
Expression Profiles and Characteristics of Apple lncRNAs in Roots, Phloem, Leaves, Flowers, and Fruit




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
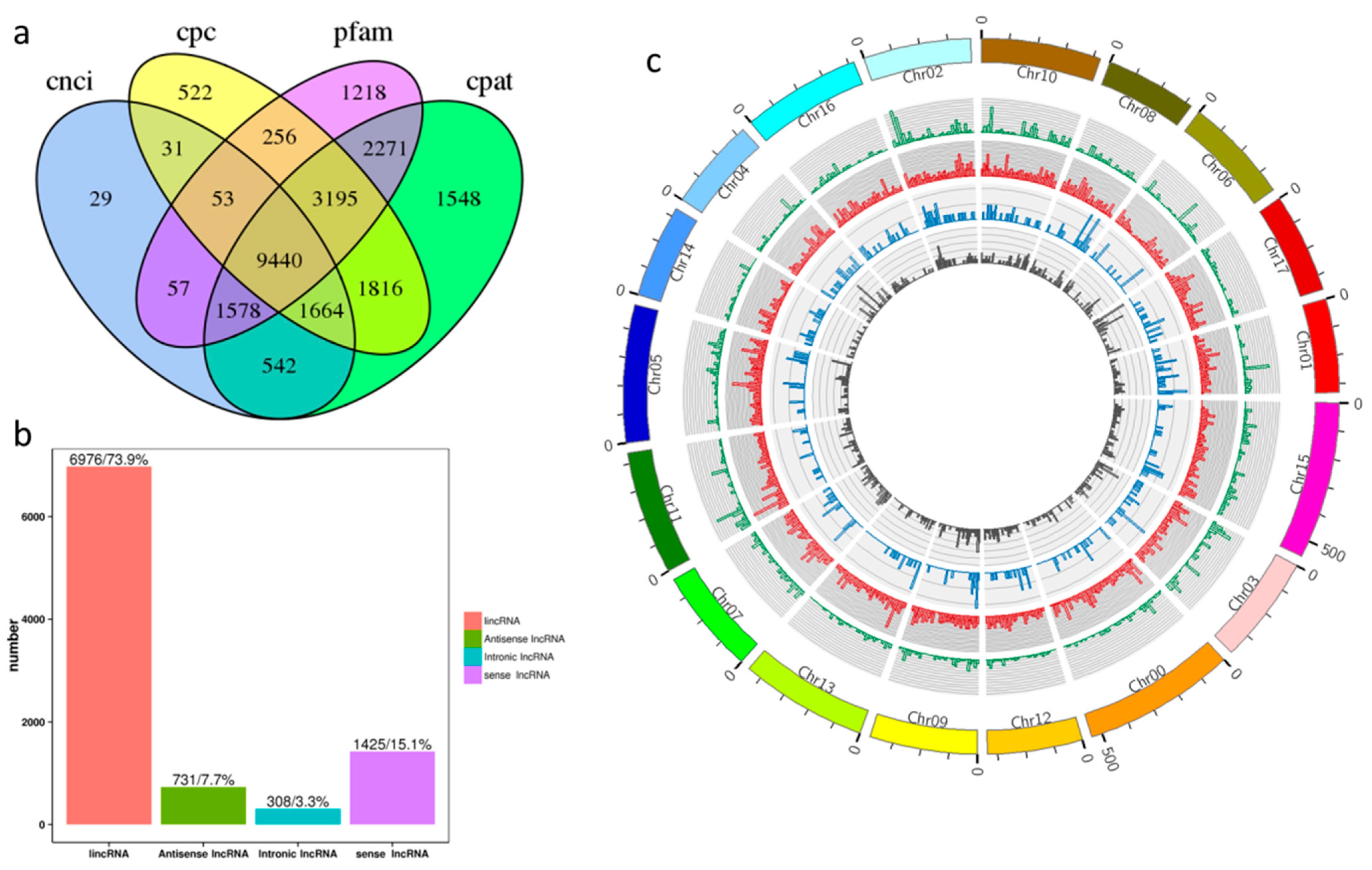
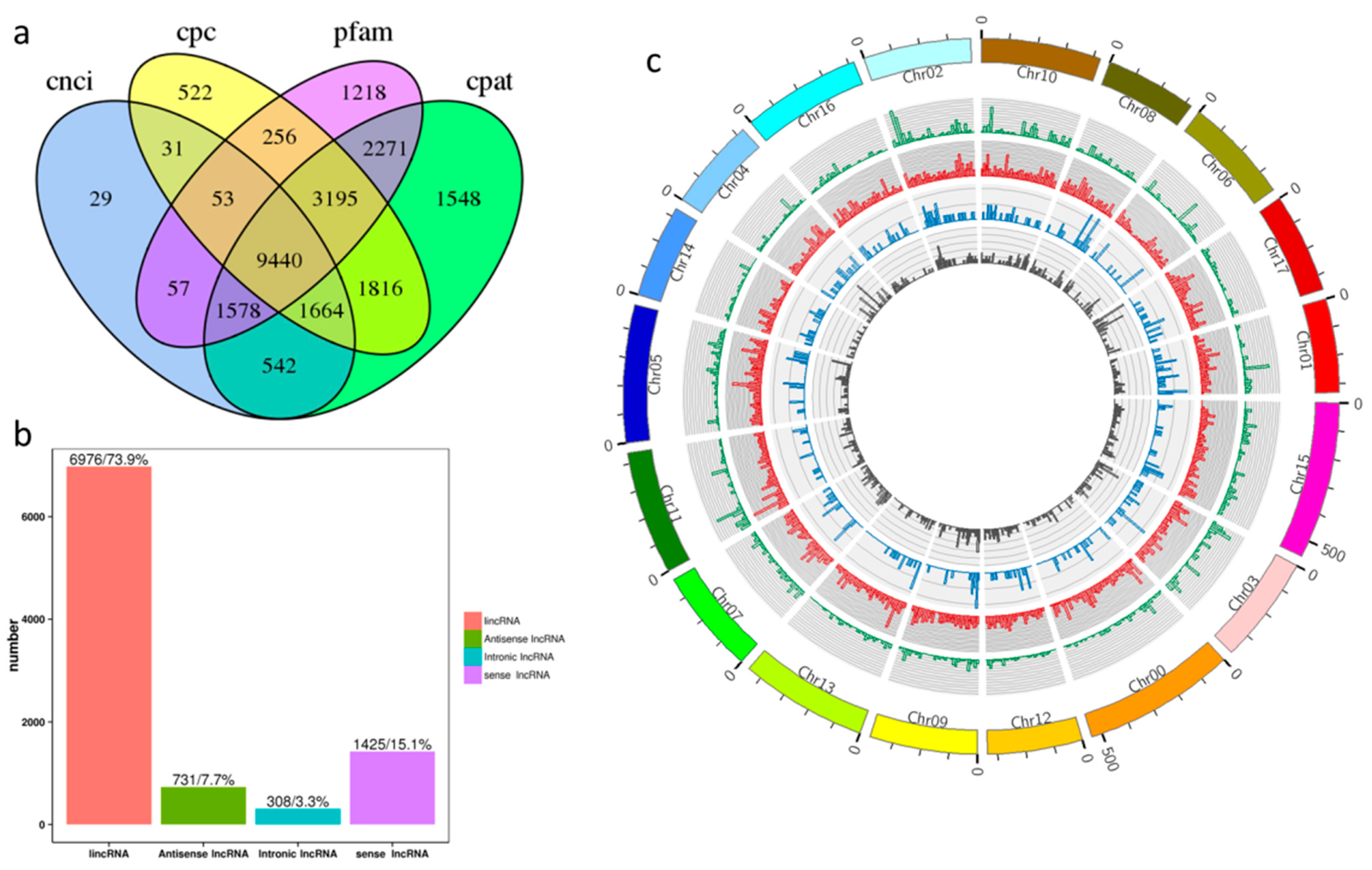
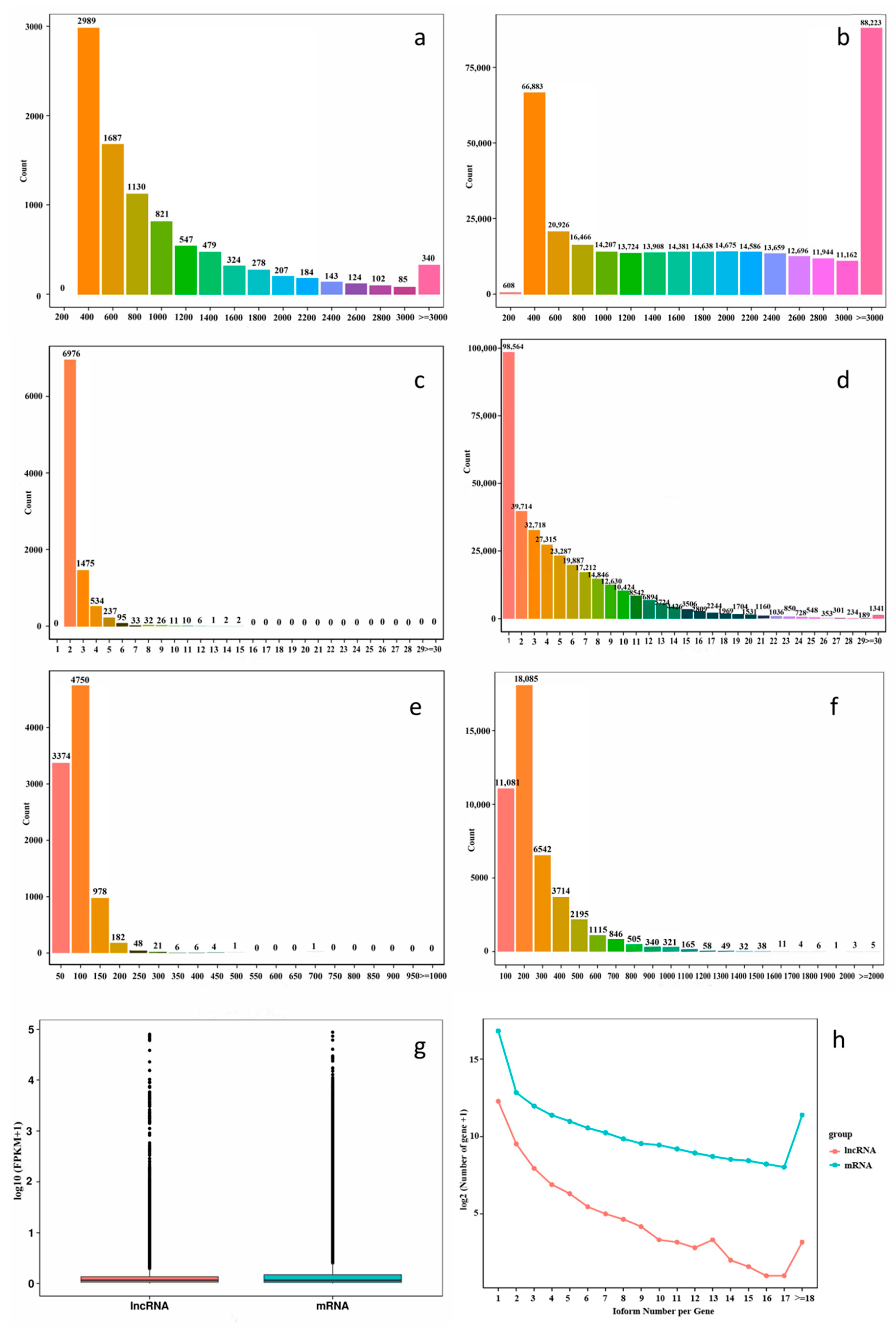
2.1. Identification and Characteristics of lncRNAs in ‘Gala’ Apples
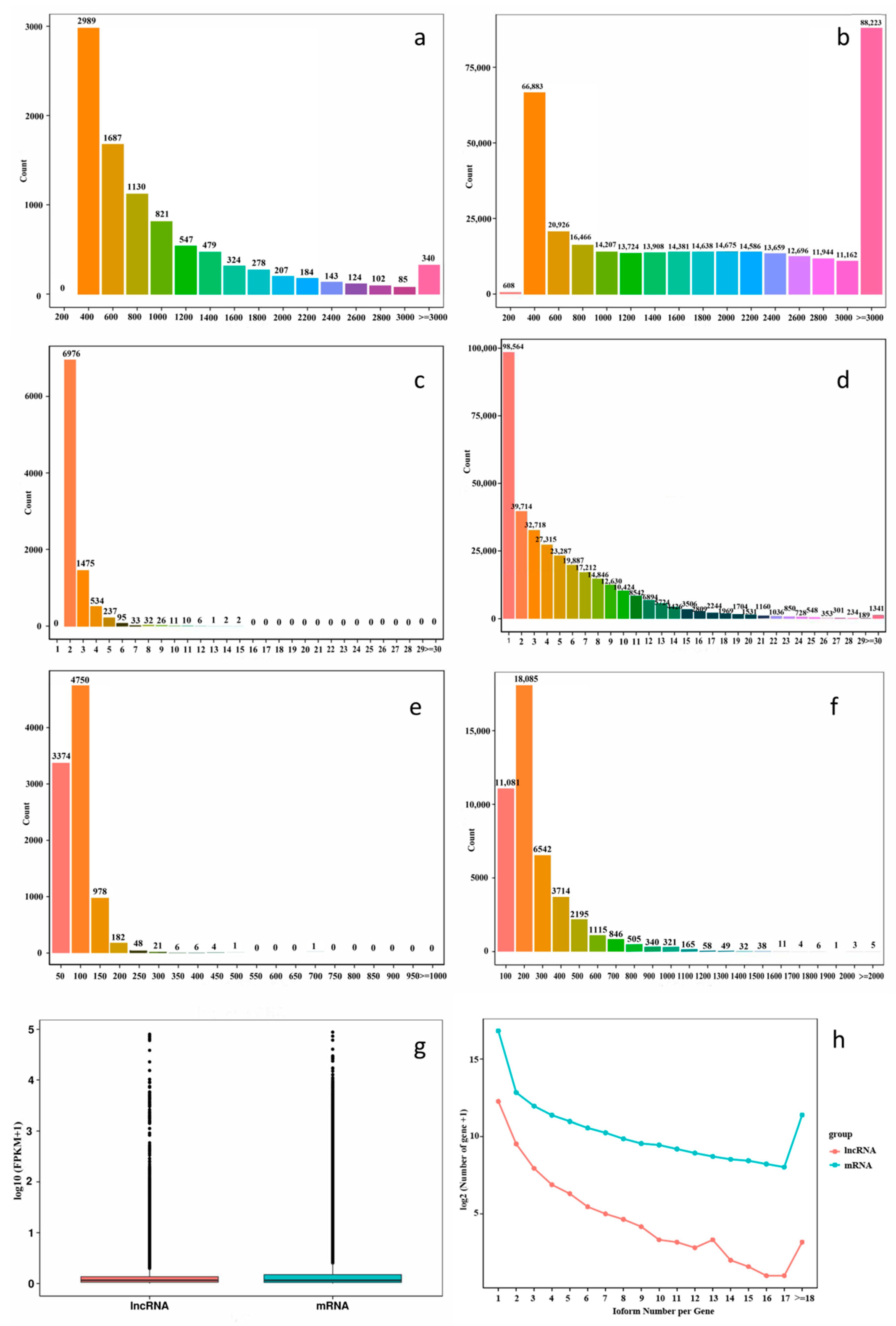
2.2. Comparative Analysis of mRNAs and lncRNAs
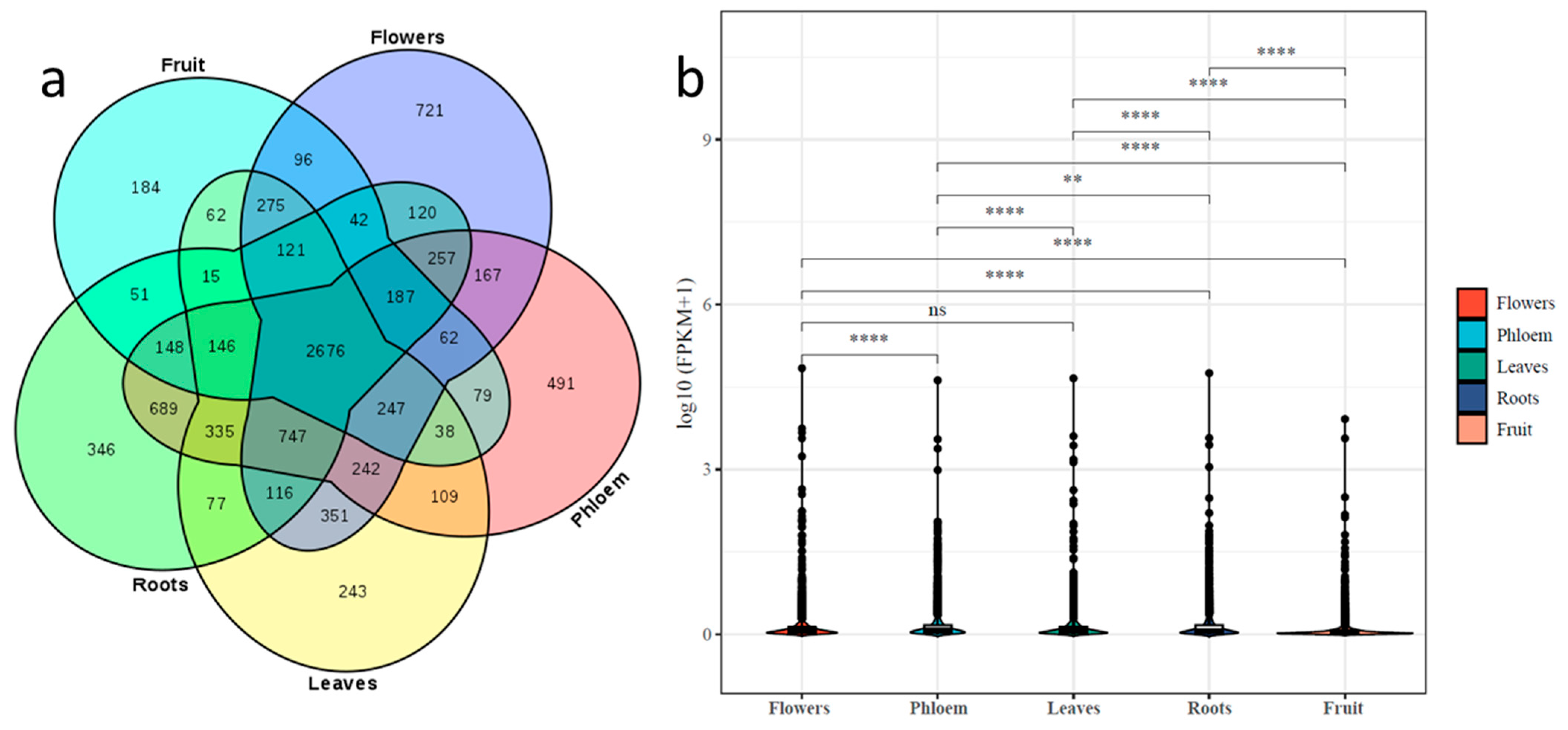
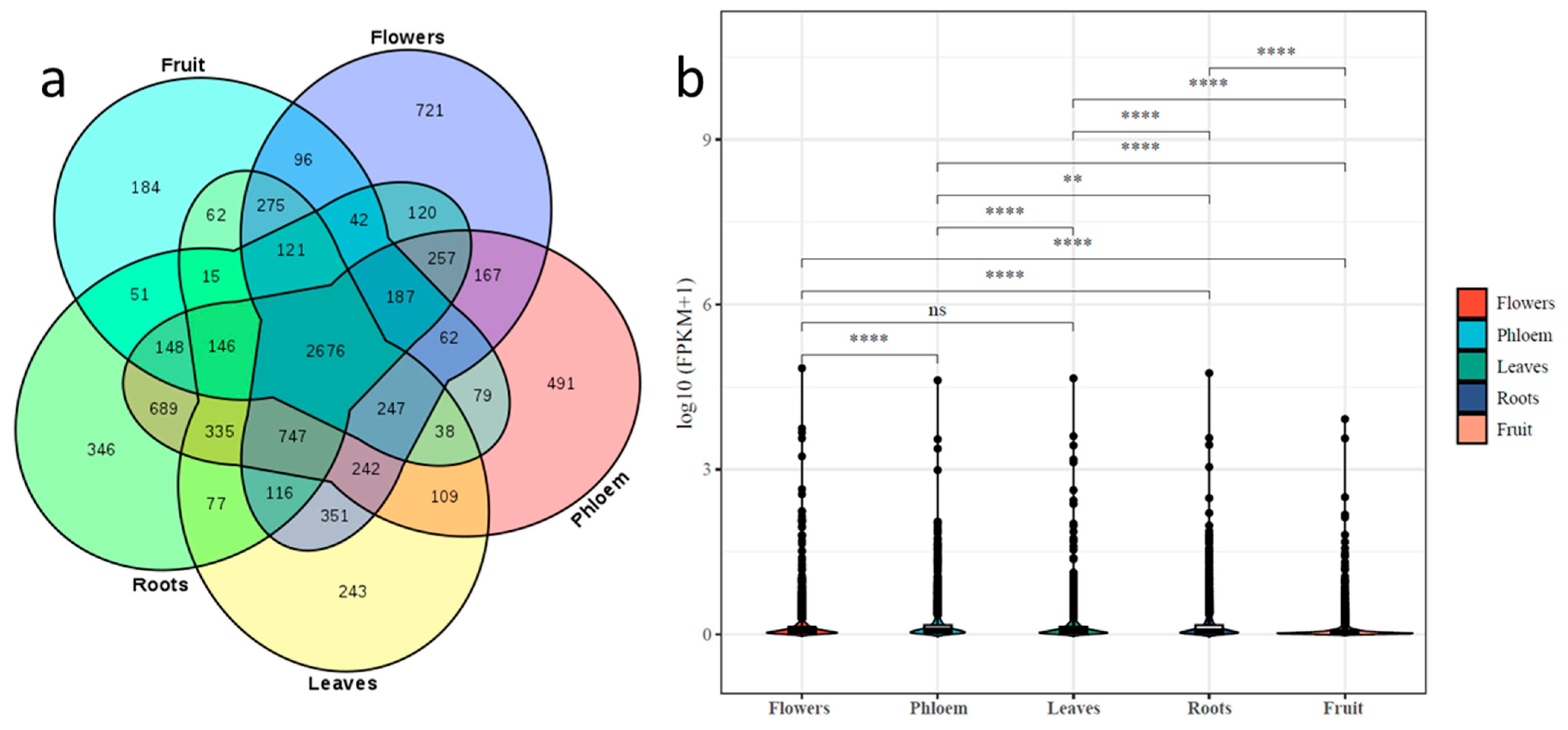
2.3. Differential Expression of lncRNAs in Tissues of ‘Gala’ Apples
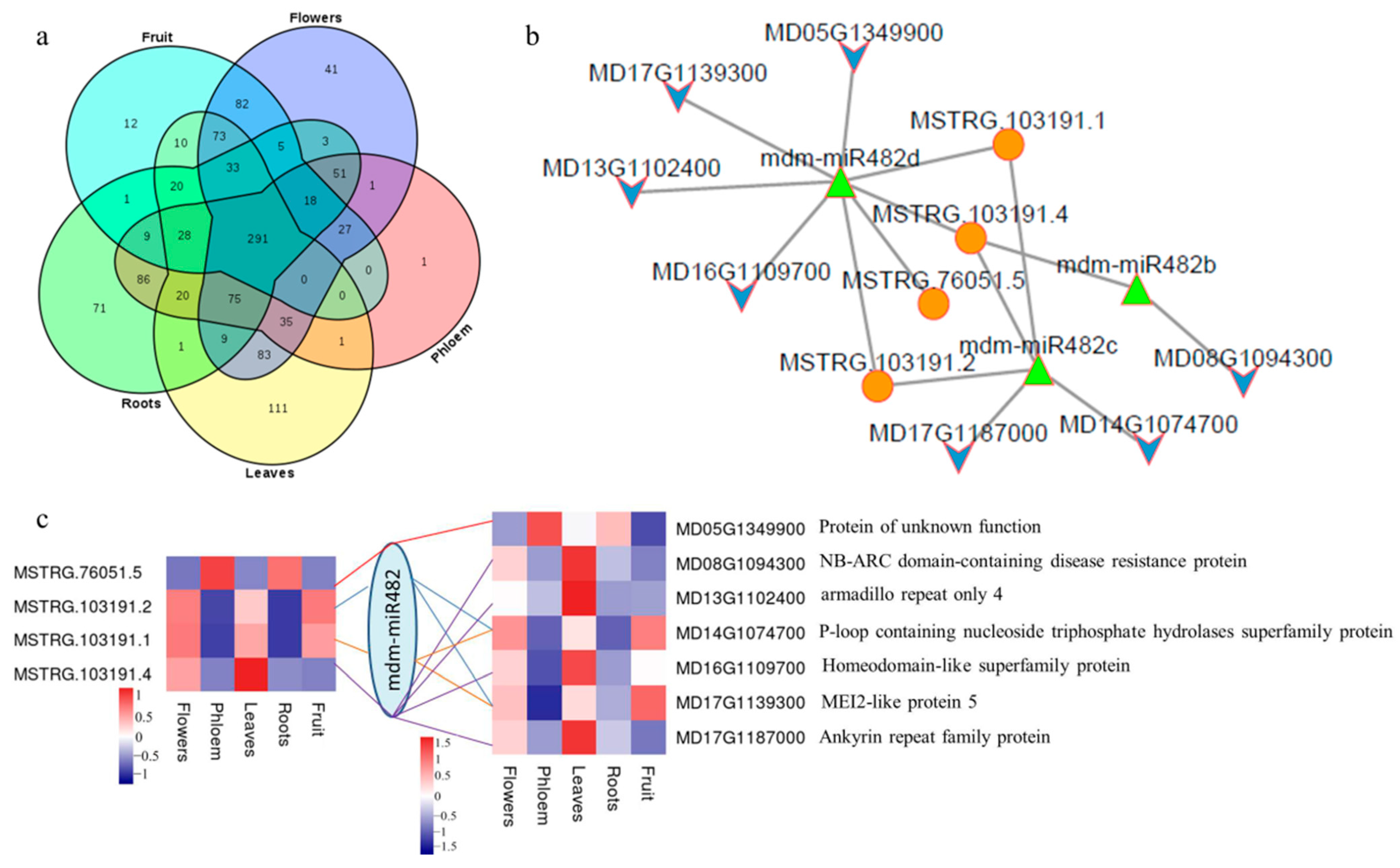
2.4. Analysis of miRNA Precursors in lncRNAs
2.5. Target Gene Prediction for lncRNAs
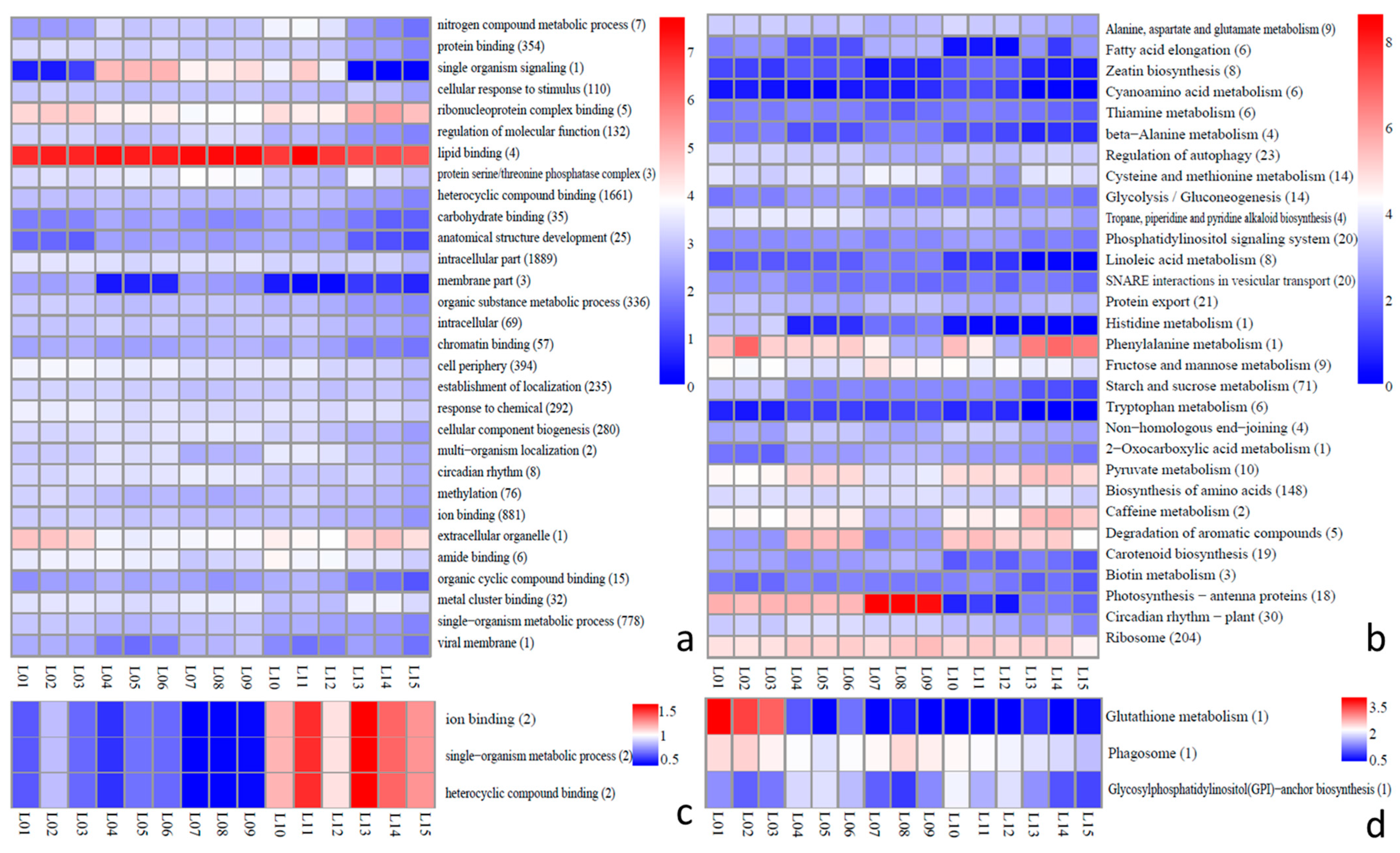
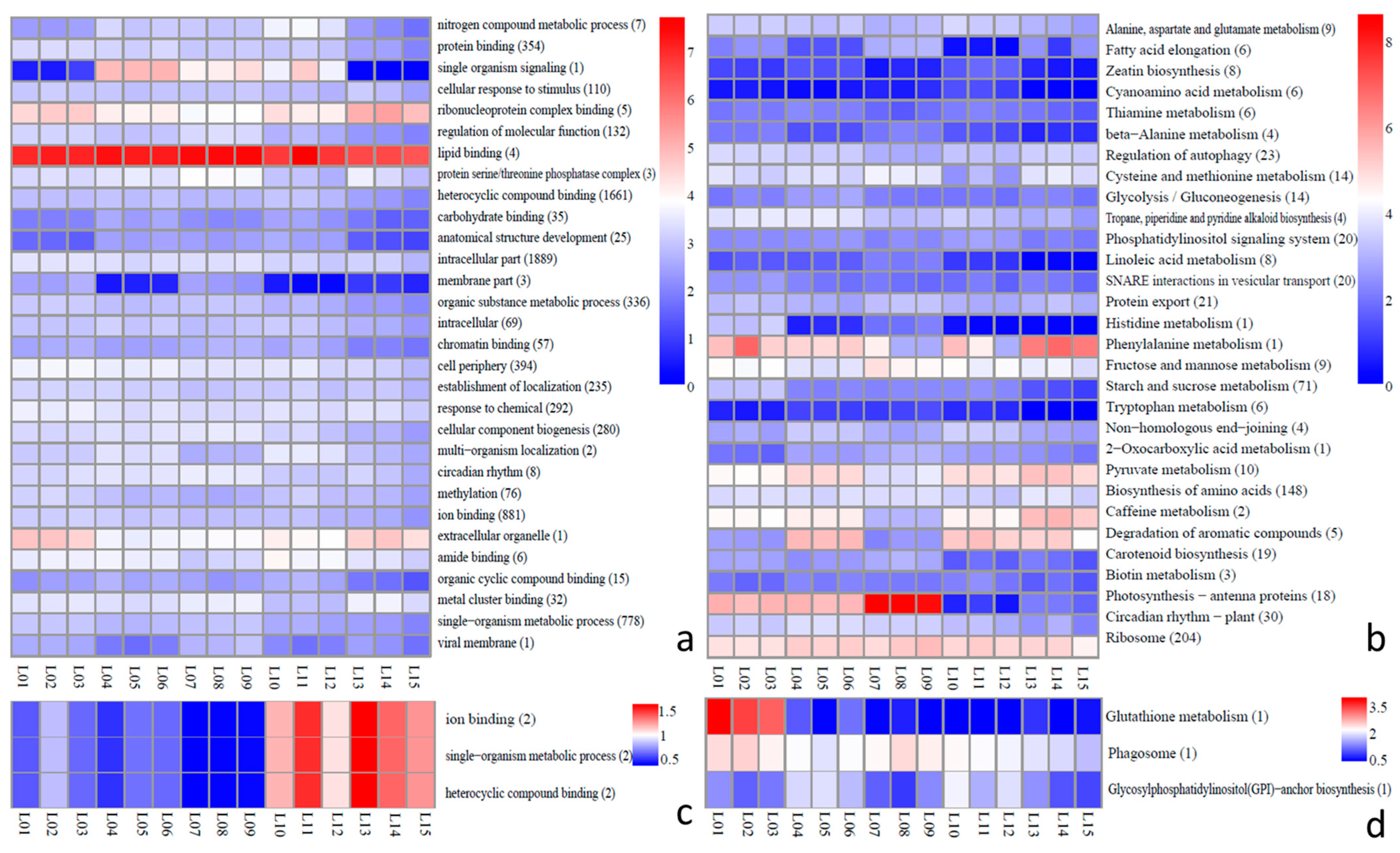
2.6. Analysis of Function of lncRNAs
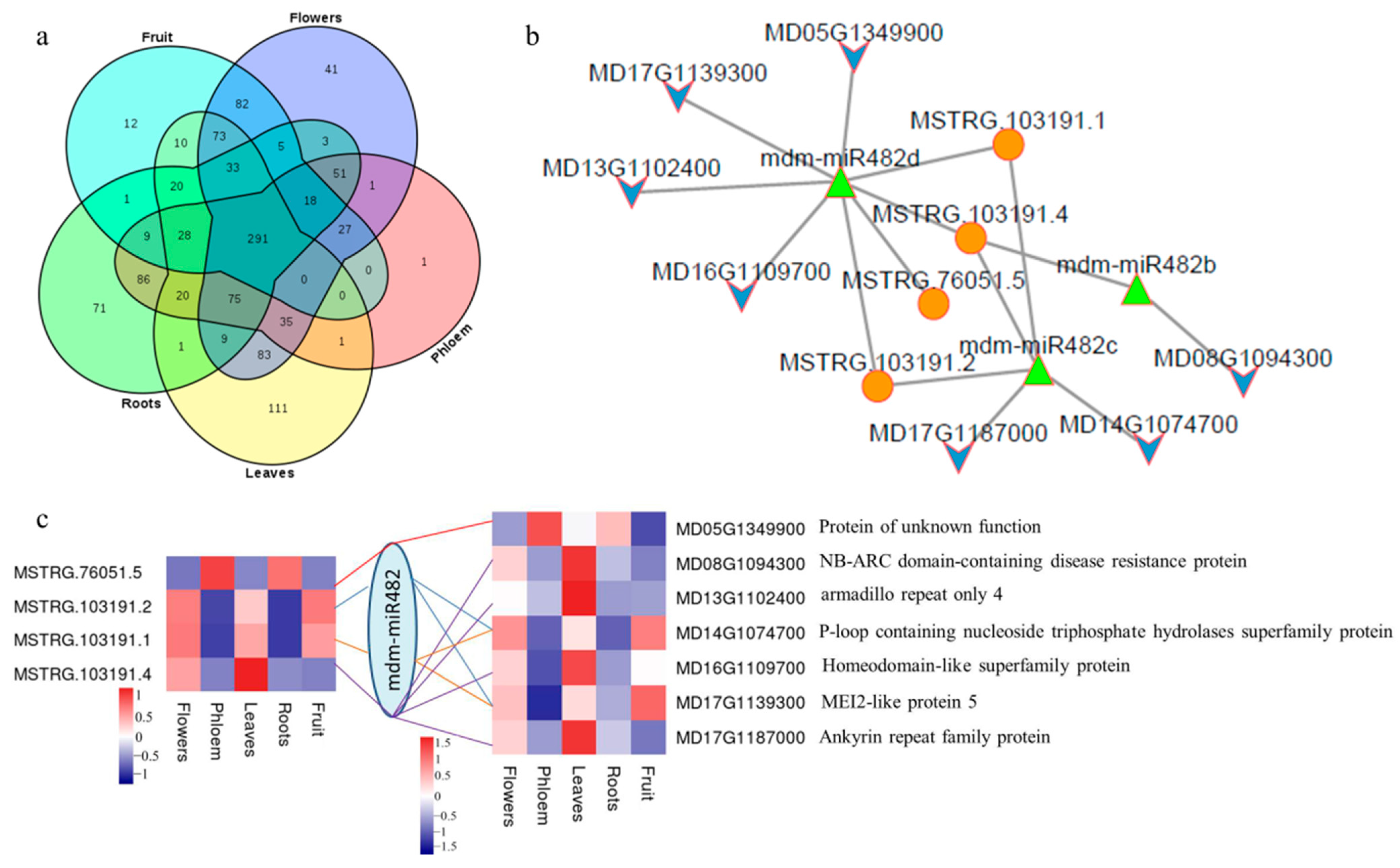
2.7. lncRNA-miRNA Interaction Network in Apples
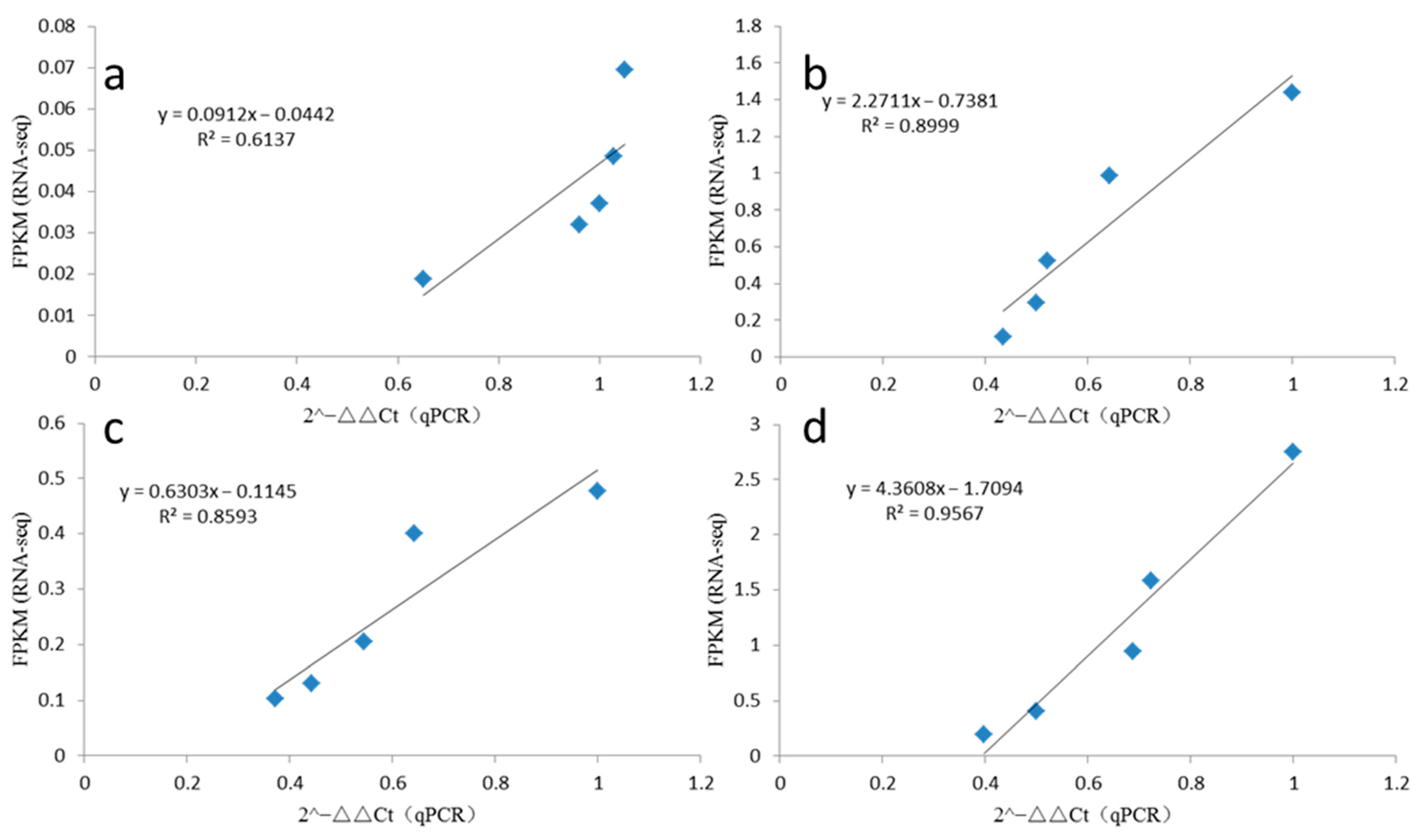
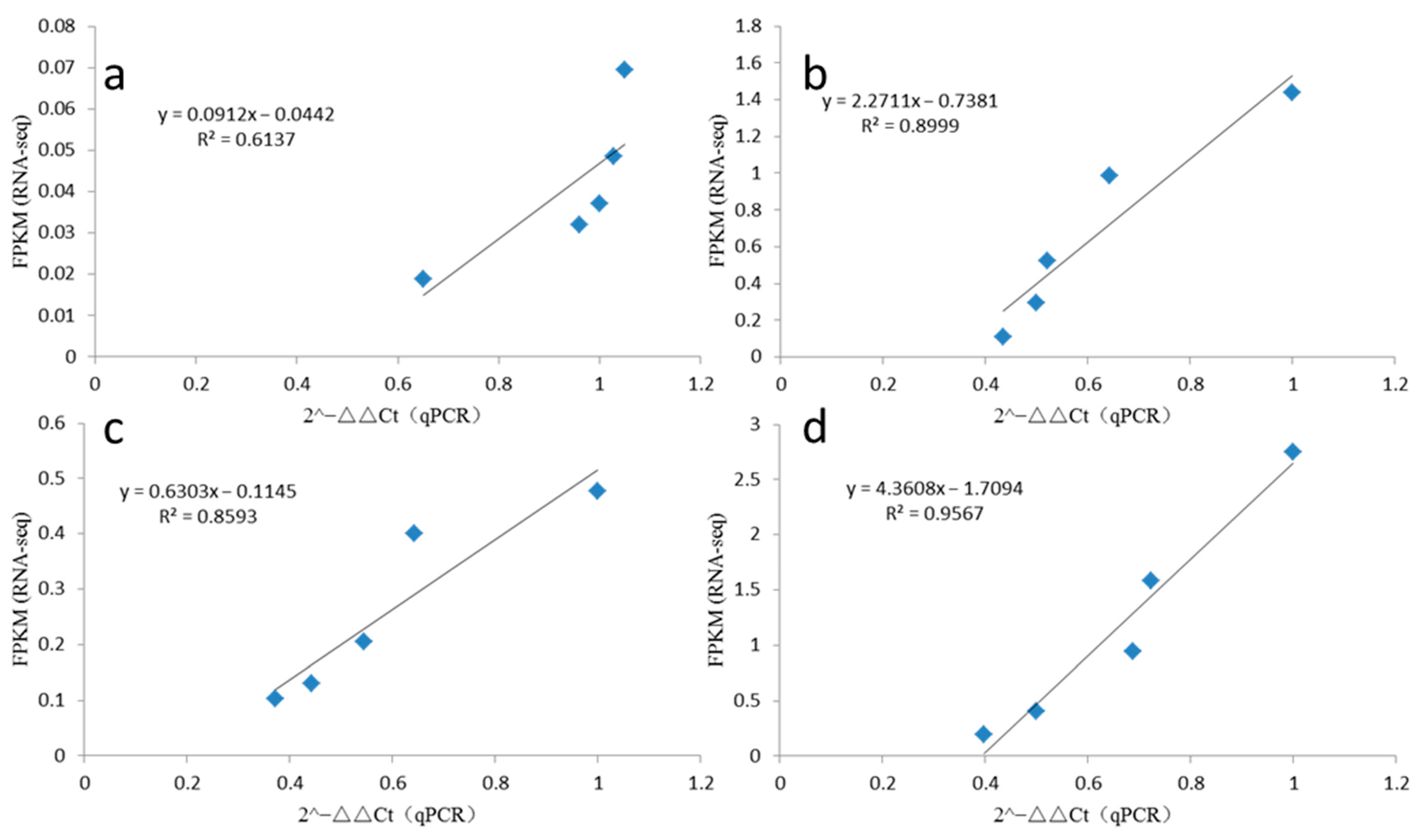
2.8. qRT-PCR Validation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Isolation, Quantification and Qualification
4.3. Library Preparation for lncRNA-Seq
4.4. Clustering and Sequencing
4.5. Quality Control
4.6. LncRNA Analysis
4.7. Quantification of Gene Expression Levels
4.8. Differential Expression Analysis
4.9. Gene Functional Annotation
- Nr (NCBI non-redundant protein sequences).
- Pfam (Protein family).
- KOG/COG (Clusters of Orthologous Groups of proteins).
- Swiss-Prot (a manually annotated and reviewed protein sequence database).
- KEGG (Kyoto Encyclopedia of Genes and Genomes).
- GO (Gene Ontology).
- GO enrichment analysis of the differentially expressed genes (DEGs) was implemented using the topGO R packages.
4.10. Prediction of miRNA Target Sites in lncRNAs
4.11. Quantitative Real-Time PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, X. Small RNAs and Their Roles in Plant Development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Liu, S.; Qi, H.; Cai, H.; Xu, M. Research Progress on Plant Long Non-Coding RNA. Plants 2020, 9, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Rinn, J. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef]
- Sun, Y.; Hao, P.; Lv, X.; Tian, J.; Wang, Y.; Zhang, X.; Xu, X.; Han, Z.; Wu, T. A long non-coding apple RNA, MSTRG.85814.11, acts as a transcriptional enhancer of SAUR32 and contributes to the Fe-deficiency response. Plant J. 2020, 103, 53–67. [Google Scholar] [CrossRef]
- Liu, D.; Mewalal, R.; Hu, R.; Tuskan, G.A.; Yang, X. New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 2017, 4, 17031. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, J.; Yang, Y.; Tan, C.; Zhu, Y.; Hu, L.; Qi, Y.; Lu, Z.J. Stress-responsive regulation of long non-coding RNA polyadenylation in Oryza sativa. Plant J. 2017, 93, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Ben Amor, B.; Wirth, S.; Merchan, F.; Laporte, P.; D’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2008, 19, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Aung, K.; Lin, S.-I.; Wu, C.-C.; Huang, Y.-T.; Su, C.-L.; Chiou, T.-J. pho2, a phosphate overaccumulator, is caused by a nonsense mutation in a microRNA399 target gene. Plant Physiol. 2006, 141, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Somoza, I.R.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Pant, B.D.; Buhtz, A.; Kehr, J.; Scheible, W.-R. MicroRNA399 is a long-distance signal for the regulation of plant phosphate homeostasis. Plant J. 2007, 53, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Bardou, F.; Ariel, F.; Simpson, C.G.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.W.; Crespi, M. Long Noncoding RNA Modulates Alternative Splicing Regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, C.C.; Rouse, D.T.; Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. The molecular basis of vernalization: The central role of FLOWERING LOCUS C (FLC). Proc. Natl. Acad. Sci. USA 2000, 97, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.-H.; Wang, M.-B. Molecular Functions of Long Non-Coding RNAs in Plants. Genes 2012, 3, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, S.; Raitskin, O.; Wu, Z.; Liu, F.; Sun, Q.; Dean, C. Functional Consequences of Splicing of the Antisense Transcript COOLAIR on FLC Transcription. Mol. Cell 2014, 54, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Campalans, A.; Kondorosi, A.; Crespi, M. Enod40, a Short Open Reading Frame–Containing mRNA, Induces Cytoplasmic Localization of a Nuclear RNA Binding Protein in Medicago truncatula. Plant Cell 2004, 16, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Dey, M.; Complainville, A.; Charon, C.; Torrizo, L.; Kondorosi, A.; Crespi, M.; Datta, S. Phytohormonal responses in enod40-overexpressing plants of Medicago truncatula and rice. Physiol. Plant. 2004, 120, 132–139. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Chua, N.-H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.-D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigó, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Karlik, E.; Gözükırmızı, N. Evaluation of Barley lncRNAs Expression Analysis in Salinity Stress. Russ. J. Genet. 2018, 54, 198–204. [Google Scholar] [CrossRef]
- Sun, Q.; Csorba, T.; Skourti-Stathaki, K.; Proudfoot, N.J.; Dean, C. R-Loop Stabilization Represses Antisense Transcription at the Arabidopsis FLC Locus. Science 2013, 340, 619–621. [Google Scholar] [CrossRef] [Green Version]
- Ariel, F.; Jegu, T.; Latrasse, D.; Romero-Barrios, N.; Christ, A.; Benhamed, M.; Crespi, M. Noncoding Transcription by Alternative RNA Polymerases Dynamically Regulates an Auxin-Driven Chromatin Loop. Mol. Cell 2014, 55, 383–396. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Liu, Q.; Li, J.; Jiang, D.; Zhou, L.; Wu, P.; Lu, S.; Li, F.; Zhu, L.; Liu, Z.; et al. Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012, 22, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating Protein-Coding and Noncoding RNA: Challenges and Ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Mujahid, H.; Hou, Y.; Nallamilli, B.R.; Peng, Z. Plant Long ncRNAs: A New Frontier for Gene Regulatory Control. Am. J. Plant Sci. 2013, 4, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Li, S.; Cui, X.; Zheng, Y. A comprehensive review of web-based resources of non-coding RNAs for plant science research. Int. J. Biol. Sci. 2018, 14, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Ausin, I.; Greenberg, M.V.C.; Simanshu, D.K.; Hale, C.J.; Vashisht, A.A.; Simon, S.A.; Lee, T.-F.; Feng, S.; Española, S.D.; Meyers, B.C.; et al. Involved in de NOVO 2-containing complex involved in RNA-directed DNA methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 8374–8381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.-T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Yang, Y.; Li, R.; Fu, D.; Wen, L.; Luo, Y.; Zhu, H. RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening. J. Exp. Bot. 2015, 66, 4483–4495. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-Wide Analysis Uncovers Regulation of Long Intergenic Noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Ma, H.; Zhang, J.; Wu, T.; Song, T.; Tian, J.; Yao, Y. Systematic identification of long noncoding RNA s expressed during light-induced anthocyanin accumulation in apple fruit. Plant J. 2019, 100, 572–590. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Z.; Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crop. Prod. 2018, 118, 334–346. [Google Scholar] [CrossRef]
- Liu, S.; Wu, L.; Qi, H.; Xu, M. LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crop. Prod. 2019, 133, 333–347. [Google Scholar] [CrossRef]
- Wierzbicki, A.T. The role of long non-coding RNA in transcriptional gene silencing. Curr. Opin. Plant Biol. 2012, 15, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.E.; Guenzl, P.M.; Barlow, D.P.; Pauler, F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhao, H.; Cui, P.; Albesher, N.; Xiong, L. A Nucleus-Localized Long Non-Coding RNA Enhances Drought and Salt Stress Tolerance. Plant Physiol. 2017, 175, 1321–1336. [Google Scholar] [CrossRef] [Green Version]
- Depège, N.; Bellafiore, S.; Rochaix, J.-D. Role of Chloroplast Protein Kinase Stt7 in LHCII Phosphorylation and State Transition in Chlamydomonas. Science 2003, 299, 1572–1575. [Google Scholar] [CrossRef] [Green Version]
- Dewir, Y.H.; Paek, K.Y. The control of in vitro flowering and association of glutathione metabolism. In Plant Tissue Culture and Applied Plant Biotechnology; Kumar, A., Roy, S., Eds.; Aavishkar Publishers: Jaipur, India, 2011; pp. 77–108. [Google Scholar]
- Kang, C.; Liu, Z. Global identification and analysis of long non-coding RNAs in diploid strawberry Fragaria vesca during flower and fruit development. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Galvão, V.C.; Zhang, Y.-C.; Horrer, D.; Zhang, T.-Q.; Hao, Y.-H.; Feng, Y.-Q.; Wang, S.; Schmid, M.; Wang, J.-W. Gibberellin Regulates the Arabidopsis Floral Transition through miR156-Targeted SQUAMOSA PROMOTER BINDING–LIKE Transcription Factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Wang, Z.-M.; Wang, M.; Wang, X.-J. Widespread Long Noncoding RNAs as Endogenous Target Mimics for MicroRNAs in Plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.L.; Li, Y.X.; Zhang, J.; Zhang, J.; Yao, Y.C. Cloning and functional assay of McmiR160 regulated leaf coloration in Malus spp. J. Beijing Univ. Agric. 2019, 34, 14–19. (In Chinese) [Google Scholar]
- Aung, B.; Gao, R.; Gruber, M.Y.; Yuan, Z.-C.; Sumarah, M.; Hannoufa, A. MsmiR156 affects global gene expression and promotes root regenerative capacity and nitrogen fixation activity in alfalfa. Transgenic Res. 2017, 26, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, K.; Xu, Z.; Liao, P.; Zhang, X.; Liu, L.; Wei, K.; Liu, D.; Li, Y.-F.; Sunkar, R.; et al. Small RNA profiles from Panax notoginseng roots differing in sizes reveal correlation between miR156 abundances and root biomass levels. Sci. Rep. 2017, 7, 9418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera-Rojas, C.H.; Rocha, G.H.B.; Polverari, L.; Brito, D.A.P.; Batista, D.S.; Notini, M.M.; Da Cruz, A.C.F.; Morea, E.G.O.; Sabatini, S.; Otoni, W.C.; et al. miR156-targeted SPL10 controls Arabidopsis root meristem activity and root-derived de novo shoot regeneration via cytokinin responses. J. Exp. Bot. 2019, 71, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Shao, C.; Wang, H.; Jin, Y. Target mimics: An embedded layer of microRNA-involved gene regulatory networks in plants. BMC Genom. 2012, 13, 197. [Google Scholar] [CrossRef] [Green Version]
- He, J.-H.; Han, Z.-P.; Zou, M.-X.; Wang, L.; Lv, Y.B.; Bin Zhou, J.; Cao, M.-R.; Li, Y.-G. Analyzing the LncRNA, miRNA, and mRNA Regulatory Network in Prostate Cancer with Bioinformatics Software. J. Comput. Biol. 2018, 25, 146–157. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, D.; Zhang, T.; Duan, A.; Zhang, J.; He, C. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Res. 2018, 25, 465–476. [Google Scholar] [CrossRef]
- Datta, R.; Paul, S. Long non-coding RNAs: Fine-tuning the developmental responses in plants. J. Biosci. 2019, 44, 77. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.C.; Asahina, M.; Liu, P.-P.; Kristof, J.R.; Coppersmith, J.L.; Pluskota, W.E.; Bassel, G.W.; Goloviznina, N.A.; Nguyen, T.T.; Martínez-Andújar, C.; et al. The microRNA156 and microRNA172 gene regulation cascades at post-germinative stages in Arabidopsis. Seed Sci. Res. 2010, 20, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.-G.; Shan, J.-X.; Shi, M.; Gao, J.-P.; Lin, H.-X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef]
- Lei, K.-J.; Lin, Y.-M.; Ren, J.; Bai, L.; Miao, Y.-C.; An, G.-Y.; Song, C.-P. Modulation of the Phosphate-Deficient Responses by MicroRNA156 and its Targeted squamosa promoter binding protein-like 3 in Arabidopsis. Plant Cell Physiol. 2015, 57, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.J.; Lin, Y.M.; An, G.Y. miR156 modulates rhizosphere acidification in response to phosphate limitation in Arabidopsis. J. Plant Res. 2015, 129, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Z.; Zhang, J.; Zhang, Y.; Han, Q.; Hu, T.; Xu, X.; Liu, H.; Li, H.; Ye, Z. Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett. 2010, 585, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- e Silva, G.F.F.; Silva, E.M.; Azevedo, M.D.S.; Guivin, M.A.C.; Ramiro, D.A.; Figueiredo, C.R.; Carrer, H.; Peres, L.E.P.; Nogueira, F.T.S. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014, 78, 604–618. [Google Scholar] [CrossRef]
- Li, K.; Wei, Y.-H.; Wang, R.-H.; Mao, J.-P.; Tian, H.-Y.; Chen, S.-Y.; Li, S.-H.; Tahir, M.-M.; Zhang, D. Mdm-MIR393b-mediated adventitious root formation by targeted regulation of MdTIR1A expression and weakened sensitivity to auxin in apple rootstock. Plant Sci. 2021, 308, 110909. [Google Scholar] [CrossRef]
- Li, X.; Chen, P.; Xie, Y.; Yan, Y.; Wang, L.; Dang, H.; Zhang, J.; Xu, L.; Ma, F.; Guan, Q. Apple SERRATE negatively mediates drought resistance by regulating MdMYB88 and MdMYB124 and microRNA biogenesis. Hortic. Res. 2020, 7, 1–11. [Google Scholar] [CrossRef]
- Xue, T.; Liu, Z.; Dai, X.; Xiang, F. Primary root growth in Arabidopsis thaliana is inhibited by the miR159 mediated repression of MYB33, MYB65 and MYB101. Plant Sci. 2017, 262, 182–189. [Google Scholar] [CrossRef]
- Matthewman, C.A.; Kawashima, C.G.; Huska, D.; Csorba, T.; Dalmay, T.; Kopriva, S. miR395 is a general component of the sulfate assimilation regulatory network in Arabidopsis. FEBS Lett. 2012, 586, 3242–3248. [Google Scholar] [CrossRef]
- Ferdous, J.; Whitford, R.; Nguyen, M.; Brien, C.; Langridge, P.; Tricker, P.J. Drought-inducible expression of Hv-miR827 enhances drought tolerance in transgenic barley. Funct. Integr. Genom. 2016, 17, 279–292. [Google Scholar] [CrossRef]
- Xu, J.; Li, C.; Cui, L.; Zhang, T.; Wu, Z.; Zhu, P.-Y.; Meng, Y.-J.; Zhang, K.-J.; Yu, X.-Q.; Lou, Q.-F.; et al. New insights into the roles of cucumber TIR1 homologs and miR393 in regulating fruit/seed set development and leaf morphogenesis. BMC Plant Biol. 2017, 17, 130. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Xu, H.F.; Wang, N.; Jiang, S.H.; Liu, J.X.; Wang, D.Y.; Zuo, W.F.; Chen, X.S. Molecular cloning and expression analysis of an auxin signaling related gene MdARF3 in red flesh apple. Acta Hortic. Sin. 2017, 44, 633–643. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.L.; Fang, Z.W.; Sun, C.; Zhang, P.; Zhang, X.; Lu, C.; Wang, S.P.; Ma, D.F.; Zhu, Y.X. Rapid identification of a stripe rust resistant gene in a space-induced wheat mutant using specific locus amplified fragment (SLAF) sequencing. Sci. Rep. 2003, 8, 3086. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Gao, Y.; Sun, S.; Li, L.; Wang, K. Expression Profiles and Characteristics of Apple lncRNAs in Roots, Phloem, Leaves, Flowers, and Fruit. Int. J. Mol. Sci. 2022, 23, 5931. https://doi.org/10.3390/ijms23115931
Wang D, Gao Y, Sun S, Li L, Wang K. Expression Profiles and Characteristics of Apple lncRNAs in Roots, Phloem, Leaves, Flowers, and Fruit. International Journal of Molecular Sciences. 2022; 23(11):5931. https://doi.org/10.3390/ijms23115931
Chicago/Turabian StyleWang, Dajiang, Yuan Gao, Simiao Sun, Lianwen Li, and Kun Wang. 2022. "Expression Profiles and Characteristics of Apple lncRNAs in Roots, Phloem, Leaves, Flowers, and Fruit" International Journal of Molecular Sciences 23, no. 11: 5931. https://doi.org/10.3390/ijms23115931
APA StyleWang, D., Gao, Y., Sun, S., Li, L., & Wang, K. (2022). Expression Profiles and Characteristics of Apple lncRNAs in Roots, Phloem, Leaves, Flowers, and Fruit. International Journal of Molecular Sciences, 23(11), 5931. https://doi.org/10.3390/ijms23115931