
TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
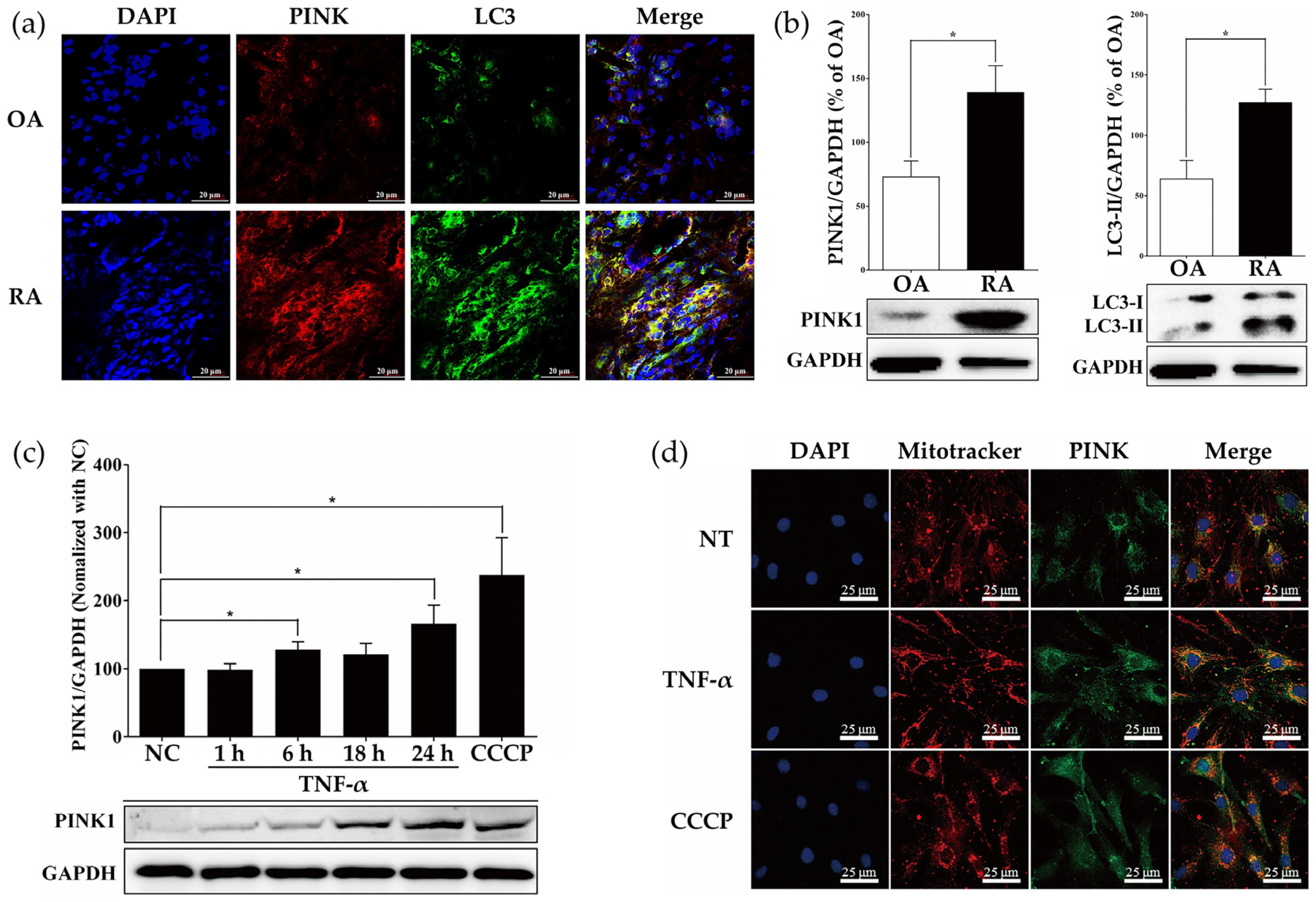
2.1. Measurement of PINK1 Expression in Synovial Tissues of Patients with Osteoarthritis (OA) and RA
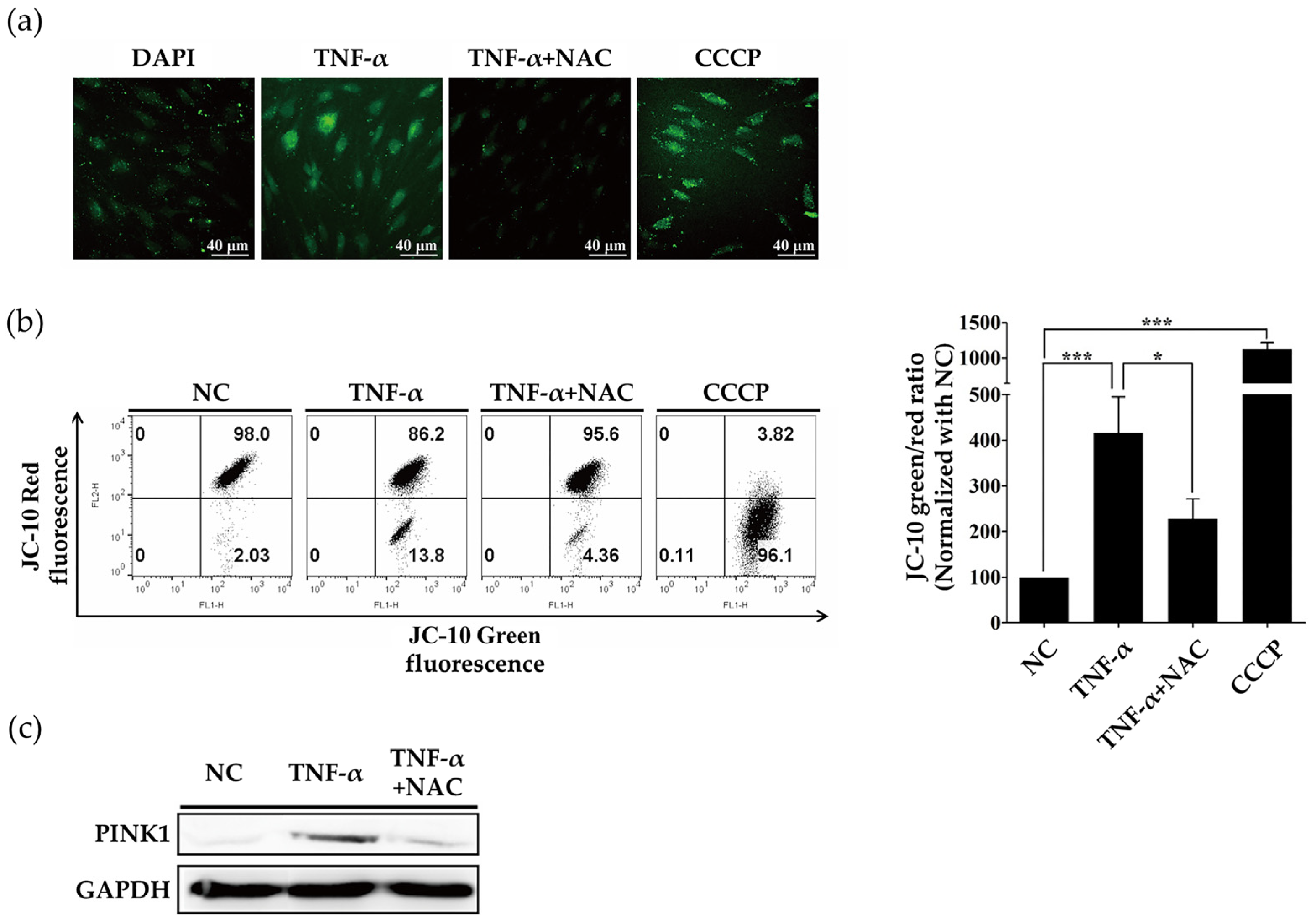
2.2. ROS Induced by TNF-α Regulate Expression of PINK1
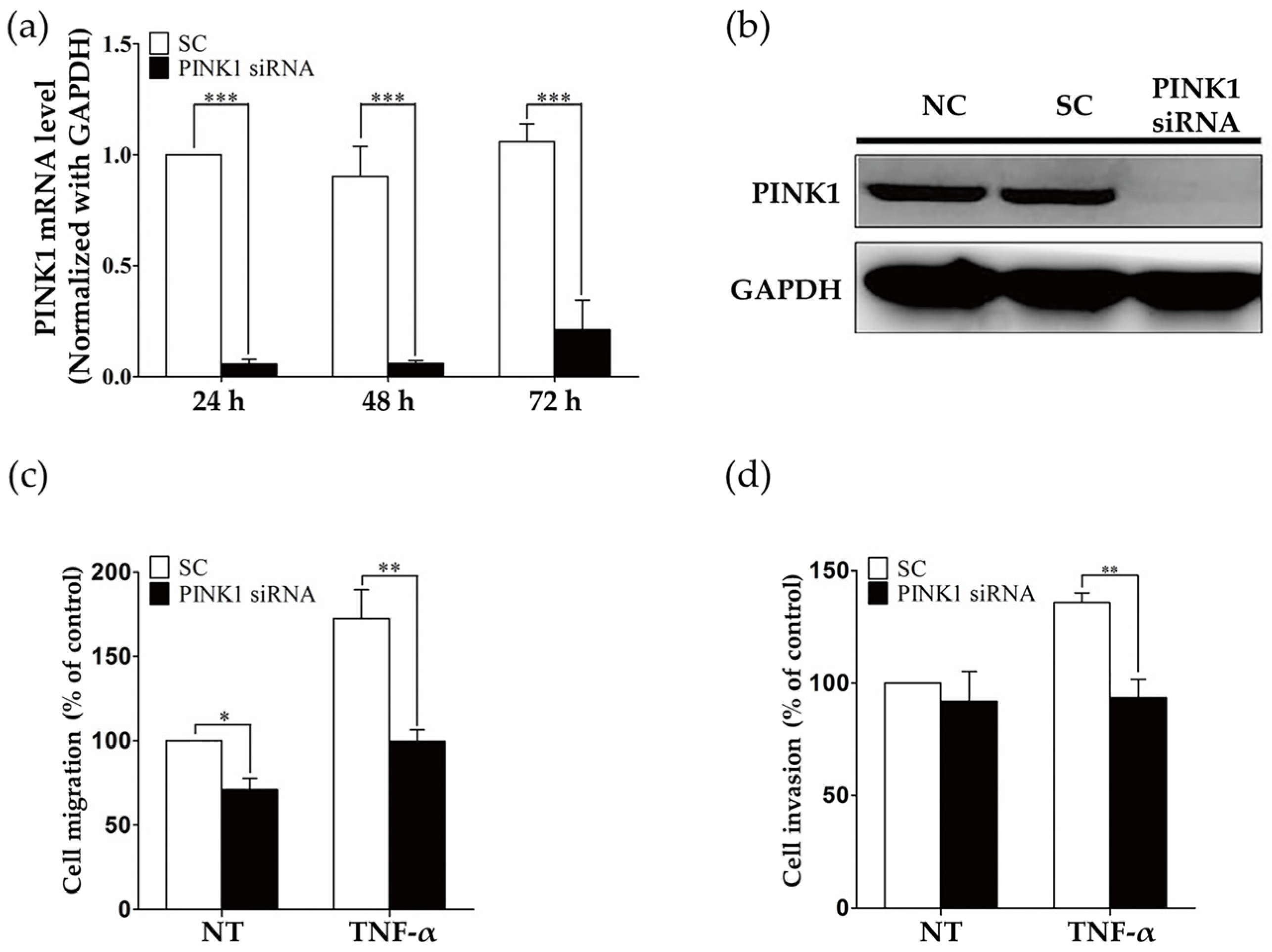
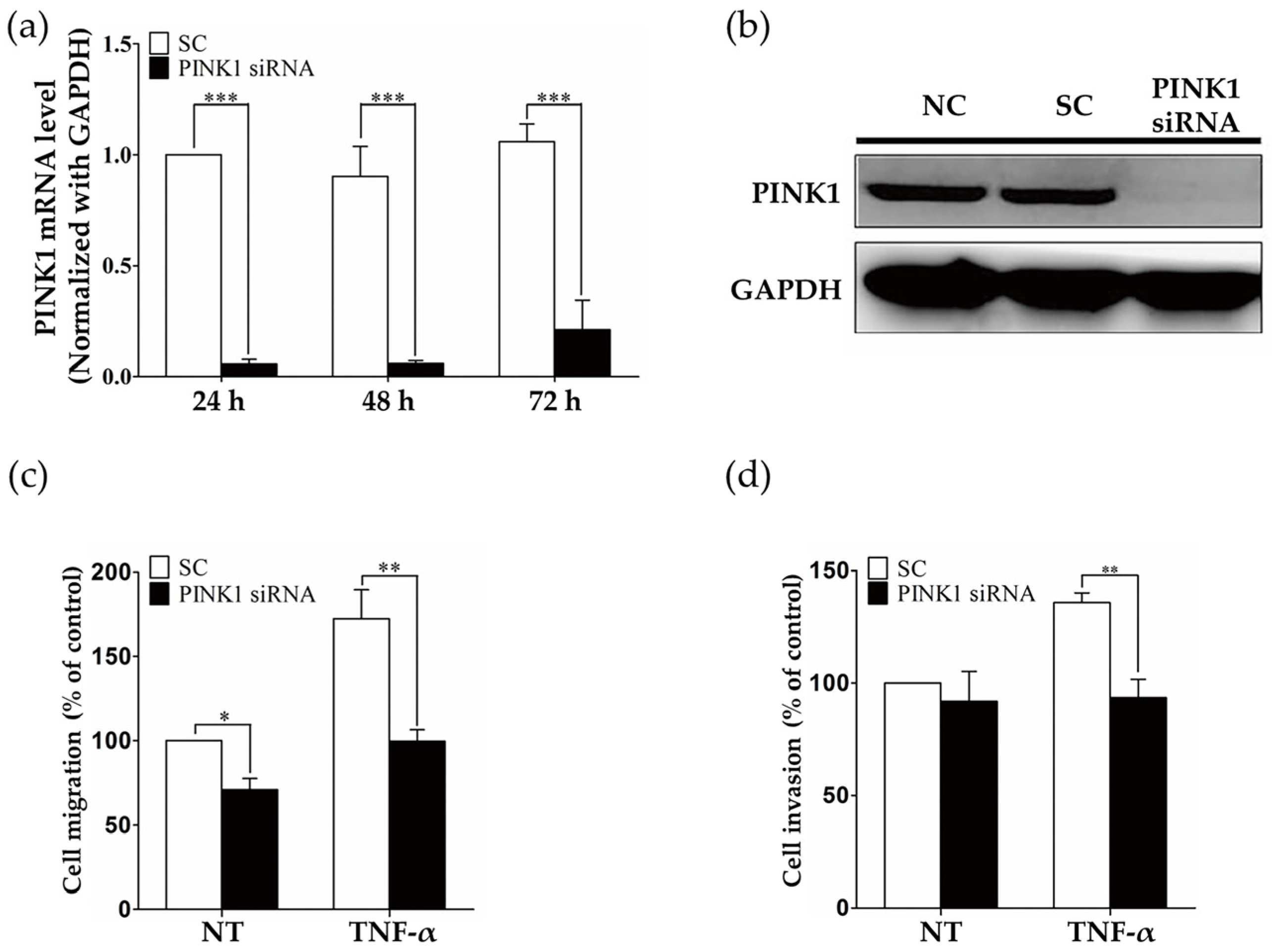
2.3. PINK1 Inhibition Decreased RASF Migration and Invasion
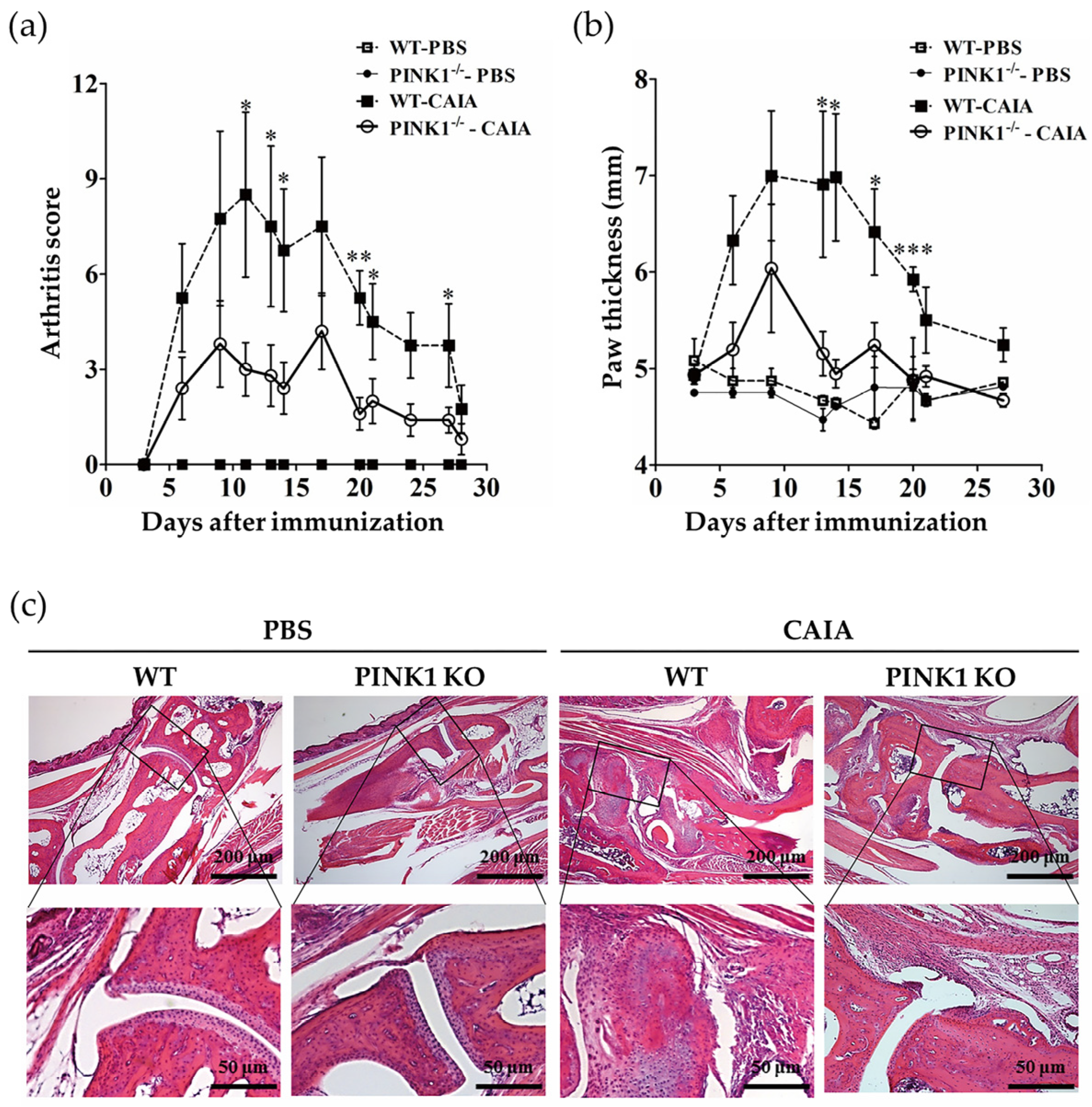
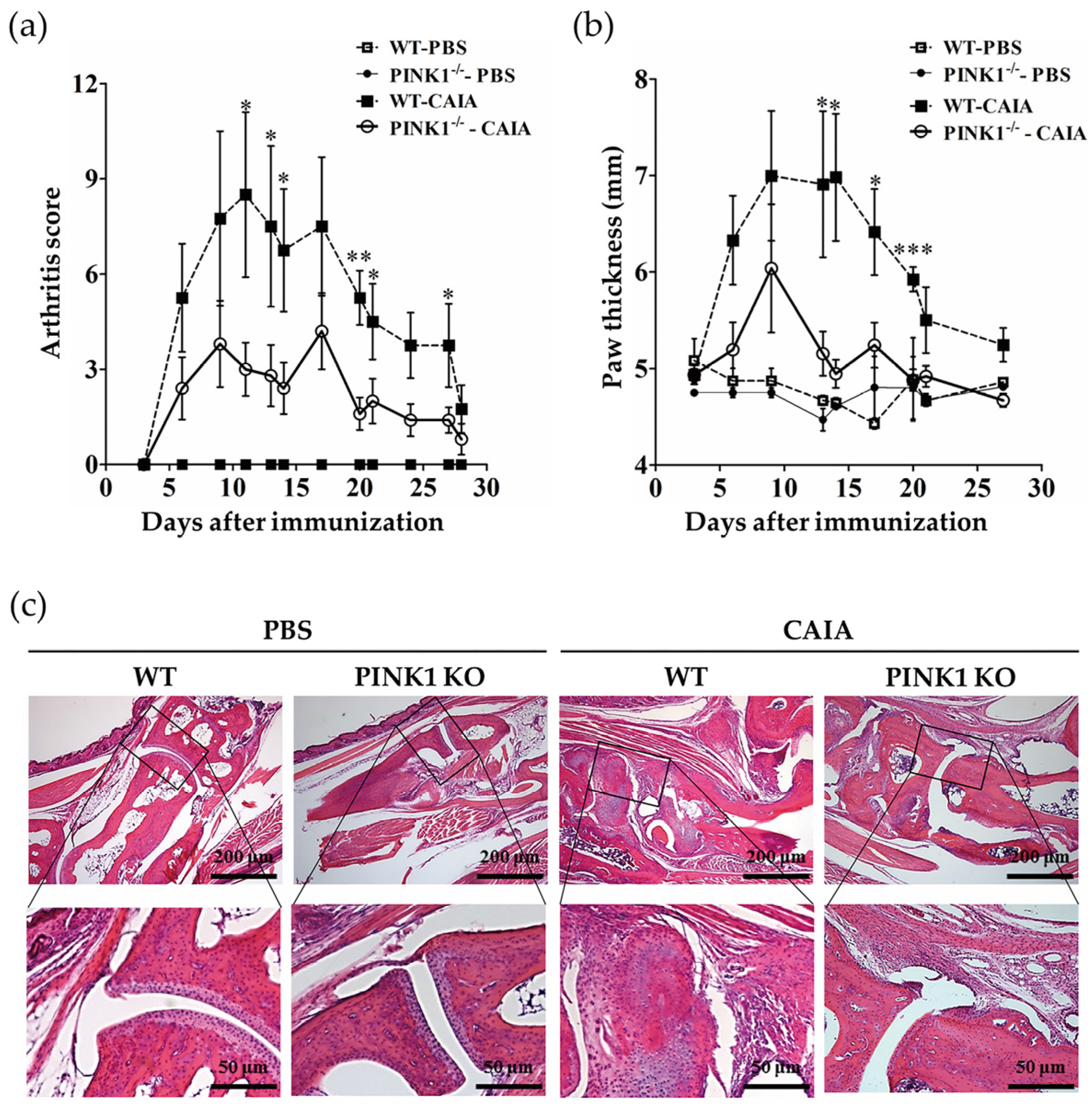
2.4. PINK1 Deficiency Ameliorates Collagen Antibody-Induced Arthritis
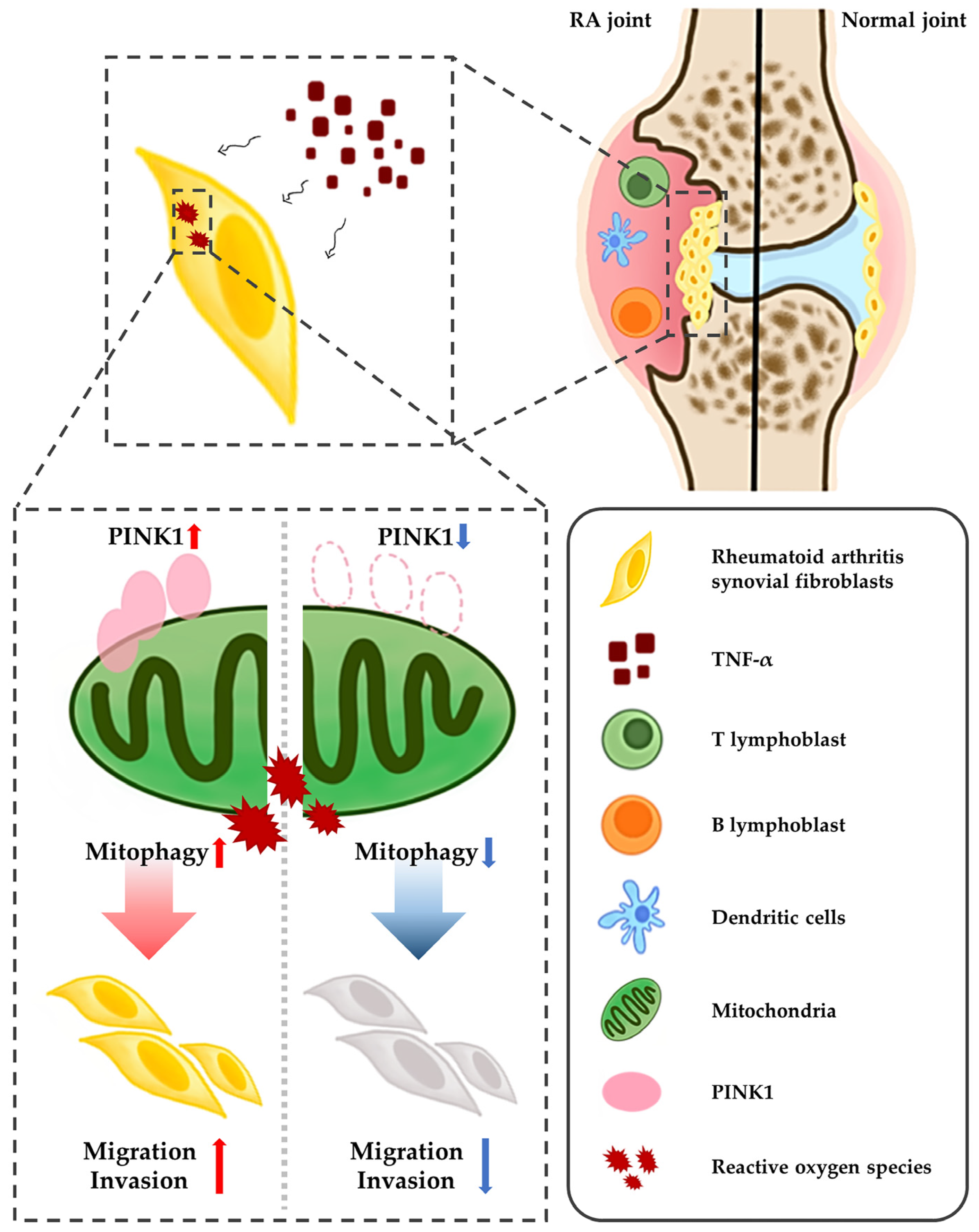
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Approvals of Animal Experiments
4.3. Mice
4.4. Western Blotting
4.5. Immunofluorescence
4.6. Measurement of Mitochondria Membrane Potential (ΔΨm)
4.7. Migration and Invasion Assay
4.8. CAIA Induction
4.9. Histopathological Examination
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| RA | Rheumatoid arthritis |
| RASFs | Rheumatoid arthritis synovial fibroblasts |
| PINK1 | PTEN-induced putative kinase1 |
| TNF-α | Tumor necrosis factor-alpha |
| CCCP | Carbonyl cyanide 3-chlorophenylhydrazone |
| NAC | N-acetylcysteine |
| CAIA | Collagen antibody-induced arthritis |
| ROS | Reactive oxygen species |
| TNF-α | Tumor necrosis factor-alpha |
References
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, N.; Schwarz-Eywill, M.; Weis, D.; Leppelmann-Jansen, P.; Lukoschek, M.; Keilholz, U.; Barth, T.F. Increased expression of integrins on fibroblast-like synoviocytes from rheumatoid arthritis in vitro correlates with enhanced binding to extracellular matrix proteins. Ann. Rheum. Dis. 1997, 56, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.S.; Kang, M.S.; Jeong, J.A.; Bae, Y.S. Semi-mature DC are immunogenic and not tolerogenic when inoculated at a high dose in collagen-induced arthritis mice. Eur. J. Immunol. 2009, 39, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.H.; Lee, J.H.; Jung, N.C.; Choi, H.J.; Song, J.Y.; Seo, H.G.; Choi, J.; Jung, S.Y.; Kang, S.; Choi, Y.S.; et al. Rosiglitazone-mediated dendritic cells ameliorate collagen-induced arthritis in mice. Biochem. Pharmacol. 2016, 115, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed. J. 2021, 44, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Panayi, G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 2001, 344, 907–916. [Google Scholar] [CrossRef]
- Mcinnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Connor, A.M.; Mahomed, N.; Gandhi, R.; Keystone, E.C.; Berger, S.A. TNFalpha modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2012, 14, R62. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcwilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Yang, J.; Wang, D.; Li, C.; Fu, Y.; Wang, H.; He, W.; Zhang, J. Mitophagy in Parkinson’s Disease: Pathogenic and Therapeutic Implications. Front. Neurol. 2017, 8, 527. [Google Scholar] [CrossRef] [Green Version]
- Beilina, A.; Van Der Brug, M.; Ahmad, R.; Kesavapany, S.; Miller, D.W.; Petsko, G.A.; Cookson, M.R. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 2005, 102, 5703–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.J.; Boussaad, I.; Jarazo, J.; Fitzgerald, J.C.; Antony, P.; Keatinge, M.; Blechman, J.; Schwamborn, J.C.; Kruger, R.; Placzek, M.; et al. PINK1 deficiency impairs adult neurogenesis of dopaminergic neurons. Sci. Rep. 2021, 11, 6617. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, J.-H.; Lee, J.-H.; Choi, H.-J.; Choi, S.-Y.; Noh, K.-E.; Jung, N.-C.; Song, J.-Y.; Choi, J.; Seo, H.G.; Jung, S.Y.; et al. TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis. Int. J. Mol. Sci. 2022, 23, 5650. https://doi.org/10.3390/ijms23105650
Nam J-H, Lee J-H, Choi H-J, Choi S-Y, Noh K-E, Jung N-C, Song J-Y, Choi J, Seo HG, Jung SY, et al. TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis. International Journal of Molecular Sciences. 2022; 23(10):5650. https://doi.org/10.3390/ijms23105650
Chicago/Turabian StyleNam, Ji-Hee, Jun-Ho Lee, Hyun-Ji Choi, So-Yeon Choi, Kyung-Eun Noh, Nam-Chul Jung, Jie-Young Song, Jinjung Choi, Han Geuk Seo, Sang Youn Jung, and et al. 2022. "TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis" International Journal of Molecular Sciences 23, no. 10: 5650. https://doi.org/10.3390/ijms23105650
APA StyleNam, J.-H., Lee, J.-H., Choi, H.-J., Choi, S.-Y., Noh, K.-E., Jung, N.-C., Song, J.-Y., Choi, J., Seo, H. G., Jung, S. Y., & Lim, D.-S. (2022). TNF-α Induces Mitophagy in Rheumatoid Arthritis Synovial Fibroblasts, and Mitophagy Inhibition Alleviates Synovitis in Collagen Antibody-Induced Arthritis. International Journal of Molecular Sciences, 23(10), 5650. https://doi.org/10.3390/ijms23105650