
Altered Blood and Brain Expression of Inflammation and Redox Genes in Alzheimer’s Disease, Common to APPV717I × TAUP301L Mice and Patients


 and
and 
Abstract
:1. Introduction
2. Results
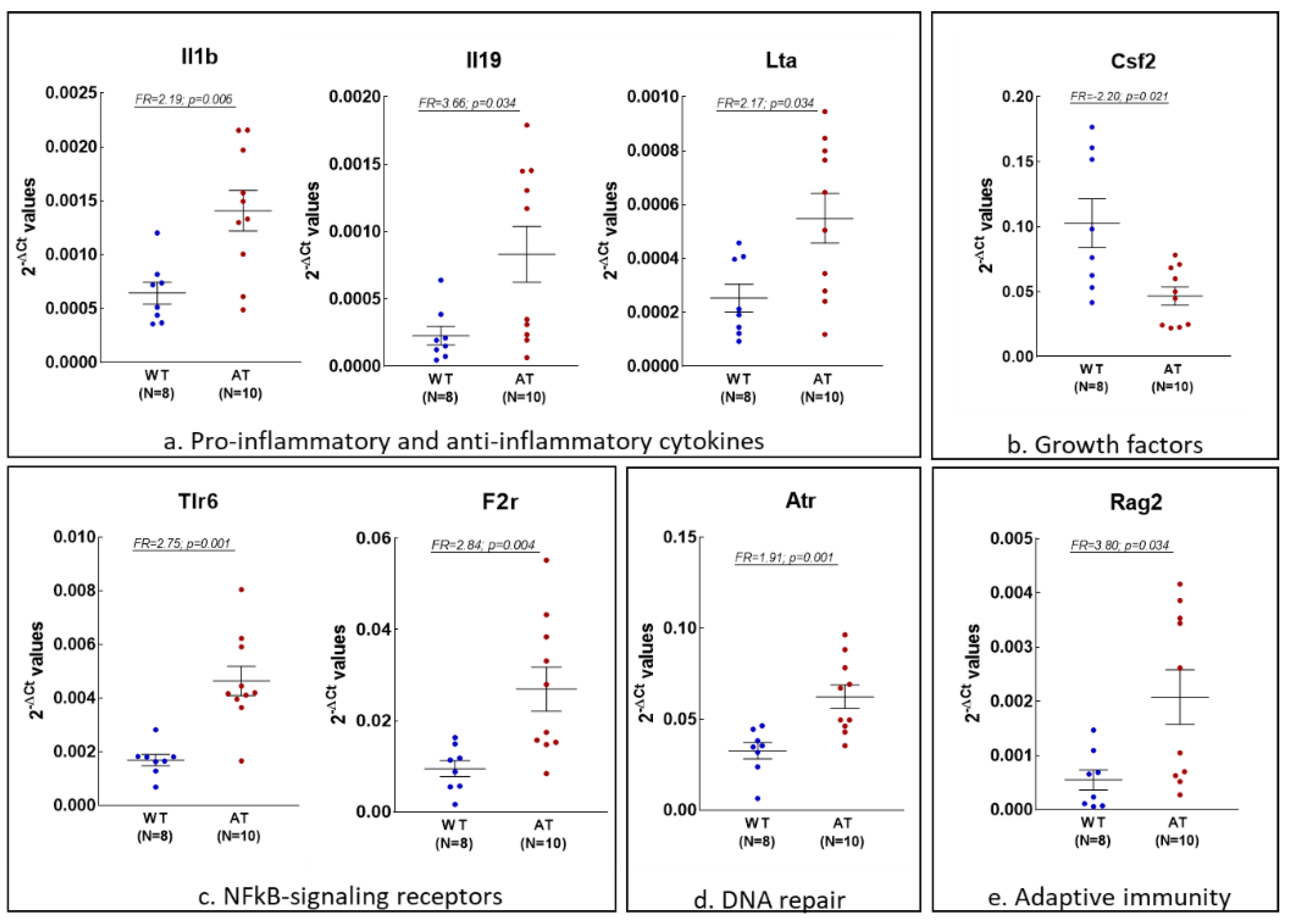
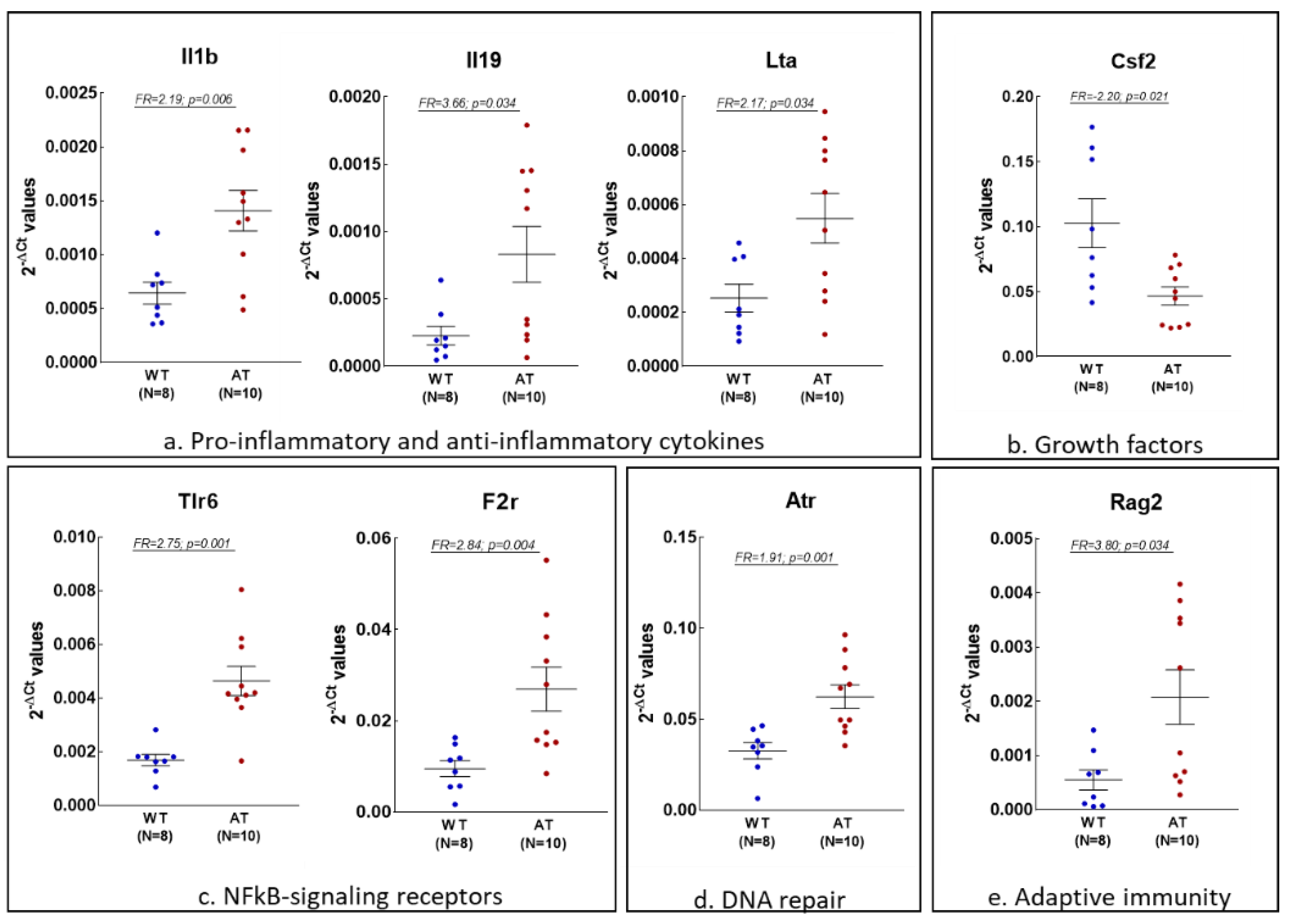
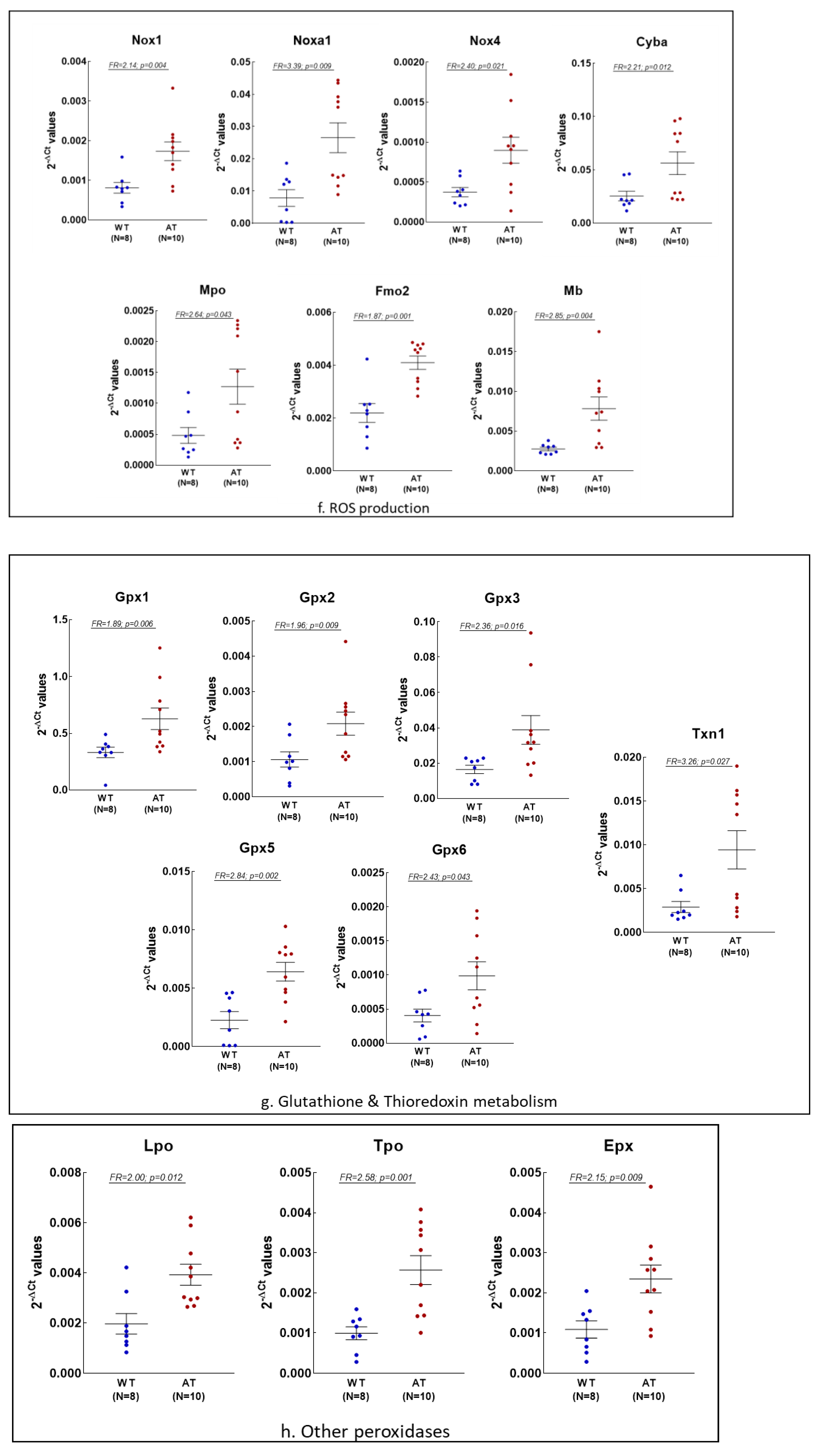
2.1. Gene Expression Changes in the Hippocampus of AT Mice
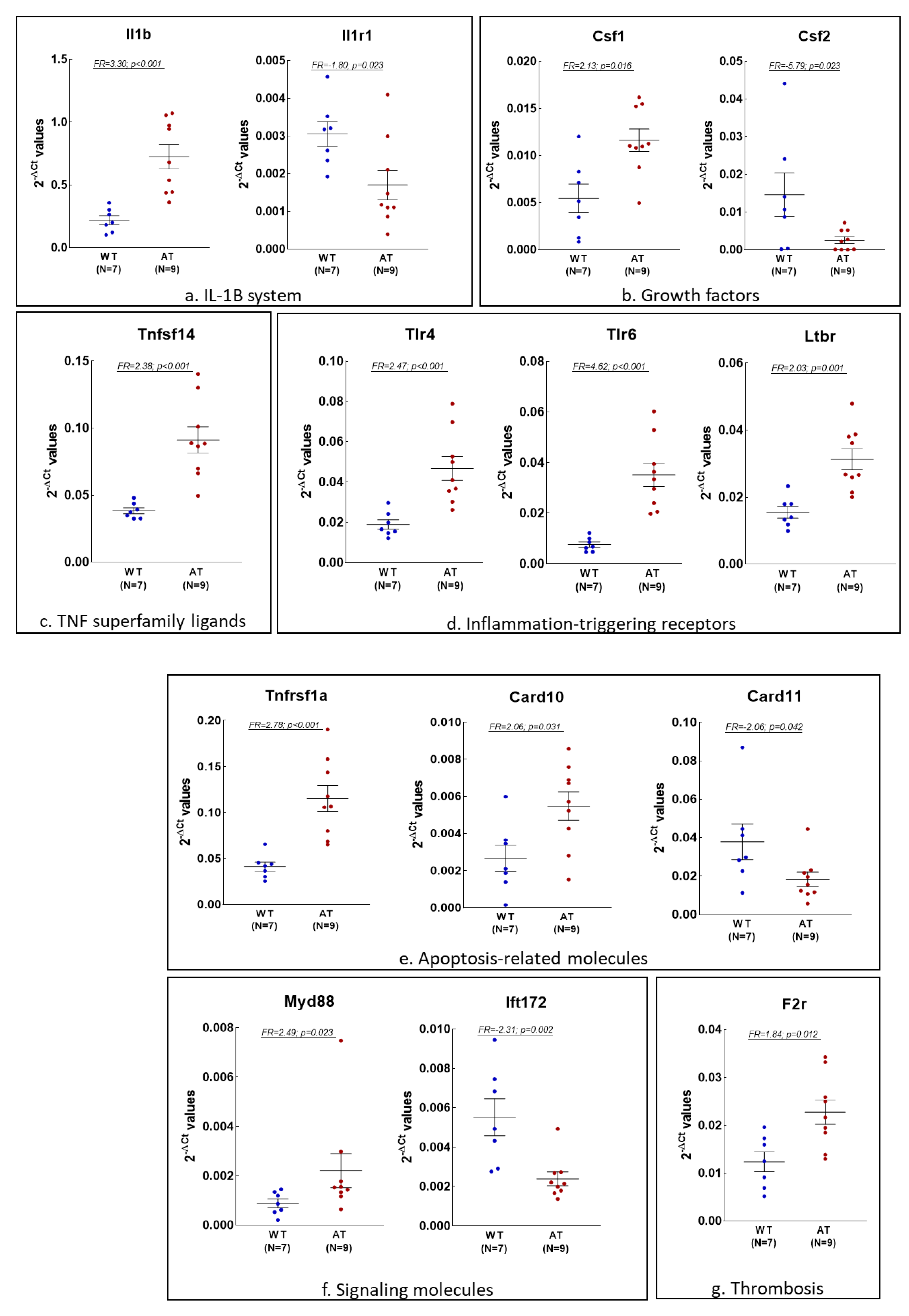
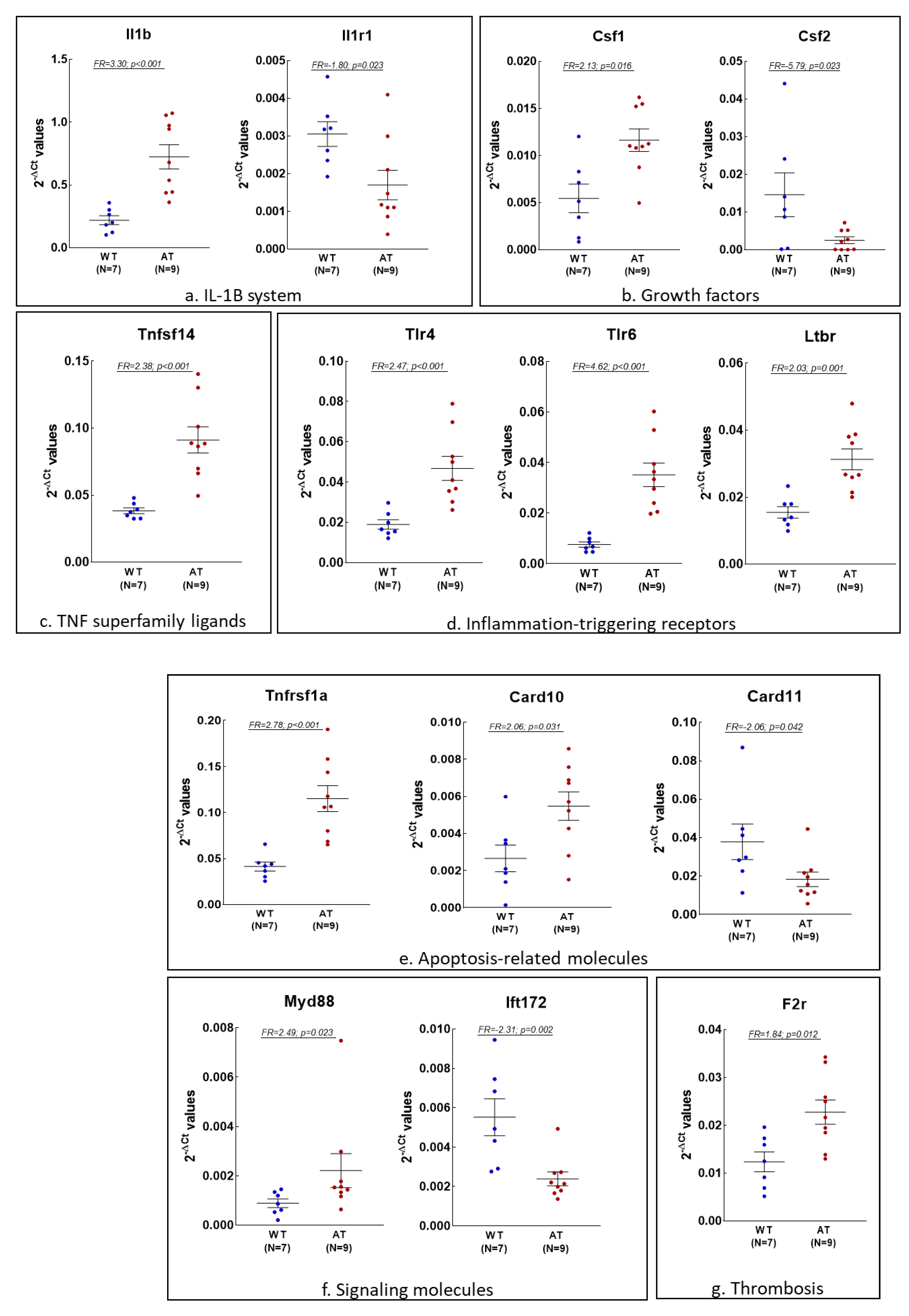
2.2. Gene Expression Changes in the Blood of AT Mice
2.3. Analysis of Inflammation and Redox Gene Changes with Higher FR Values
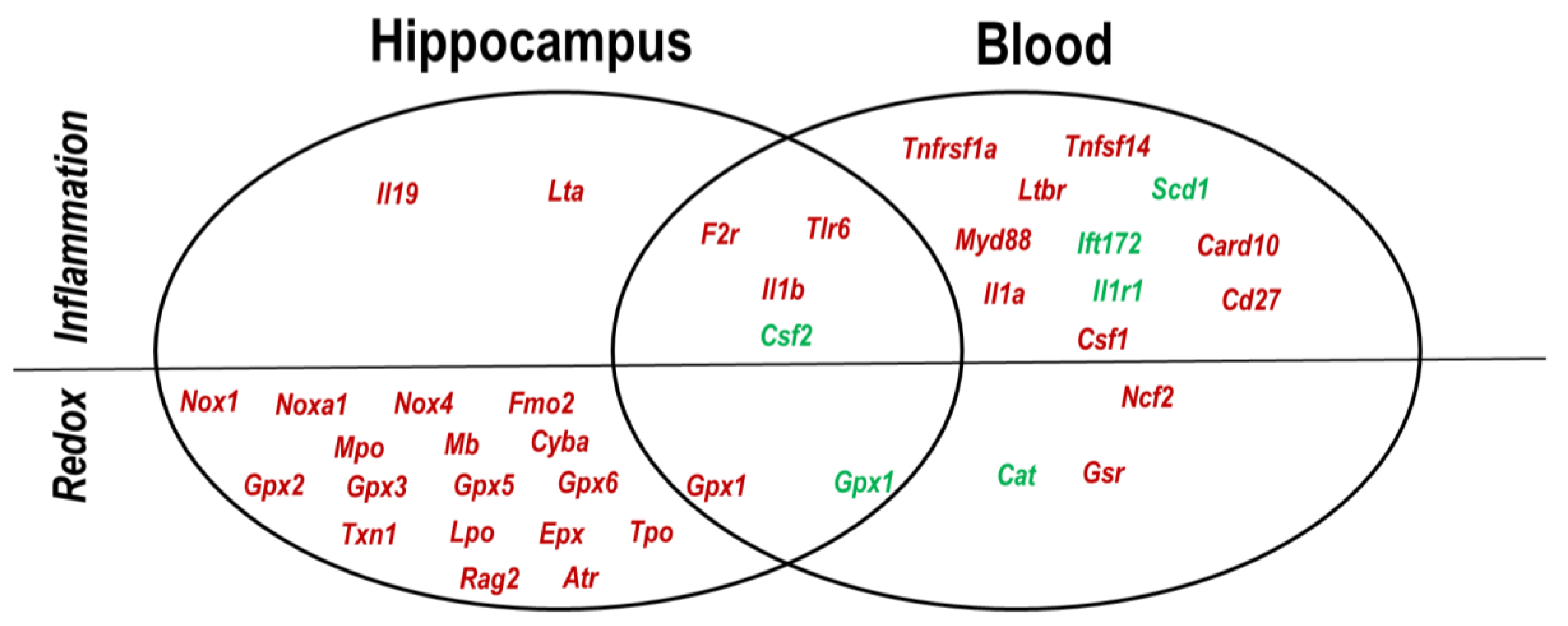
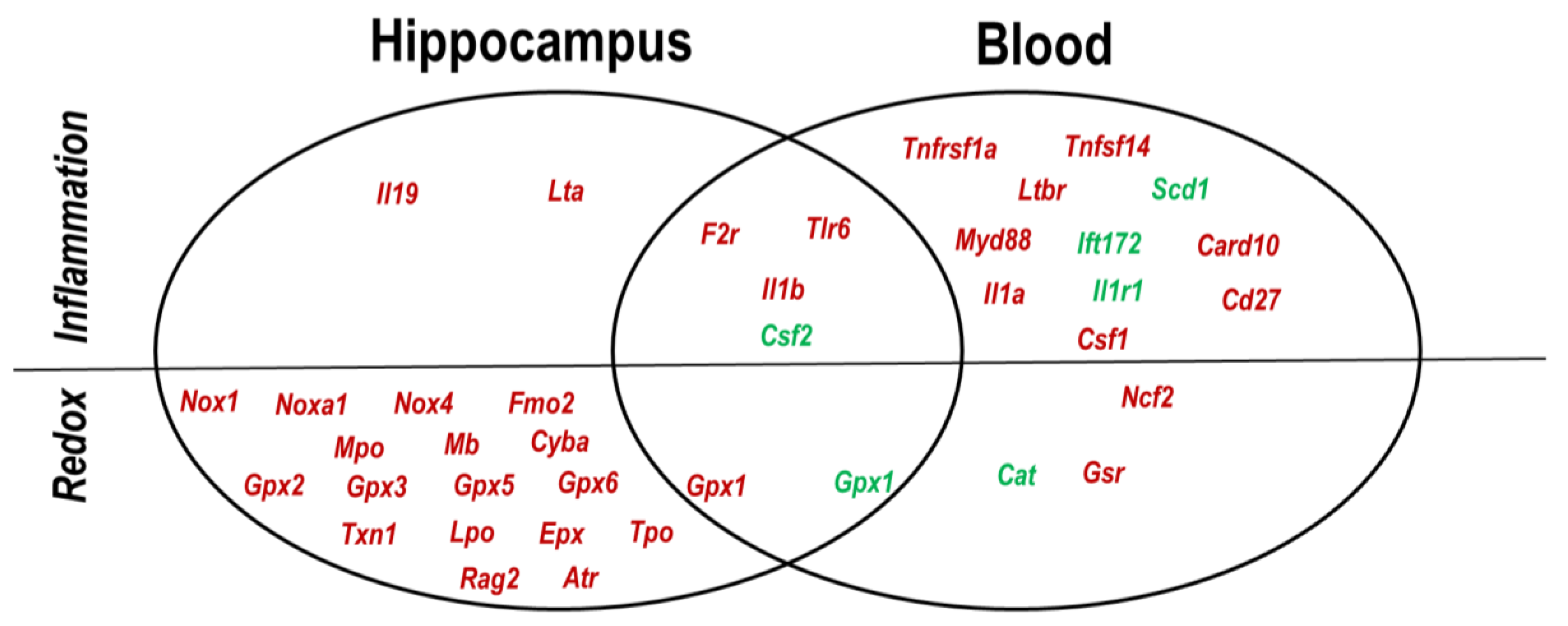
2.4. Hippocampus–Blood Comparison
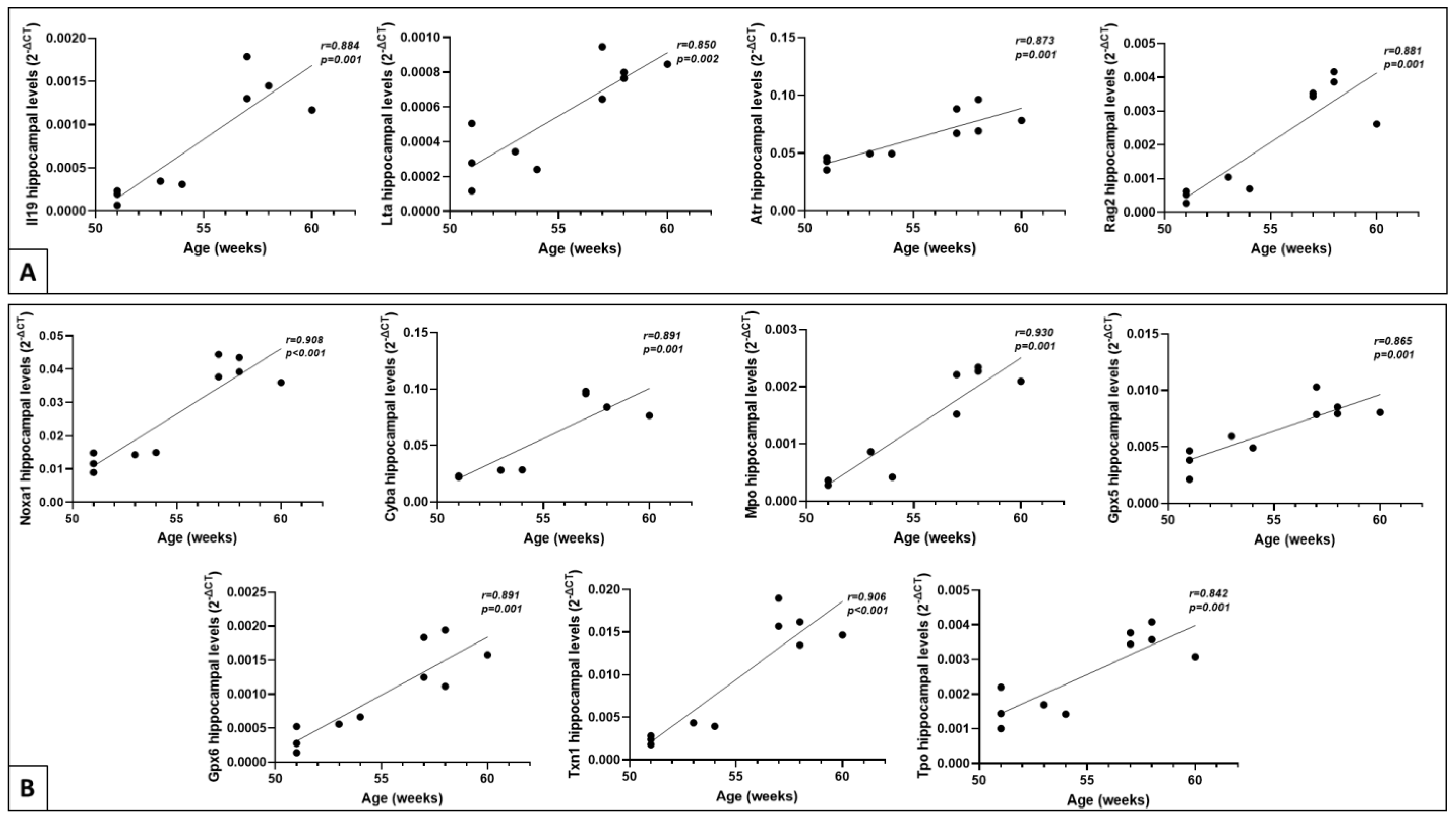
2.5. Correlation of Gene Expression Levels with Age
2.6. Comparison of Dysregulated Inflammation and Redox Genes in AT Mice vs. AD Patients
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Blood and Hippocampi Collection
4.3. Gene Expression Analysis
4.4. GEO Data Mining
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Di Lorenzo, R.; Frangipane, F.; Maletta, R.; Puccio, G.; et al. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hashimoto, M.; Fukuhara, R.; Ishikawa, T.; Yatabe, Y.; Kaneda, K.; Yuuki, S.; Honda, K.; Matsuzaki, S.; Tsuyuguchi, A.; et al. Relationship between Dementia Severity and Behavioural and Psychological Symptoms in Early-Onset Alzheimer’s Disease. Psychogeriatrics 2015, 15, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia Prevention, Intervention, and Care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Hasana, S.; Hossain, M.F.; Islam, M.S.; Behl, T.; Perveen, A.; Hafeez, A.; Ashraf, G.M. Molecular Genetics of Early- and Late-Onset Alzheimer’s Disease. Curr. Gene Ther. 2021, 21, 43–52. [Google Scholar] [CrossRef]
- Abondio, P.; Sarno, S.; Giuliani, C.; Laganà, V.; Maletta, R.; Bernardi, L.; Bruno, F.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Amyloid Precursor Protein A713T Mutation in Calabrian Patients with Alzheimer’s Disease: A Population Genomics Approach to Estimate Inheritance from a Common Ancestor. Biomedicines 2021, 10, 20. [Google Scholar] [CrossRef]
- Harwood, J.C.; Leonenko, G.; Sims, R.; Escott-Price, V.; Williams, J.; Holmans, P. Defining Functional Variants Associated with Alzheimer’s Disease in the Induced Immune Response. Brain Commun. 2021, 3, fcab083. [Google Scholar] [CrossRef]
- Wee, J.J.; Kumar, S. Prediction of Hub Genes of Alzheimer’s Disease Using a Protein Interaction Network and Functional Enrichment Analysis. Genom. Inform. 2020, 18, e39. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Status and Future Directions of Clinical Trials in Alzheimer’s Disease. Int. Rev. Neurobiol. 2020, 154, 3–50. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Caruso, A.; Nicoletti, F.; Gaetano, A.; Scaccianoce, S. Risk Factors for Alzheimer’s Disease: Focus on Stress. Front. Pharmacol. 2019, 10, 976. [Google Scholar] [CrossRef]
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA Damage to Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal. 2021, 34, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on MicroRNAs. Oxidative Med. Cell. Longev. 2020, 2020, 2638130. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The Role and Consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. DNA Damage-induced Neuroinflammation in Neurodegenerative Disease. Alzheimer’s Dement. 2021, 17, e055175. [Google Scholar] [CrossRef]
- Canugovi, C.; Misiak, M.; Ferrarelli, L.K.; Croteau, D.L.; Bohr, V.A. The Role of DNA Repair in Brain Related Disease Pathology. DNA Repair 2013, 12, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Akkaya, Ç.; Yavuzer, S.S.; Yavuzer, H.; Erkol, G.; Bozluolcay, M.; Dinçer, Y. DNA Damage, DNA Susceptibility to Oxidation and Glutathione Redox Status in Patients with Alzheimer’s Disease Treated with and without Memantine. J. Neurol. Sci. 2017, 378, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic Acid Oxidation: An Early Feature of Alzheimer’s Disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.R.T.; Santos, A.C.F.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.L.; Campos, A.H.J.F.M.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of Oxidative DNA Damage, Cell-Cycle Regulation and Neuronal Death May Influence the Clinical Manifestation of Alzheimer’s Disease. PLoS ONE 2014, 9, e99897. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, E.; Dobre, M.; Cucos, C.A.; Rojo, A.I.; Jiménez-Villegas, J.; Capetillo-Zarate, E.; Matute, C.; Piñol-Ripoll, G.; Manda, G.; Cuadrado, A. Whole Blood Expression Pattern of Inflammation and Redox Genes in Mild Alzheimer’s Disease. J. Inflamm. Res. 2021, 14, 6085–6102. [Google Scholar] [CrossRef]
- Kodamullil, A.T.; Iyappan, A.; Karki, R.; Madan, S.; Younesi, E.; Hofmann-Apitius, M. Of Mice and Men: Comparative Analysis of Neuro-Inflammatory Mechanisms in Human and Mouse Using Cause-and-Effect Models. J. Alzheimer’s Dis. 2017, 59, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 Deficiency Replicates Transcriptomic Changes in Alzheimer’s Patients and Worsens APP and TAU Pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid Activates GSK-3beta to Aggravate Neuronal Tauopathy in Bigenic Mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-Regulated Glutathione Recycling Independent of Biosynthesis Is Critical for Cell Survival during Oxidative Stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Samaey, C.; Schreurs, A.; Stroobants, S.; Balschun, D. Early Cognitive and Behavioral Deficits in Mouse Models for Tauopathy and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 335. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Z.-P.; Huang, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J.; Yu, Y.-N. ATM and ATR: Sensing DNA Damage. World J. Gastroenterol. 2004, 10, 155–160. [Google Scholar] [CrossRef]
- Sin, Y.; Tanaka, K.; Saijo, M. The C-Terminal Region and SUMOylation of Cockayne Syndrome Group B Protein Play Critical Roles in Transcription-Coupled Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, W.; Byrum, J.N.; Sapkota, H.; Rahman, N.S.; Cail, R.C.; Zhao, S.; Schatz, D.G.; Rodgers, K.K. Spatio-Temporal Regulation of RAG2 Following Genotoxic Stress. DNA Repair 2015, 27, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Malynn, B.A.; Ma, A. Preserving Immune Homeostasis with A20. Adv. Immunol. 2020, 148, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Klohs, J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegener. Dis. 2019, 19, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Basu, A.; Krady, J.K.; Levison, S.W. Interleukin-1: A Master Regulator of Neuroinflammation. J. Neurosci. Res. 2004, 78, 151–156. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain Interleukin 1 and S-100 Immunoreactivity Are Elevated in Down Syndrome and Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, C.W.; Patel, A.J. Regulation of Beta-Amyloid Precursor Protein Isoform MRNAs by Transforming Growth Factor-Beta 1 and Interleukin-1 Beta in Astrocytes. Brain Res. Mol. Brain Res. 1993, 19, 251–256. [Google Scholar] [CrossRef]
- Liao, Y.-F.; Wang, B.-J.; Cheng, H.-T.; Kuo, L.-H.; Wolfe, M.S. Tumor Necrosis Factor-Alpha, Interleukin-1beta, and Interferon-Gamma Stimulate Gamma-Secretase-Mediated Cleavage of Amyloid Precursor Protein through a JNK-Dependent MAPK Pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained Interleukin-1β Overexpression Exacerbates Tau Pathology despite Reduced Amyloid Burden in an Alzheimer’s Mouse Model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, G. Interleukin-19: Multiple Roles in Immune Regulation and Disease. Cytokine Growth Factor Rev. 2010, 21, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Parajuli, B.; Wang, Y.; Azuma, Y.-T.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-19 Acts as a Negative Autocrine Regulator of Activated Microglia. PLoS ONE 2015, 10, e0118640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridwan, S.; Bauer, H.; Frauenknecht, K.; von Pein, H.; Sommer, C.J. Distribution of Granulocyte-Monocyte Colony-Stimulating Factor and Its Receptor α-Subunit in the Adult Human Brain with Specific Reference to Alzheimer’s Disease. J. Neural Transm. 2012, 119, 1389–1406. [Google Scholar] [CrossRef]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O’Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D.; et al. Safety and Efficacy of Sargramostim (GM-CSF) in the Treatment of Alzheimer’s Disease. Alzheimer’s Dement. 2021, 7, e12158. [Google Scholar] [CrossRef]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-Cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [Green Version]
- Trieb, K.; Ransmayr, G.; Sgonc, R.; Lassmann, H.; Grubeck-Loebenstein, B. APP Peptides Stimulate Lymphocyte Proliferation in Normals, but Not in Patients with Alzheimer’s Disease. Neurobiol. Aging 1996, 17, 541–547. [Google Scholar] [CrossRef]
- Späni, C.; Suter, T.; Derungs, R.; Ferretti, M.T.; Welt, T.; Wirth, F.; Gericke, C.; Nitsch, R.M.; Kulic, L. Reduced β-Amyloid Pathology in an APP Transgenic Mouse Model of Alzheimer’s Disease Lacking Functional B and T Cells. Acta Neuropathol. Commun. 2015, 3, 71. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, T.P.; Nosalski, R.; Szczepaniak, P.; Budzyn, K.; Osmenda, G.; Skiba, D.; Sagan, A.; Wu, J.; Vinh, A.; Marvar, P.J.; et al. Role of Chemokine RANTES in the Regulation of Perivascular Inflammation, T-Cell Accumulation, and Vascular Dysfunction in Hypertension. FASEB J. 2016, 30, 1987–1999. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. RANTES Upregulation in the Alzheimer’s Disease Brain: A Possible Neuroprotective Role. Neurobiol. Aging 2010, 31, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH Oxidase in Brain Injury and Neurodegenerative Disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal Expression of Myeloperoxidase Is Increased in Alzheimer’s Disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Colton, C.A.; Vitek, M.P.; Wink, D.A.; Xu, Q.; Cantillana, V.; Previti, M.L.; Van Nostrand, W.E.; Weinberg, J.B.; Dawson, H. NO Synthase 2 (NOS2) Deletion Promotes Multiple Pathologies in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2006, 103, 12867–12872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The Physiology and Pathophysiology of Nitric Oxide in the Brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Apelt, J.; Schliebs, R. Fibrillary Beta-Amyloid Deposits Are Closely Associated with Atrophic Nitric Oxide Synthase (NOS)-Expressing Neurons but Do Not Upregulate the Inducible NOS in Transgenic Tg2576 Mouse Brain with Alzheimer Pathology. Neurosci. Lett. 2001, 302, 73–76. [Google Scholar] [CrossRef]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread Peroxynitrite-Mediated Damage in Alzheimer’s Disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Katsuse, O.; Iseki, E. Nitric Oxide Pathways in Alzheimer’s Disease and Other Neurodegenerative Dementias. Neurol. Res. 2004, 26, 563–566. [Google Scholar] [CrossRef]
- Jia, J.; Zeng, X.; Xu, G.; Wang, Z. The Potential Roles of Redox Enzymes in Alzheimer’s Disease: Focus on Thioredoxin. ASN Neuro 2021, 13, 1759091421994351. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The Emerging Role of Glutathione in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels--a Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Angiulli, F.; Conti, E.; Zoia, C.P.; Da Re, F.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics 2021, 11, 1525. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s Disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef] [PubMed]
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.M.; Meijboom, F.L.B. Animal to Human Translation: A Systematic Scoping Review of Reported Concordance Rates. J. Transl. Med. 2019, 17, 223. [Google Scholar] [CrossRef] [Green Version]
- Sultan, F.A. Dissection of Different Areas from Mouse Hippocampus. Bio-Protocol 2013, 3, e955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawiec, J.A.; Chen, H.; Alom-Ruiz, S.; Jaye, M. Modified PAXgene Method Allows for Isolation of High-Integrity Total RNA from Microlitre Volumes of Mouse Whole Blood. Lab. Anim. 2009, 43, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. MiRDeepFinder: A MiRNA Analysis Tool for Deep Sequencing of Plant Small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | AT vs. WT (FC > |2|) | GEODATASET Human Brain | Significance | Ref |
|---|---|---|---|---|
| Il19 | ↑FR = 3.66; p =0.034 | ↑7 AD vs. 4 CTRL (FCX) | FR = 1.80; p =0.03 | [GSE185909] |
| F2r | ↑FR = 2.84; p =0.004 | ↑7 incipient AD vs. 9 CTRL (HP) | FR = 2; p =0.004 | [GSE1297] |
| ↑7 severe AD vs. 9 CTRL (HP) | FR = 2.03; p =0.002 | [GSE1297] | ||
| Tlr6 | ↑FR = 2.75; p =0.001 | ↑ 7 AD vs. 4 CTRL (FCX) | FR = 1.62; p =0.024 | [GSE185909] |
| Il1b | ↑FR = 2.90; p =0.006 | ↓7 severe AD vs. 8 CTRL (GM) | FR = −10.86; p =0.00000698 | [GSE28146] |
| Lta | ↑FR = 2.17; p =0.034 | ↑7 AD vs. 4 CTRL (FCX) | FR = 1.61; p =0.024 | [GSE185909] |
| ↑7 incipient AD vs. 8 CTRL (GM) | FR = 3.51; p =0.002 | [GSE28146] | ||
| ↑8 AD vs. 8 CTRL (NCX) | FR = 1.57; p =0.05 | [GSE37264] | ||
| Csf2 | ↓FR = −2.20; p =0.021 | ↑7 incipient AD vs. 9 CTRL (HP) | FR = 1.98; p =0.02 | [GSE1297] |
| ↑7 severe AD vs. 9 CTRL (HP) | FR = 2.41; p =0.00009546 | [GSE1297] | ||
| Gpx3 | ↑FR = 2.36; p =0.016 | ↑ 36 AD vs. 16 CTRL (EC) | FR = 1.66; p =0.000025 * | [GSE118553] |
| Nox1 | ↑FR = 2.14; p =0.004 | ↑ 7 incipient AD vs. 9 CTRL (HP) | FR = 1.66; p =0.04 | [GSE1297] |
| Mice Blood (9 AT vs. 7 WT) | Human Blood (38 AD vs. 38 CTRL) | ||||
|---|---|---|---|---|---|
| Gene | FR | p-Value | Gene | FR | p-Value |
| Ltbr | 2.03 | 0.001 | LTBR | 1.54 | 0.001 |
| F2r | 1.84 | 0.012 | F2R | 1.53 | 0.006 |
| Il1b | 3.30 | <0.001 | IL1B | 1.79 | <0.001 |
| Csf1 | 2.13 | 0.016 | CSF1 | 1.76 | <0.001 |
| Gsr | 2.23 | <0.001 | GSR | 3.93 | <0.001 |
| Mice Blood Data | Human Blood Data [24] | ||||||
|---|---|---|---|---|---|---|---|
| 21 AT vs. 12 WT | 7 AT vs. 8 WT | 38 MCI vs. 38 CTRL | |||||
| 48.5 ± 4.9 weeks | 37.1 ± 0.8 weeks | ||||||
| Gene | FR | p-value | FR | p-value | Gene | FR | p-value |
| Gsr | 2.08 | <0.001 | 1.70 | 0.001 | GSR | 3.93 | <0.001 |
| Osgin1 | 2.72 | <0.001 | 1.69 | 0.001 | OSGIN1 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucos, C.A.; Milanesi, E.; Dobre, M.; Musat, I.A.; Manda, G.; Cuadrado, A. Altered Blood and Brain Expression of Inflammation and Redox Genes in Alzheimer’s Disease, Common to APPV717I × TAUP301L Mice and Patients. Int. J. Mol. Sci. 2022, 23, 5799. https://doi.org/10.3390/ijms23105799
Cucos CA, Milanesi E, Dobre M, Musat IA, Manda G, Cuadrado A. Altered Blood and Brain Expression of Inflammation and Redox Genes in Alzheimer’s Disease, Common to APPV717I × TAUP301L Mice and Patients. International Journal of Molecular Sciences. 2022; 23(10):5799. https://doi.org/10.3390/ijms23105799
Chicago/Turabian StyleCucos, Catalina Anca, Elena Milanesi, Maria Dobre, Ioana Andreea Musat, Gina Manda, and Antonio Cuadrado. 2022. "Altered Blood and Brain Expression of Inflammation and Redox Genes in Alzheimer’s Disease, Common to APPV717I × TAUP301L Mice and Patients" International Journal of Molecular Sciences 23, no. 10: 5799. https://doi.org/10.3390/ijms23105799
APA StyleCucos, C. A., Milanesi, E., Dobre, M., Musat, I. A., Manda, G., & Cuadrado, A. (2022). Altered Blood and Brain Expression of Inflammation and Redox Genes in Alzheimer’s Disease, Common to APPV717I × TAUP301L Mice and Patients. International Journal of Molecular Sciences, 23(10), 5799. https://doi.org/10.3390/ijms23105799