
Optimized Heterologous Expression and Efficient Purification of a New TRAIL-Based Antitumor Fusion Protein SRH–DR5-B with Dual VEGFR2 and DR5 Receptor Specificity

 ,
,  ,
,  ,
,
Abstract
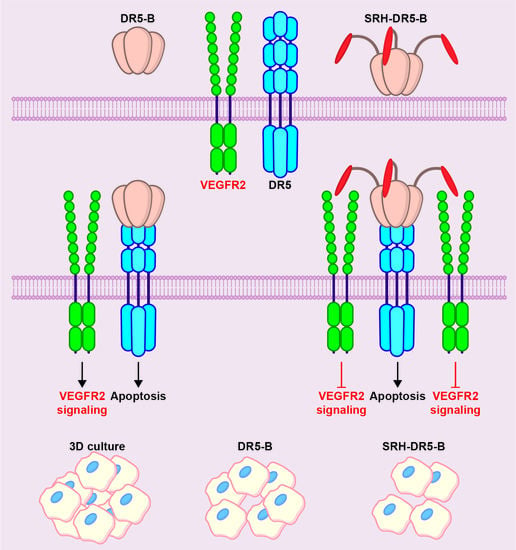
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
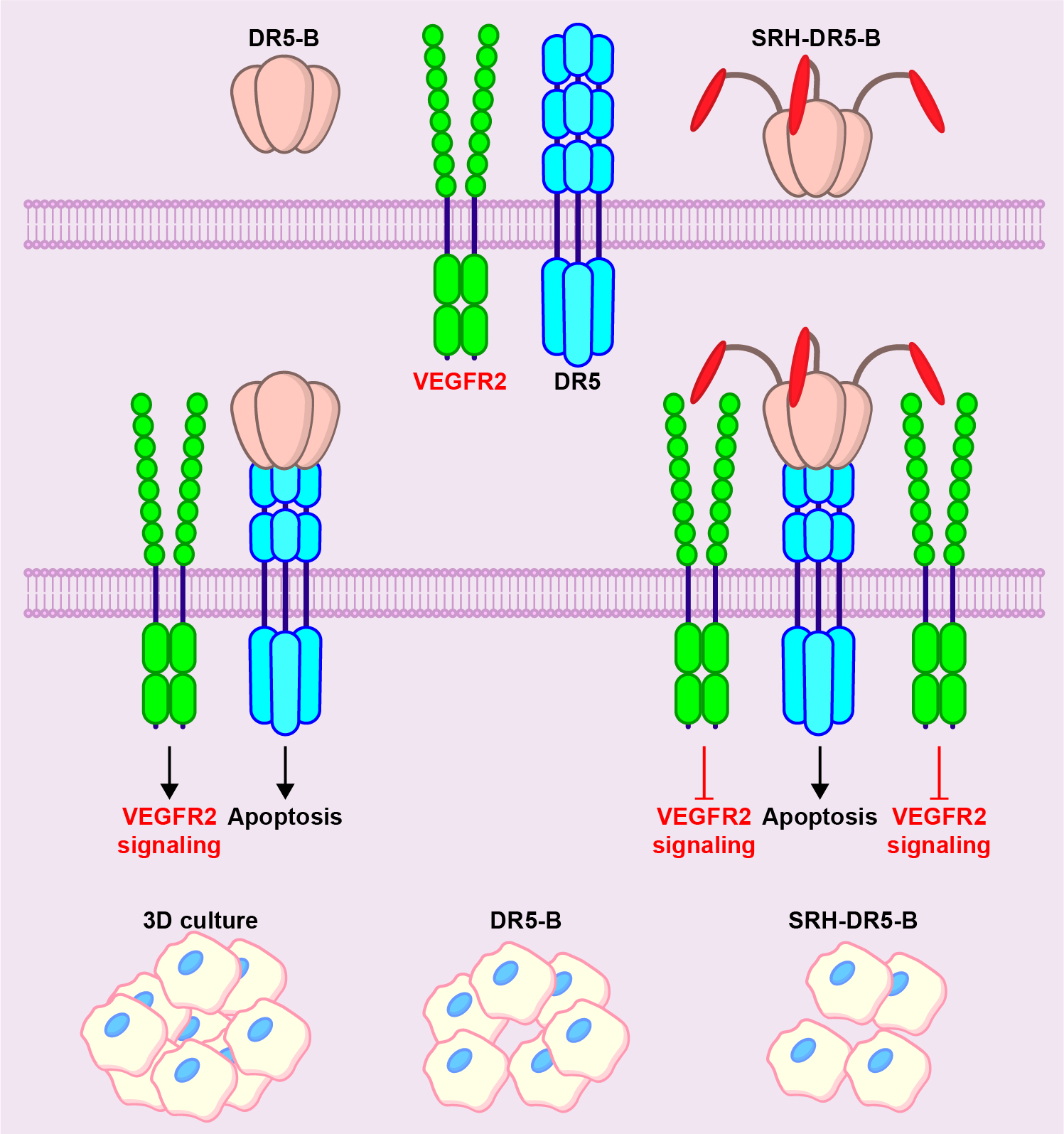
2.1. Heterologous Expression of HRH–DR5-B Fusion in Escherichia coli Strains
2.2. His/Ser Substitution at P2 Position after N-terminal Met Prevents the Degradation of HRH–DR5-B
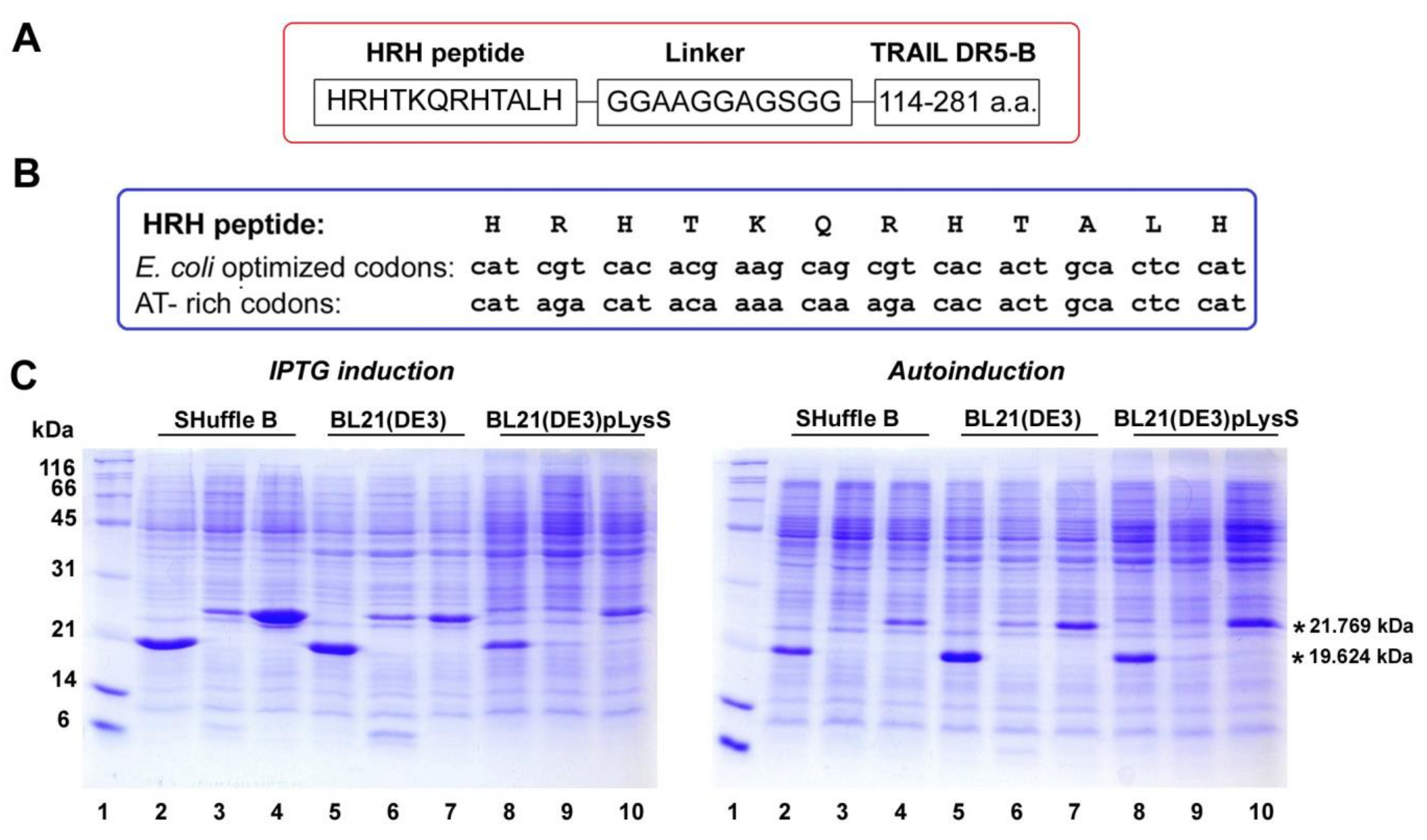
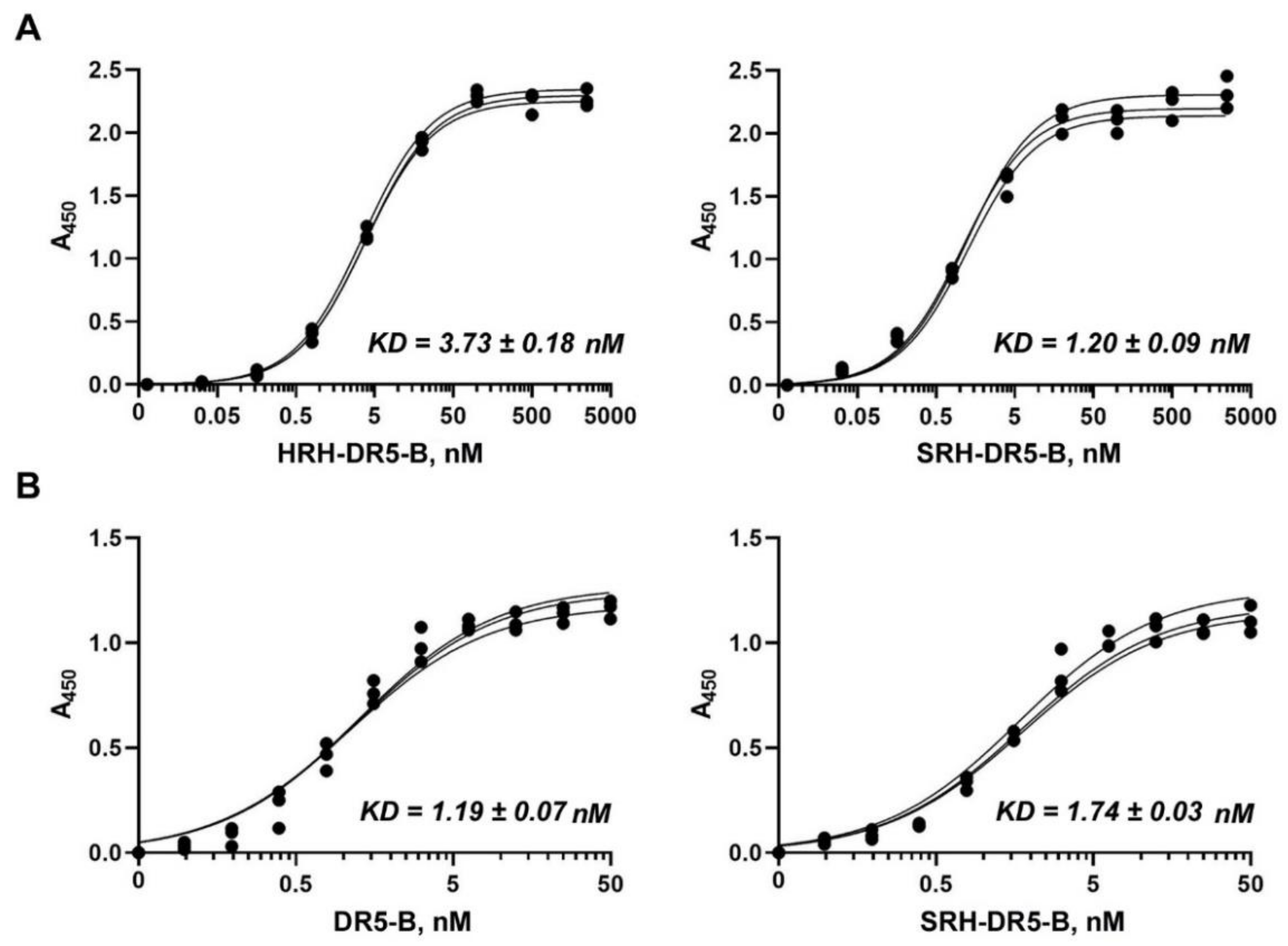
2.3. SRH–DR5-B Showed High Affinity for DR5 and VEGFR2 Receptors
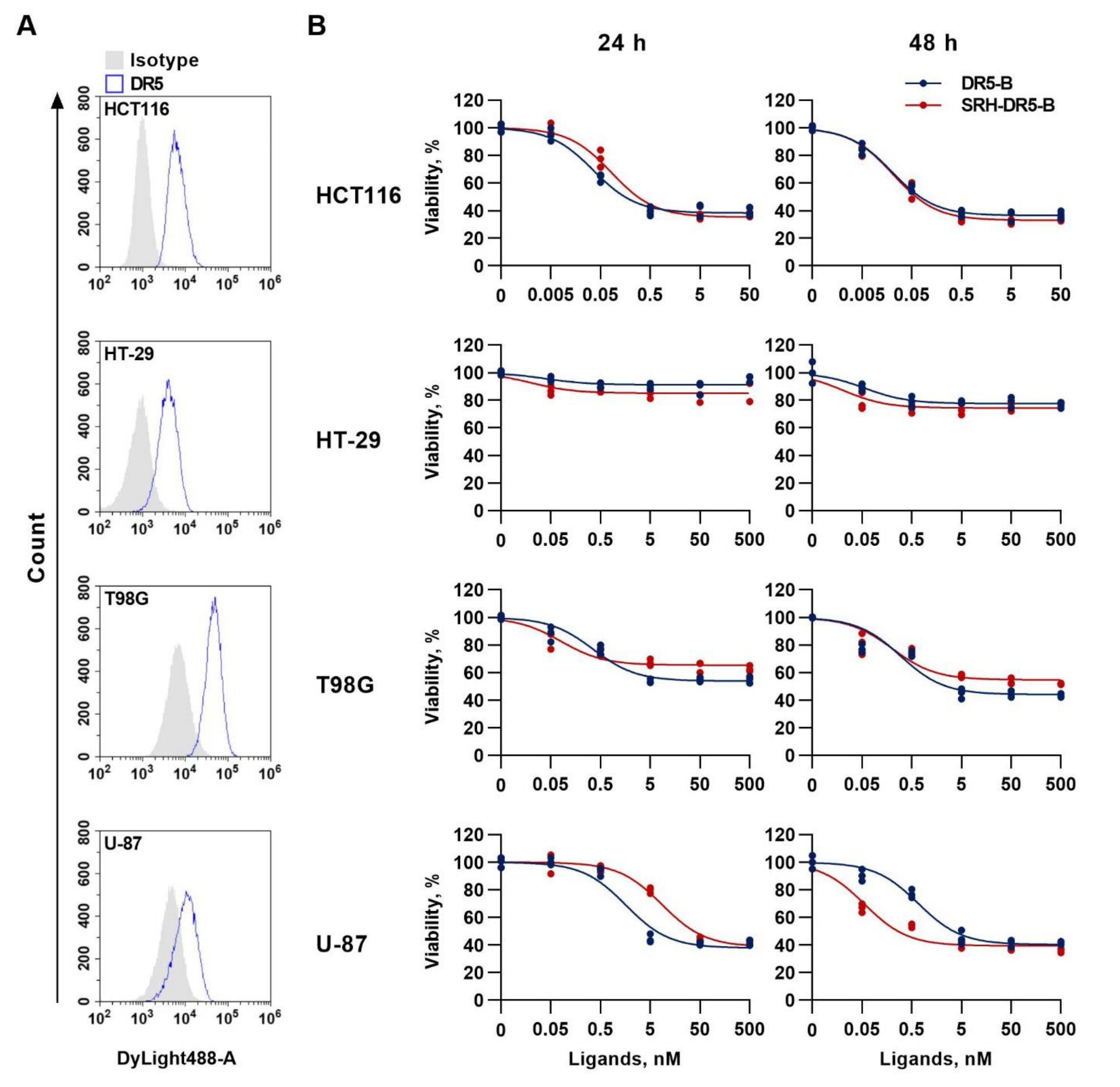
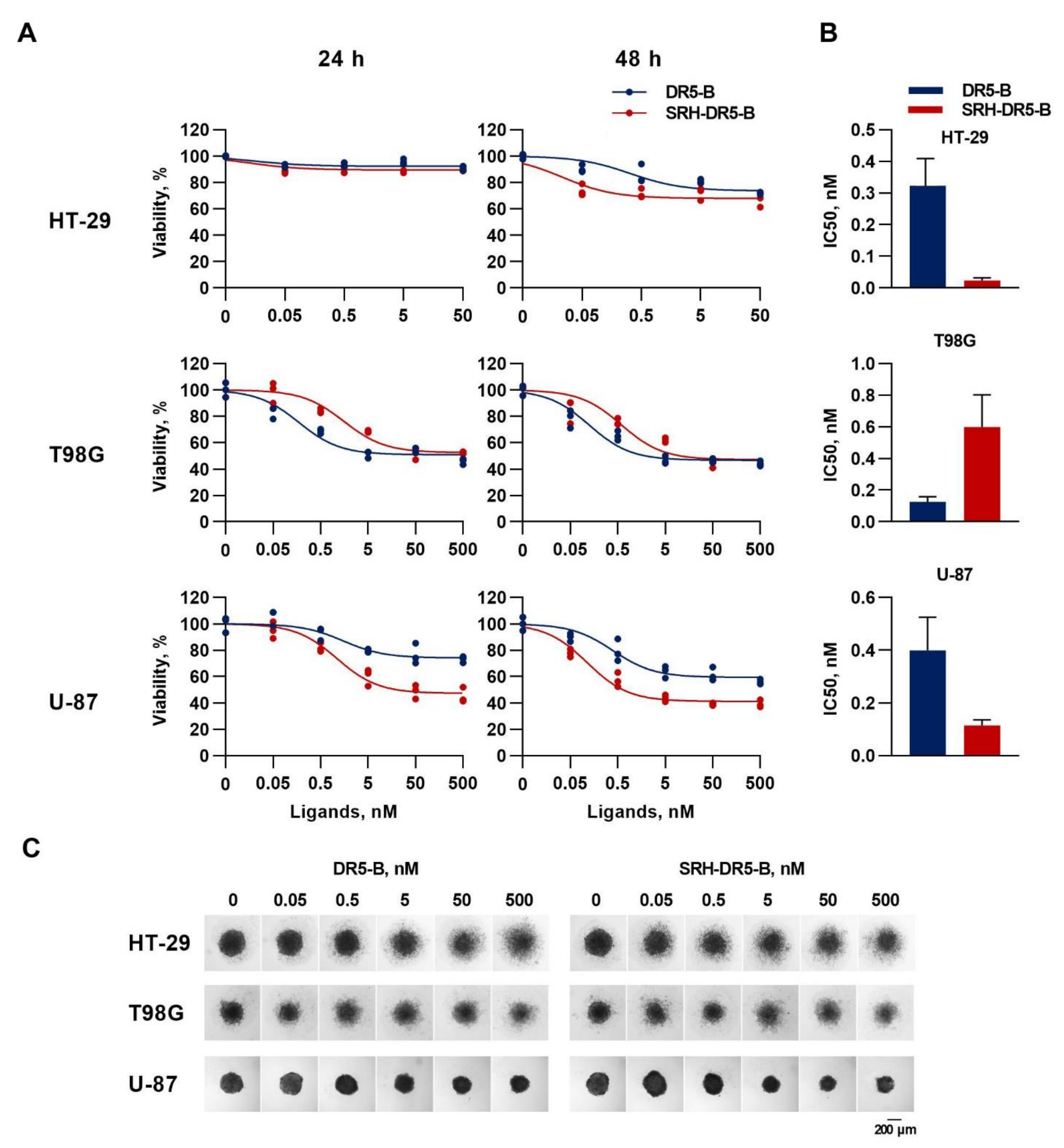
2.4. The Cytotoxic Activity of SRH–DR5-B in 2D and 3D Tumor Cell Models
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Lines
4.2. Construction of Plasmid Vectors for the Expression of HRH–DR5-B and SRH–DR5-B Fusions
4.3. Expression of Recombinant Proteins in E. coli Strains
4.4. Purification of Recombinant Proteins DR5-B, HRH–DR5-B and SRH–DR5-B
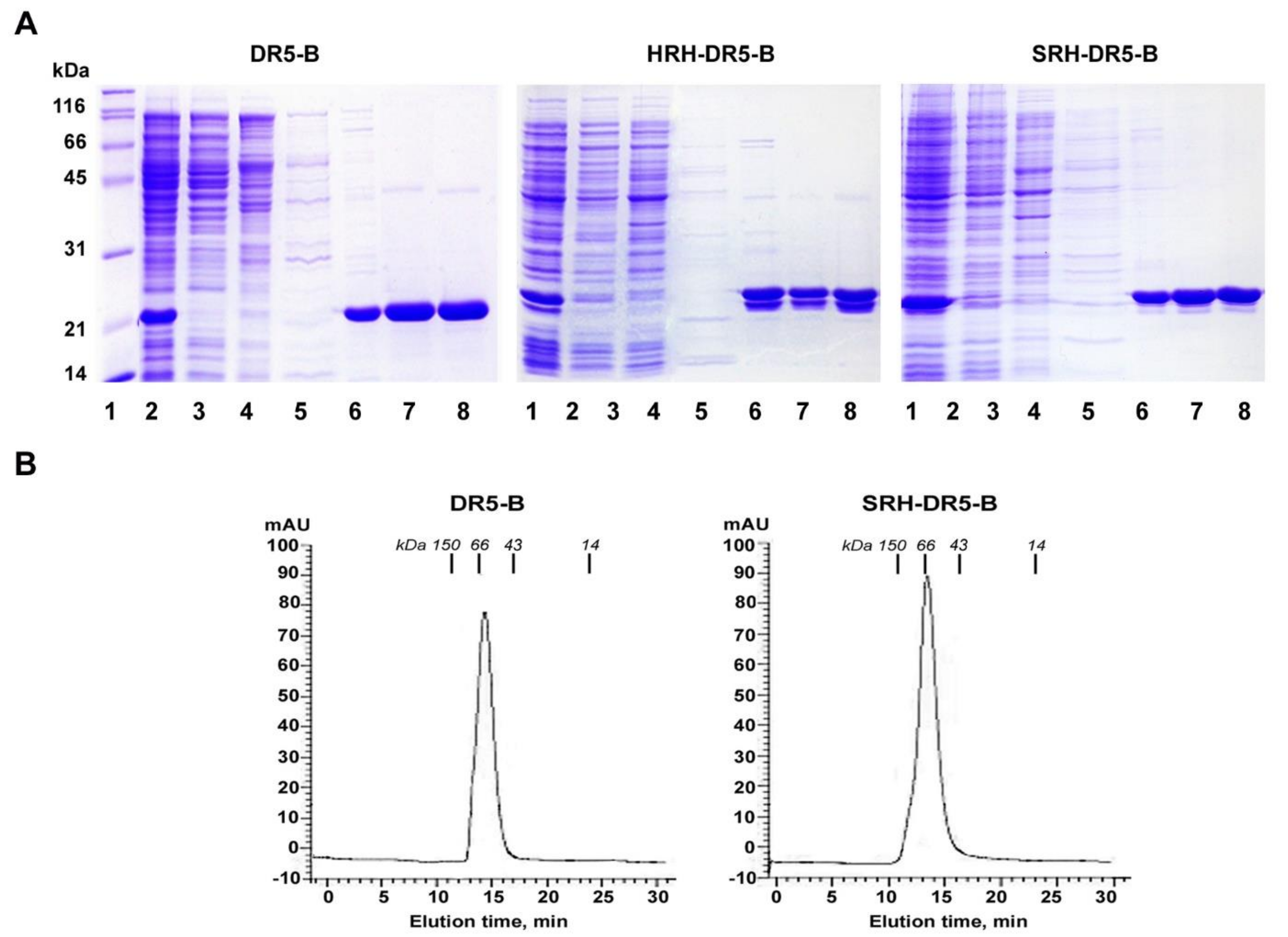
4.5. Size Exclusion Chromatography
4.6. Cell Culture and Multicellular Tumor Spheroids Formation
4.7. Cytotoxicity Evaluation of Purified Proteins
4.8. ELISA
4.9. Flow Cytometry
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TRAIL | TNF-related apoptosis inducing ligand |
| TNF | Tumor necrosis factor |
| DR4 | death receptor 4 (also known as TRAIL-R1, TRAIL receptor 1) |
| DR5 | death receptor 5 (also known as TRAIL-R2, TRAIL receptor 2) |
| DcR1 | decoy receptor 1 |
| DcR2 | decoy receptor 2 |
| OPG | osteoprotegerin |
| DR5-B | DR5-specific TRAIL mutant variant |
| VEGF | Vascular endothelial growth factor sub-family |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR1 | VEGF receptor 1 |
| VEGFR2 | VEGF receptor 2 |
| EGFR | epidermal growth factor receptor |
| EpCAM | epithelial cell adhesion molecule |
| GFP | green fluorescent protein |
| BFPs | bifunctional proteins |
| VAS-TRAIL | TRAIL fusion protein with vasostatin |
| E. coli | Escherichia coli |
| KD | dissociation constant |
| SEC | size exclusion chromatography |
| FPLC | fast protein liquid chromatography |
| ELISA | enzyme-linked immunosorbent assay |
| FACS | fluorescence activated cell sorting |
| PEG | polyethylene glycol |
| DMSO | dimethylsulfoxide |
| IPTG | isopropylthio-β-galactoside |
| TB | Terrific broth medium |
| PBS | phosphate-buffered saline |
| OPD | o-phenylenediamine dihydrochloride |
| NME | N-terminal methionine excision |
| His | histidine |
| Ser | serine |
| Met | methionine |
| Gly | glycine |
| Ala | alanine |
| Pro | proline |
| Thr | threonine |
| Val | valine |
| Cys | cysteine |
| A | adenine |
| T | thymine |
| G | guanine |
| C | cytosine |
References
- Ferrer-Miralles, N.; Domingo-Espín, J.; Corchero, J.L.; Vázquez, E.; Villaverde, A. Microbial Factories for Recombinant Pharmaceuticals. Microb. Cell Factories 2009, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Puetz, J.; Wurm, F.M. Recombinant Proteins for Industrial versus Pharmaceutical Purposes: A Review of Process and Pricing. Processes 2019, 7, 476. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and Antitumor Activity of Recombinant Soluble Apo2 Ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- van Roosmalen, I.A.M.; Quax, W.J.; Kruyt, F.A.E. Two Death-Inducing Human TRAIL Receptors to Target in Cancer: Similar or Distinct Regulation and Function? Biochem. Pharmacol. 2014, 91, 447–456. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I Dose-Escalation Study of Recombinant Human Apo2L/TRAIL, a Dual Proapoptotic Receptor Agonist, in Patients with Advanced Cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Lim, B.; Greer, Y.; Lipkowitz, S.; Takebe, N. Novel Apoptosis-Inducing Agents for the Treatment of Cancer, a New Arsenal in the Toolbox. Cancers 2019, 11, 1087. [Google Scholar] [CrossRef]
- Holland, P.M. Death Receptor Agonist Therapies for Cancer, Which Is the Right TRAIL? Cytokine Growth Factor Rev. 2014, 25, 185–193. [Google Scholar] [CrossRef]
- Wajant, H. Molecular Mode of Action of TRAIL Receptor Agonists—Common Principles and Their Translational Exploitation. Cancers 2019, 11, 954. [Google Scholar] [CrossRef]
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto Better TRAILs for Cancer Treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef]
- Zhu, A.; Wang, X.; Huang, M.; Chen, C.; Yan, J.; Xu, Q.; Wei, L.; Huang, X.; Zhu, H.; Yi, C. Generation of a Novel TRAIL Mutant by Proline to Arginine Substitution Based on Codon Bias and Its Antitumor. Eff. Mol. Med. Rep. 2017, 16, 4973–4979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gasparian, M.E.; Ostapchenko, V.G.; Yagolovich, A.V.; Tsygannik, I.N.; Chernyak, B.V.; Dolgikh, D.A.; Kirpichnikov, M.P. Overexpression and Refolding of Thioredoxin/TRAIL Fusion from Inclusion Bodies and Further Purification of TRAIL after Cleavage by Enteropeptidase. Biotechnol. Lett. 2007, 29, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Sadeghnezhad, G.; Romão, E.; Bernedo-Navarro, R.; Massa, S.; Khajeh, K.; Muyldermans, S.; Hassania, S. Identification of New DR5 Agonistic Nanobodies and Generation of Multivalent Nanobody Constructs for Cancer Treatment. Int. J. Mol. Sci. 2019, 20, 4818. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Yang, H.; Shi, Q.; Fan, Q.; Wan, L.; Lu, X. Targeted Delivery to Tumor-Associated Pericytes via an Affibody with High Affinity for PDGFRβ Enhances the In Vivo Antitumor Effects of Human TRAIL. Theranostics 2017, 7, 2261–2276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lei, Q.; Shen, C.; Wang, N. NCTR25 Fusion Facilitates the Formation of TRAIL Polymers That Selectively Activate TRAIL Receptors with Higher Potency and Efficacy than TRAIL. Cancer Chemother. Pharmacol. 2021, 88, 289–306. [Google Scholar] [CrossRef]
- Madhumathi, J.; Sridevi, S.; Verma, R.S. Novel TNF-Related Apoptotic-Inducing Ligand-Based Immunotoxin for Therapeutic Targeting of CD25 Positive Leukemia. Target. Oncol. 2016, 11, 535–547. [Google Scholar] [CrossRef]
- Brin, E.; Wu, K.; Dagostino, E.; Kuo, M.M.-C.; He, Y.; Shia, W.-J.; Chen, L.-C.; Stempniak, M.; Hickey, R.; Almassy, R.; et al. TRAIL Stabilization and Cancer Cell Sensitization to Its Pro-Apoptotic Activity Achieved through Genetic Fusion with Arginine Deiminase. Oncotarget 2018, 9, 36914–36928. [Google Scholar] [CrossRef]
- de Bruyn, M.; Bremer, E.; Helfrich, W. Antibody-Based Fusion Proteins to Target Death Receptors in Cancer. Cancer Lett. 2013, 332, 175–183. [Google Scholar] [CrossRef]
- Hartung, F.; Krüwel, T.; Shi, X.; Pfizenmaier, K.; Kontermann, R.; Chames, P.; Alves, F.; Pardo, L.A. A Novel Anti-Kv10.1 Nanobody Fused to Single-Chain TRAIL Enhances Apoptosis Induction in Cancer Cells. Front. Pharmacol. 2020, 11, 686. [Google Scholar] [CrossRef]
- Schneider, B.; Münkel, S.; Krippner-Heidenreich, A.; Grunwald, I.; Wels, W.S.; Wajant, H.; Pfizenmaier, K.; Gerspach, J. Potent Antitumoral Activity of TRAIL through Generation of Tumor-Targeted Single-Chain Fusion Proteins. Cell Death Dis. 2010, 1, e68. [Google Scholar] [CrossRef]
- Moorthy, N.K.; Seifert, O.; Eisler, S.; Weirich, S.; Kontermann, R.E.; Rehm, M.; Fullstone, G. Low-Level Endothelial TRAIL-Receptor Expression Obstructs the CNS-Delivery of Angiopep-2 Functionalised TRAIL-Receptor Agonists for the Treatment of Glioblastoma. Molecules 2021, 26, 7582. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.C.; Buchanan, F.G.; Cheng, D.; Solomon, L.R.; Xiao, Y.; Xue, J.; Tahir, S.K.; Smith, M.L.; Zhang, H.; Widomski, D.; et al. Hexavalent TRAIL Fusion Protein Eftozanermin Alfa Optimally Clusters Apoptosis-Inducing TRAIL Receptors to Induce On-Target Antitumor Activity in Solid Tumors. Cancer Res. 2021, 81, 3402–3414. [Google Scholar] [CrossRef] [PubMed]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Mousa, S. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef]
- Chen, H.X.; Cleck, J.N. Adverse Effects of Anticancer Agents That Target the VEGF Pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar] [CrossRef]
- Rozga, P.; Kloska, D.; Pawlak, S.; Teska-Kaminska, M.; Galazka, M.; Bukato, K.; Pieczykolan, A.; Jaworski, A.; Molga-Kaczmarska, A.; Kopacz, A.; et al. Novel Engineered TRAIL-based Chimeric Protein Strongly Inhibits Tumor Growth and Bypasses TRAIL Resistance. Int. J. Cancer 2020, 147, 1117–1130. [Google Scholar] [CrossRef]
- Zhang, Y.; He, B.; Liu, K.; Ning, L.; Luo, D.; Xu, K.; Zhu, W.; Wu, Z.; Huang, J.; Xu, X. A Novel Peptide Specifically Binding to VEGF Receptor Suppresses Angiogenesis In Vitro and In Vivo. Signal Transduct. Target. Ther. 2017, 2, 17010. [Google Scholar] [CrossRef]
- Gasparian, M.E.; Chernyak, B.V.; Dolgikh, D.A.; Yagolovich, A.V.; Popova, E.N.; Sycheva, A.M.; Moshkovskii, S.A.; Kirpichnikov, M.P. Generation of New TRAIL Mutants DR5-A and DR5-B with Improved Selectivity to Death Receptor 5. Apoptosis 2009, 14, 778–787. [Google Scholar] [CrossRef]
- Yagolovich, A.V.; Artykov, A.A.; Dolgikh, D.A.; Kirpichnikov, M.P.; Gasparian, M.E. A New Efficient Method for Production of Recombinant Antitumor Cytokine TRAIL and Its Receptor-Selective Variant DR5-B. Biochemistry 2019, 84, 627–636. [Google Scholar] [CrossRef]
- Voges, D.; Watzele, M.; Nemetz, C.; Wizemann, S.; Buchberger, B. Analyzing and Enhancing MRNA Translational Efficiency in an Escherichia coli In Vitro Expression System. Biochem. Biophys. Res. Commun. 2004, 318, 601–614. [Google Scholar] [CrossRef]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and Effects of N-Terminal Codon Bias in Bacterial Genes. Science 2013, 342, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, P.T. N-Terminal Methionine Processing. Curr. Protoc. Protein Sci. 2017, 88, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Meinnel, T.; Serero, A.; Giglione, C. Impact of the N-Terminal Amino Acid on Targeted Protein Degradation. Biol. Chem. 2006, 387, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Hirel, P.H.; Schmitter, M.J.; Dessen, P.; Fayat, G.; Blanquet, S. Extent of N-Terminal Methionine Excision from Escherichia coli Proteins Is Governed by the Side-Chain Length of the Penultimate Amino Acid. Proc. Natl. Acad. Sci. USA 1989, 86, 8247–8251. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.; Mignone, V.; Arruda, M.; Alcobia, D.; Hill, S.; Kilpatrick, L.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.M.; Clark, R.P.; Chong, D.C.; Citrin, K.M.; Wylie, L.A.; Bautch, V.L. Dynamic Alterations in Decoy VEGF Receptor-1 Stability Regulate Angiogenesis. Nat. Commun. 2017, 8, 15699. [Google Scholar] [CrossRef] [PubMed]
- Mesti, T.; Savarin, P.; Triba, M.N.; Le Moyec, L.; Ocvirk, J.; Banissi, C.; Carpentier, A.F. Metabolic Impact of Anti-Angiogenic Agents on U87 Glioma Cells. PLoS ONE 2014, 9, e99198. [Google Scholar] [CrossRef]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef]
- Du, Y.; Xu, J. Engineered Bifunctional Proteins for Targeted Cancer Therapy: Prospects and Challenges. Adv. Mater. 2021, 33, 2103114. [Google Scholar] [CrossRef]
- Kretz, A.-L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. TRAILblazing Strategies for Cancer Treatment. Cancers 2019, 11, 456. [Google Scholar] [CrossRef]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist Antibodies to Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) as Anti-Angiogenic Agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Lipinszki, Z.; Vernyik, V.; Farago, N.; Sari, T.; Puskas, L.G.; Blattner, F.R.; Posfai, G.; Gyorfy, Z. Enhancing the Translational Capacity of E. coli by Resolving the Codon Bias. ACS Synth. Biol. 2018, 7, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Al-Hawash, A.B.; Zhang, X.; Ma, F. Strategies of Codon Optimization for High-Level Heterologous Protein Expression in Microbial Expression Systems. Gene Rep. 2017, 9, 46–53. [Google Scholar] [CrossRef]
- Bivona, L.; Zou, Z.; Stutzman, N.; Sun, P.D. Influence of the second amino acid on recombinant protein expression. Protein Expr. Purif. 2010, 74, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Gapizov, S.S.; Petrovskaya, L.E.; Shingarova, L.N.; Svirschevskaya, E.V.; Dolgikh, D.A.; Kirpichnikov, M.P. The Effect of TNF and VEGF on the Properties of Ea.Hy926 Endothelial Cells in a Model of Multi-Cellular Spheroids. Acta Nat. 2018, 10, 34–42. [Google Scholar] [CrossRef]
- de Looff, M.; de Jong, S.; Kruyt, F.A.E. Multiple Interactions between Cancer Cells and the Tumor Microenvironment Modulate TRAIL Signaling: Implications for TRAIL Receptor Targeted Therapy. Front. Immunol. 2019, 10, 1530. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.U.; Kim, J.; Chung, S.W.; Lee, N.K.; Park, J.; Kweon, S.; Cho, Y.S.; Kim, H.R.; Lim, S.M.; Park, J.W.; et al. Dual Mechanistic TRAIL Nanocarrier Based on PEGylated Heparin Taurocholate and Protamine Which Exerts Both Pro-Apoptotic and Anti-Angiogenic Effects. J. Control. Release 2021, 336, 181–191. [Google Scholar] [CrossRef]
- Fan, J.; Wang, Z.; Huang, L.; Shen, Y. Efficient Refolding of the Bifunctional Therapeutic Fusion Protein VAS-TRAIL by a Triple Agent Solution. Protein Expr. Purif. 2016, 125, 68–73. [Google Scholar] [CrossRef]
- Artykov, A.A.; Yagolovich, A.V.; Dolgikh, D.A.; Kirpichnikov, M.P.; Trushina, D.B.; Gasparian, M.E. Death Receptors DR4 and DR5 Undergo Spontaneous and Ligand-Mediated Endocytosis and Recycling Regardless of the Sensitivity of Cancer Cells to TRAIL. Front. Cell Dev. Biol. 2021, 9, 733688. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yagolovich, A.V.; Artykov, A.A.; Isakova, A.A.; Vorontsova, Y.V.; Dolgikh, D.A.; Kirpichnikov, M.P.; Gasparian, M.E. Optimized Heterologous Expression and Efficient Purification of a New TRAIL-Based Antitumor Fusion Protein SRH–DR5-B with Dual VEGFR2 and DR5 Receptor Specificity. Int. J. Mol. Sci. 2022, 23, 5860. https://doi.org/10.3390/ijms23115860
Yagolovich AV, Artykov AA, Isakova AA, Vorontsova YV, Dolgikh DA, Kirpichnikov MP, Gasparian ME. Optimized Heterologous Expression and Efficient Purification of a New TRAIL-Based Antitumor Fusion Protein SRH–DR5-B with Dual VEGFR2 and DR5 Receptor Specificity. International Journal of Molecular Sciences. 2022; 23(11):5860. https://doi.org/10.3390/ijms23115860
Chicago/Turabian StyleYagolovich, Anne V., Artem A. Artykov, Alina A. Isakova, Yekaterina V. Vorontsova, Dmitry A. Dolgikh, Mikhail P. Kirpichnikov, and Marine E. Gasparian. 2022. "Optimized Heterologous Expression and Efficient Purification of a New TRAIL-Based Antitumor Fusion Protein SRH–DR5-B with Dual VEGFR2 and DR5 Receptor Specificity" International Journal of Molecular Sciences 23, no. 11: 5860. https://doi.org/10.3390/ijms23115860
APA StyleYagolovich, A. V., Artykov, A. A., Isakova, A. A., Vorontsova, Y. V., Dolgikh, D. A., Kirpichnikov, M. P., & Gasparian, M. E. (2022). Optimized Heterologous Expression and Efficient Purification of a New TRAIL-Based Antitumor Fusion Protein SRH–DR5-B with Dual VEGFR2 and DR5 Receptor Specificity. International Journal of Molecular Sciences, 23(11), 5860. https://doi.org/10.3390/ijms23115860