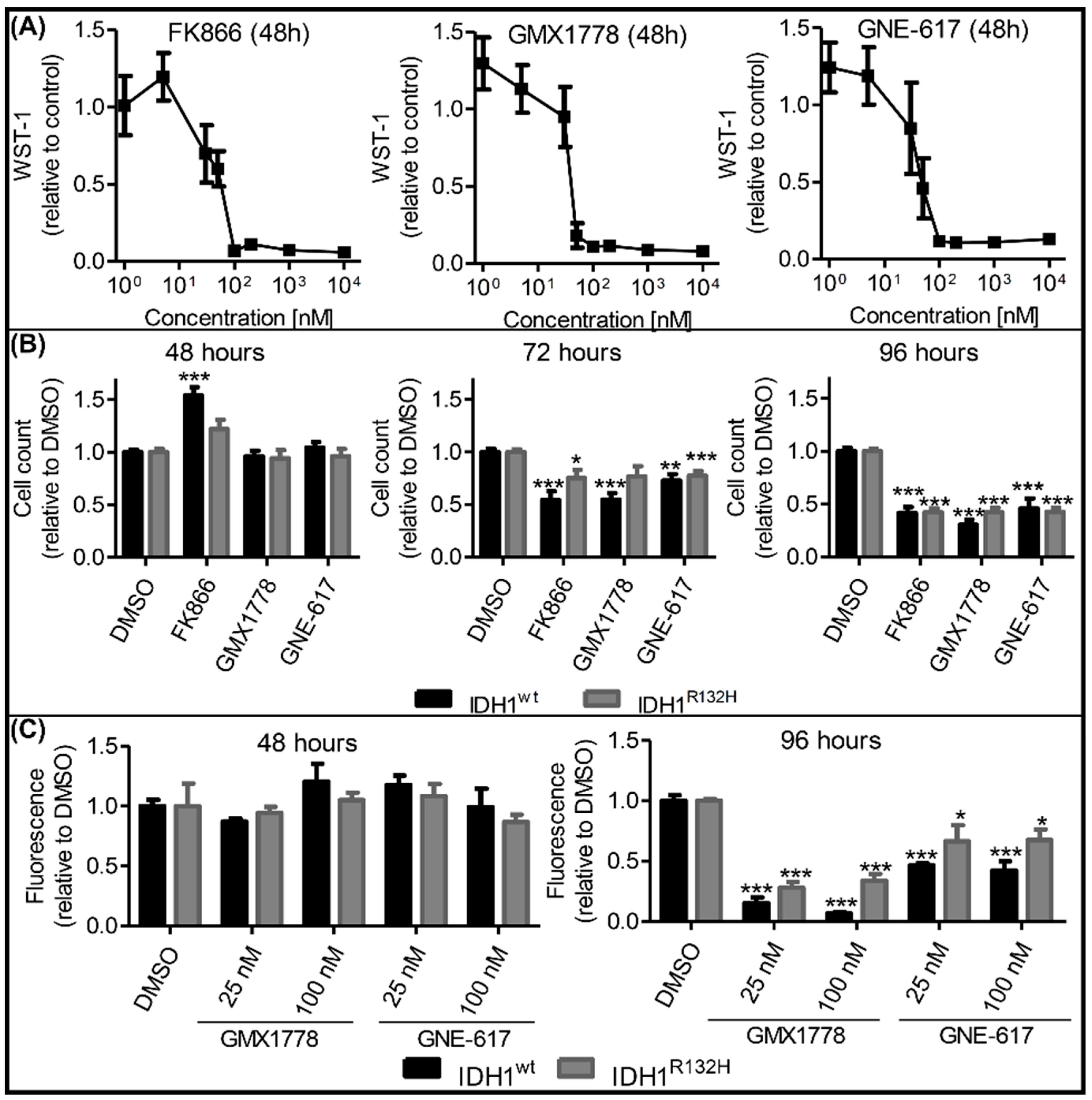
2.4. Effects of NAMPT Small-Molecule Inhibitors on Cell Viability Were Independent of IDH1 Status
2.5. Combinatorial esiNAMPT and GMX1778 Treatment Indicated Unspecific Effects of Small Molecule NAMPT Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Genome Editing Using CRISPR/Cas9
4.3. Quantification of NAD+/NADH and NADP+/NADPH
4.4. RNA Extraction, Protein Extraction and Western Blotting
4.5. Real-Time Quantitative PCR (RT-qPCR)
4.6. esiRNA Knockdown
4.7. NAMPT Inhibition Using Small-Molecule Inhibitors
4.8. WST-1 Assay
4.9. Combinatorial esiNAMPT and GMX1778 Treatment
4.10. Measurement of Cell Viability with CyQUANT® Direct Cell Proliferation Assay
4.11. Measurement of Cell Viability with Automated Picture Analysis
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, A.L.; Colman, H. Glioma biology and molecular markers. Cancer Res. Treat. 2015, 163, 15–30. [Google Scholar]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes Enhance the Invasion Potential of Glioblastoma Stem-Like Cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2018, 54, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.-N.; Gittleman, H.; Ostrom, Q.T.; Hung, K.; Sloan, A.E.; Hsieh, Y.; Kruchko, C.; Rogers, L.R.; Wang, Y.G.; Chiou, H.-Y.; et al. Comparative Brain and Central Nervous System Tumor Incidence and Survival between the United States and Taiwan Based on Population-Based Registry. Front. Public Health 2016, 4, 151. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, W.; Jin, G. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Hartong, D.T.; Dange, M.; McGee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Wahab, O.A.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the R Enantiomer of 2-Hydroxyglutarate Linked to EglN Activation. Nature 2013, 483, 484–488. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.; Bernard, A.; Hau, A.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Biedermann, J.; Preussler, M.; Conde, M.; Peitzsch, M.; Richter, S.; Wiedemuth, R.; Abou-El-Ardat, K.; Krüger, A.; Meinhardt, M.; Schackert, G.; et al. Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin. Cancers 2019, 11, 2028. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1R132mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr. Opin. Gastroenterol. 2007, 23, 164–170. [Google Scholar] [CrossRef]
- O’Brien, T.; Oeh, J.; Xiao, Y.; Liang, X.; Vanderbilt, A.; Qin, A.; Yang, L.; Lee, L.B.; Ly, J.; Cosino, E.; et al. Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia 2013, 15, 1314–1329. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Roulston, A.; Belec, L.; Billot, X.; Marcellus, R.; Bédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of Tumor Cell Lines and Patient-Derived Tumors on the NAD Salvage Pathway Renders Them Sensitive to NAMPT Inhibition with GNE-618. Neoplasia 2013, 15, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Umesh, S.; Thota, B.; Tandon, A.; Pandey, P.; Hegde, A.S.; Balasubramaniam, A.; Chandramouli, B.A.; Santosh, V.; Rao, M.R.S.; et al. PBEF1/NAmPRTase/Visfatin: A potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol. Ther. 2008, 7, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, R.E.; Mayhall, K.; Maxwell, N.M.; Kandil, E.; Coppola, D. Nicotinamide Phosphoribosyltransferase in Malignancy: A Review. Genes Cancer 2013, 4, 447–456. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef]
- Piaskowski, S.; Bienkowski, M.; Stoczynska-Fidelus, E.; Stawski, R.; Sieruta, M.; Szybka, M.; Papierz, W.; Wolanczyk, M.; Jaskolski, D.J.; Liberski, P.P.; et al. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br. J. Cancer 2011, 104, 968–970. [Google Scholar] [CrossRef]
- Golebiewska, A.; Hau, A.C.; Oudin, A.; Stieber, D.; Yabo, Y.A.; Baus, V.; Barthelemy, V.; Klein, E.; Bougnaud, S.; Keunen, O.; et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 2020, 140, 919–949. [Google Scholar] [CrossRef]
- Klink, B.; Miletic, H.; Stieber, D.; Huszthy, P.C.; Valenzuela, J.A.C.; Balss, J.; Wang, J.; Schubert, M.; Sakariassen, P.Ø.; Sundstrøm, T.; et al. A Novel, Diffusely Infiltrative Xenograft Model of Human Anaplastic Oligodendroglioma with Mutations in FUBP1, CIC, and IDH1. PLoS ONE 2013, 8, e59773. [Google Scholar] [CrossRef]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Töpfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro. Oncol. 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Chen, J.; Qiu, J.; Huang, K.; Wu, M.; Xia, C. IDH1 R132H Mutation Enhances Cell Migration by Activating AKT-mTOR Signaling Pathway, but Sensitizes Cells to 5-FU Treatment as NADPH and GSH Are Reduced. PLoS ONE 2017, 12, e0169038. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed]
- Garrett-Bakelman, F.E.; Melnick, A.M. Differentiation therapy for IDH1/2 mutant malignancies. Cell Res. 2013, 23, 975–977. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tiburcio, P.D.B.; Gillespie, D.L.; Jensen, R.L.; Huang, L.E. Extracellular glutamate and IDH1(R132H) inhibitor promote glioma growth by boosting redox potential. J. Neurooncol. 2020, 146, 427–437. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, Y.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, E8247–E8256. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Peinado-Serrano, J.; Carnero, A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Zhang, Y.; Shu, C.; Tsujiuchi, T.; Banu, M.A.; Garcia, F.; Roth, K.A.; Bruce, J.N.; et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat. Commun. 2017, 8, 1067. [Google Scholar] [CrossRef]
- Dalamaga, M.; Christodoulatos, G.S.; Mantzoros, C.S. The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: Diagnostic and therapeutic perspectives and challenges. Metabolism 2018, 82, 72–87. [Google Scholar] [CrossRef]
- Bowlby, S.C.; Thomas, M.J.; D’Agostino, R.B.; Kridel, S.J. Nicotinamide Phosphoribosyl Transferase (Nampt) Is Required for De Novo Lipogenesis in Tumor Cells. PLoS ONE 2012, 7, e40195. [Google Scholar] [CrossRef] [PubMed]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates Reactive Oxygen Species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, P.-F.; Zhao, H.-Y.; Zhang, F.-C.; Zhao, W.-H.; Feng, M. Inhibitor of Nicotinamide Phosphoribosyltransferase Sensitizes Glioblastoma Cells to Temozolomide via Activating ROS/JNK Signaling Pathway. BioMed. Res. Int. 2016, 2016, 1450843. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, R.; Willemse, M.; Haeger, A.; Attanasio, F.; Guneri, T.; Schwab, A.; Stock, C.M.; Buccione, R.; Fransen, J.A.M.; Wieringa, B. Intracellular NAD(H) levels control motility and invasion of glioma cells. Cell. Mol. Life Sci. 2013, 70, 2175–2190. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar]
- Tan, B.; Young, D.A.; Lu, Z.H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.-S.; et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+biosynthesis, in human cancer cells: Metabolic basis and potential clinical implications. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef]
- Lozada-Delgado, E.L.; Grafals-Ruiz, N.; Vivas-Mejía, P.E. RNA interference for glioblastoma therapy: Innovation ladder from the bench to clinical trials. Life Sci. 2017, 188, 26–36. [Google Scholar] [CrossRef]
- Tietze, S.; Schau, I.; Michen, S.; Ennen, F.; Janke, A.; Schackert, G.; Aigner, A.; Appelhans, D.; Temme, A. A Poly(Propyleneimine) Dendrimer-Based Polyplex-System for Single-Chain Antibody-Mediated Targeted Delivery and Cellular Uptake of SiRNA. Small 2017, 13, 1700072. [Google Scholar] [CrossRef]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Contr. Release 2017, 262, 247–258. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clausing, M.; William, D.; Preussler, M.; Biedermann, J.; Grützmann, K.; Richter, S.; Buchholz, F.; Temme, A.; Schröck, E.; Klink, B. Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model. Int. J. Mol. Sci. 2022, 23, 5787. https://doi.org/10.3390/ijms23105787
Clausing M, William D, Preussler M, Biedermann J, Grützmann K, Richter S, Buchholz F, Temme A, Schröck E, Klink B. Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model. International Journal of Molecular Sciences. 2022; 23(10):5787. https://doi.org/10.3390/ijms23105787
Chicago/Turabian StyleClausing, Maximilian, Doreen William, Matthias Preussler, Julia Biedermann, Konrad Grützmann, Susan Richter, Frank Buchholz, Achim Temme, Evelin Schröck, and Barbara Klink. 2022. "Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model" International Journal of Molecular Sciences 23, no. 10: 5787. https://doi.org/10.3390/ijms23105787
APA StyleClausing, M., William, D., Preussler, M., Biedermann, J., Grützmann, K., Richter, S., Buchholz, F., Temme, A., Schröck, E., & Klink, B. (2022). Different Effects of RNAi-Mediated Downregulation or Chemical Inhibition of NAMPT in an Isogenic IDH Mutant and Wild-Type Glioma Cell Model. International Journal of Molecular Sciences, 23(10), 5787. https://doi.org/10.3390/ijms23105787