
Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes



Abstract
:1. Introduction
2. Results
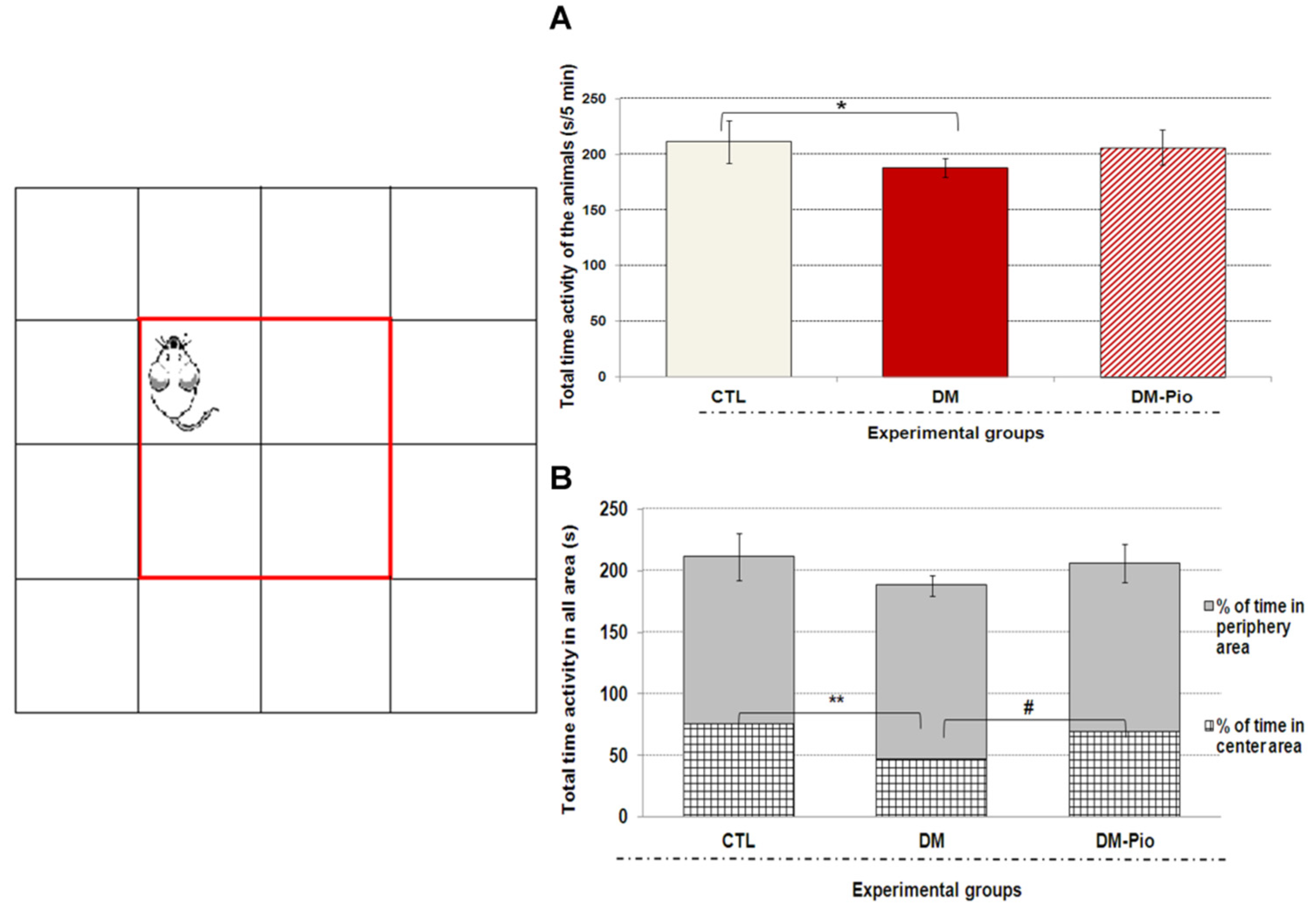
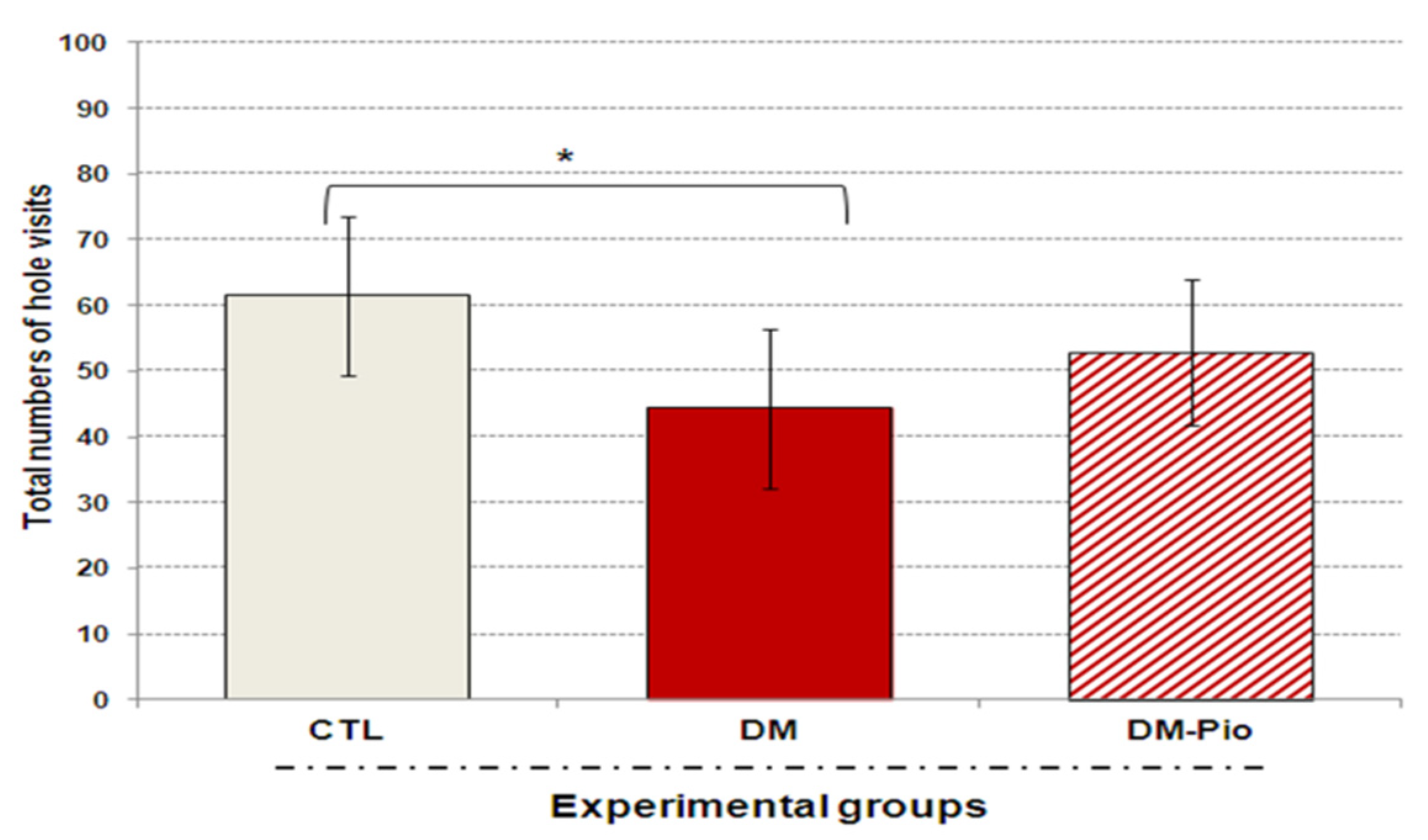
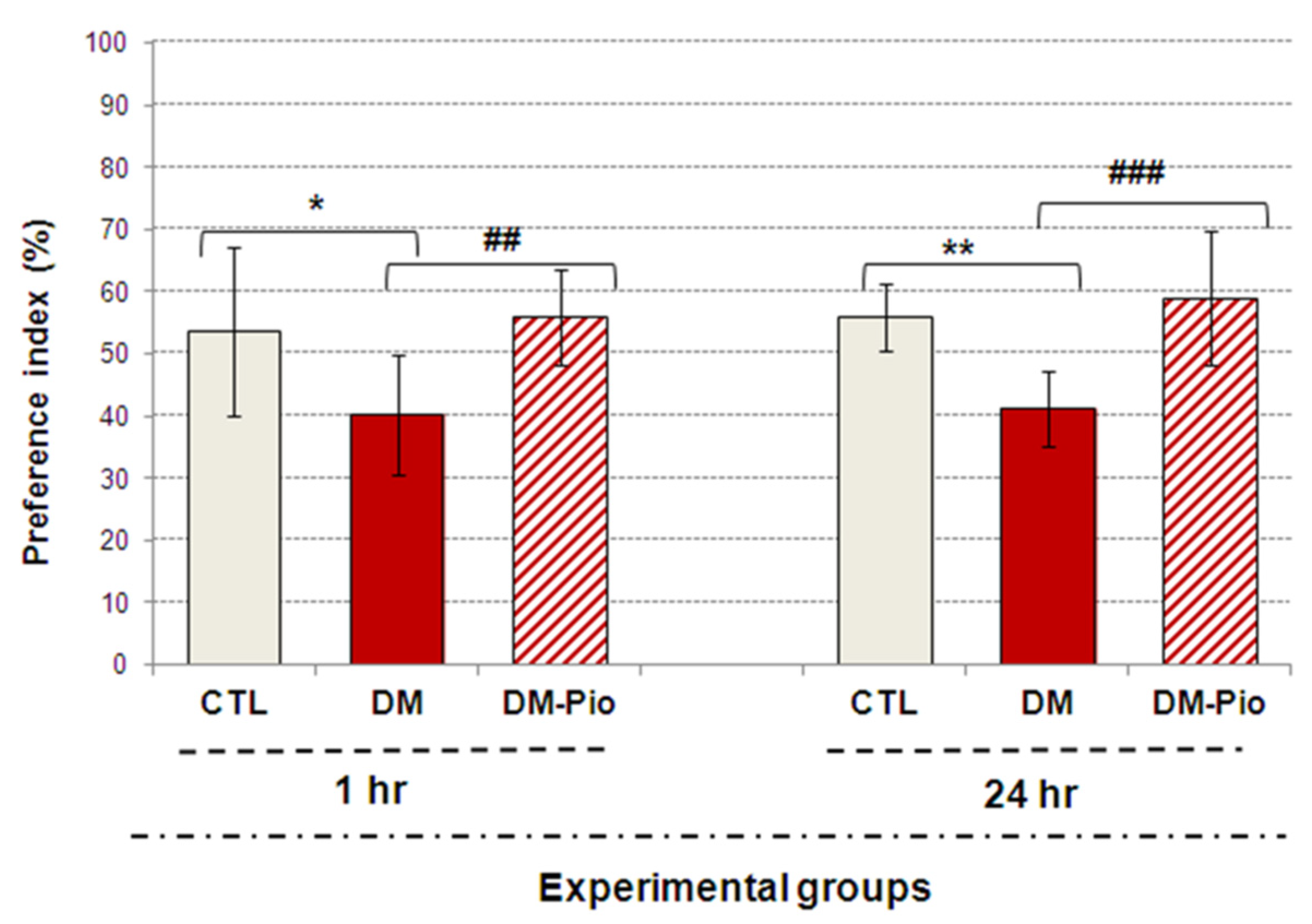
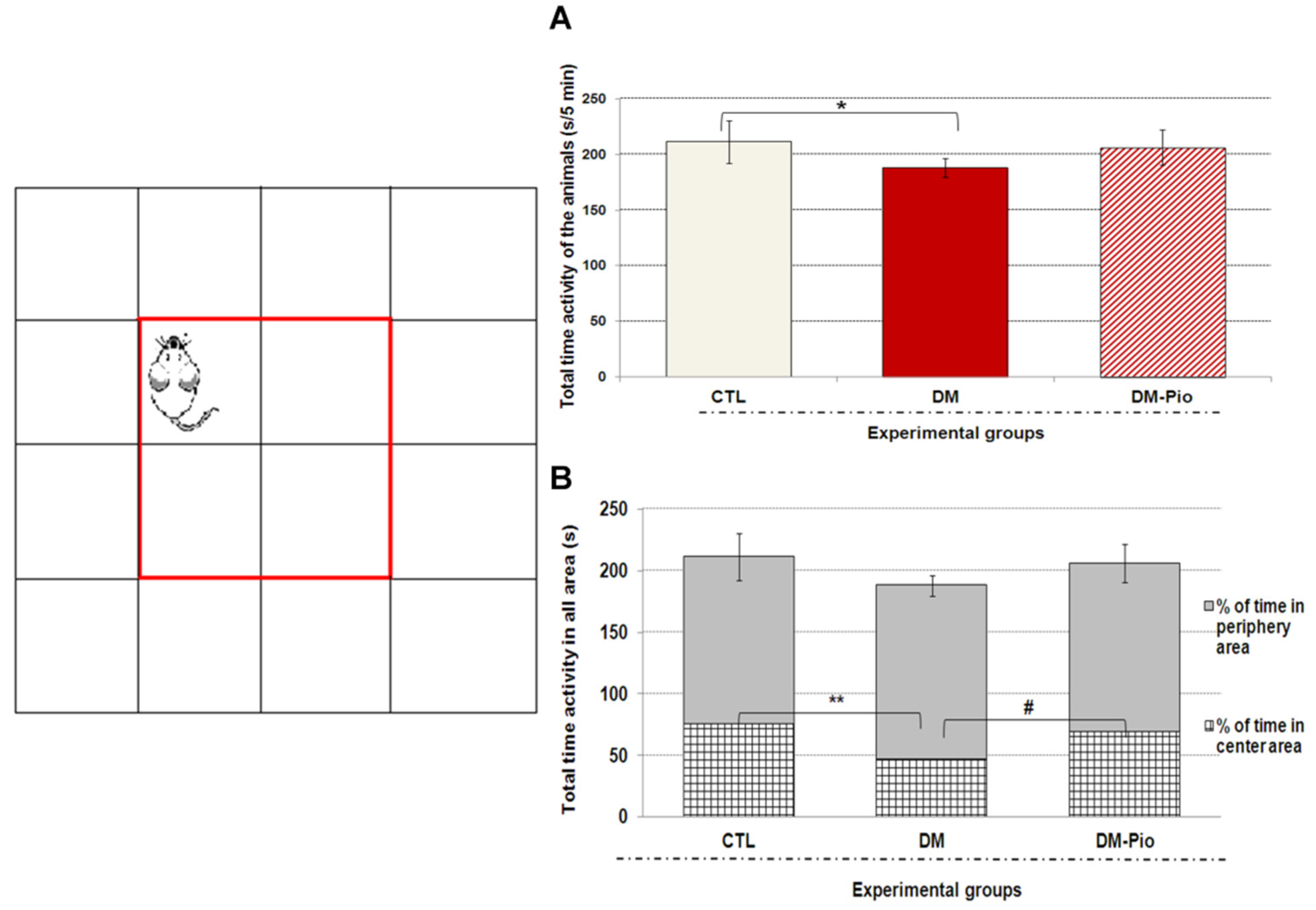
2.1. Effect of Pioglitazone on Exploratory Activity and Memory of Diabetic Mice
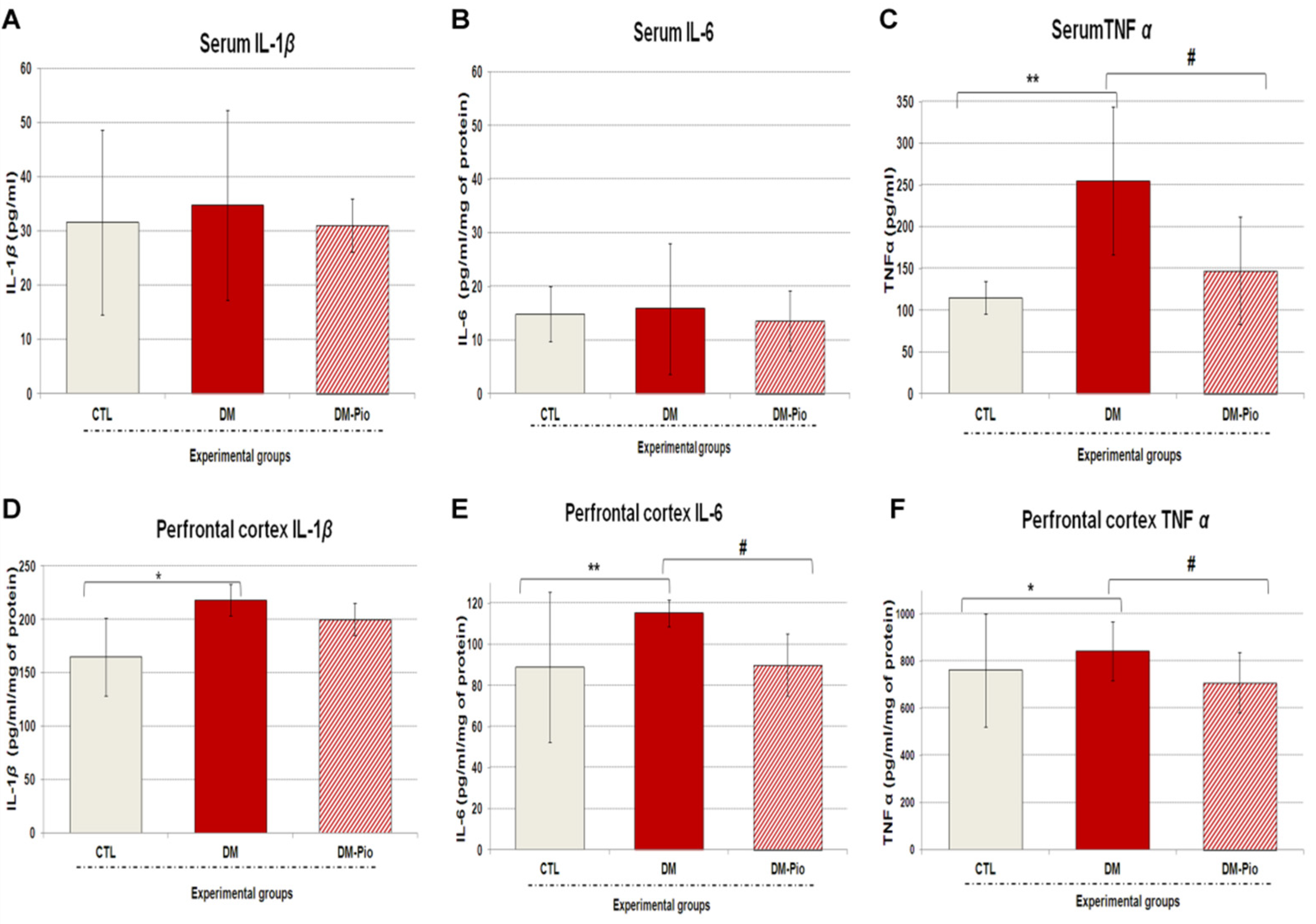
2.2. Effect of Pioglitazone on the Level of Peripheral and Central Pro-Inflammatory Cytokines in Diabetes Mice
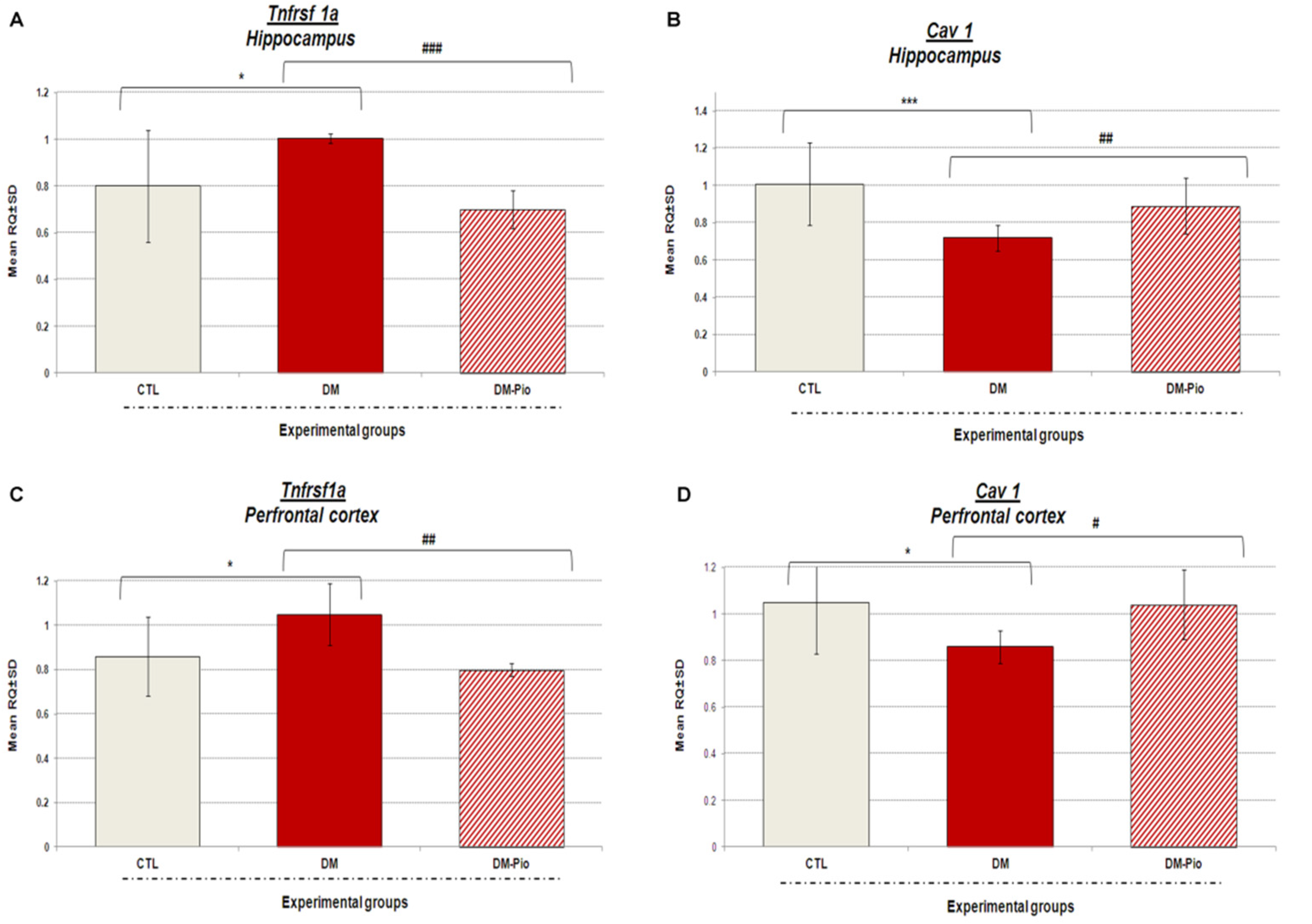
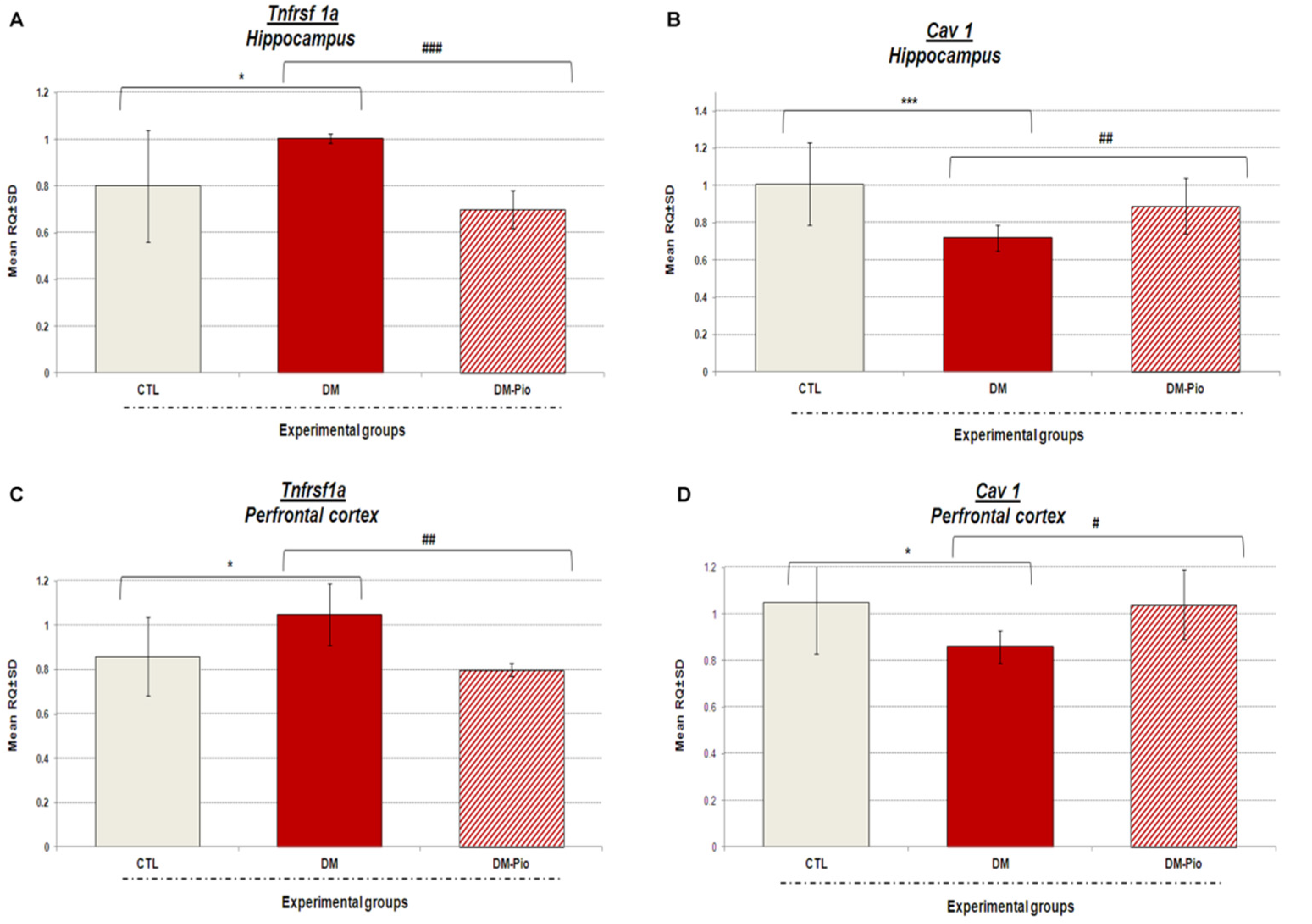
2.3. Changes in Tnfrsf1a and Cav1 Gene Expression in the Hippocampus and Prefrontal Cortex of Diabetes Mice in Response to Treatment with Pio
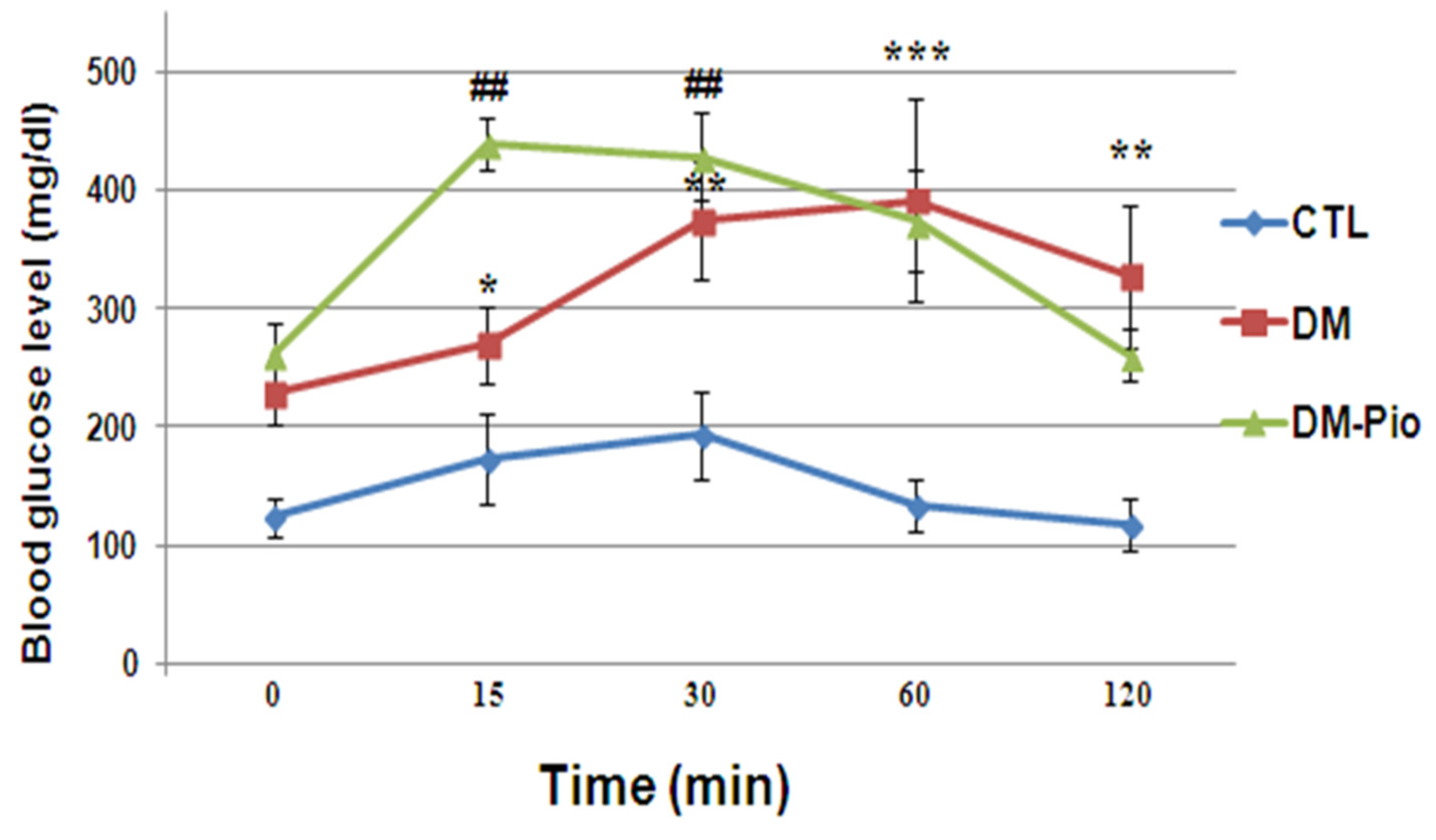
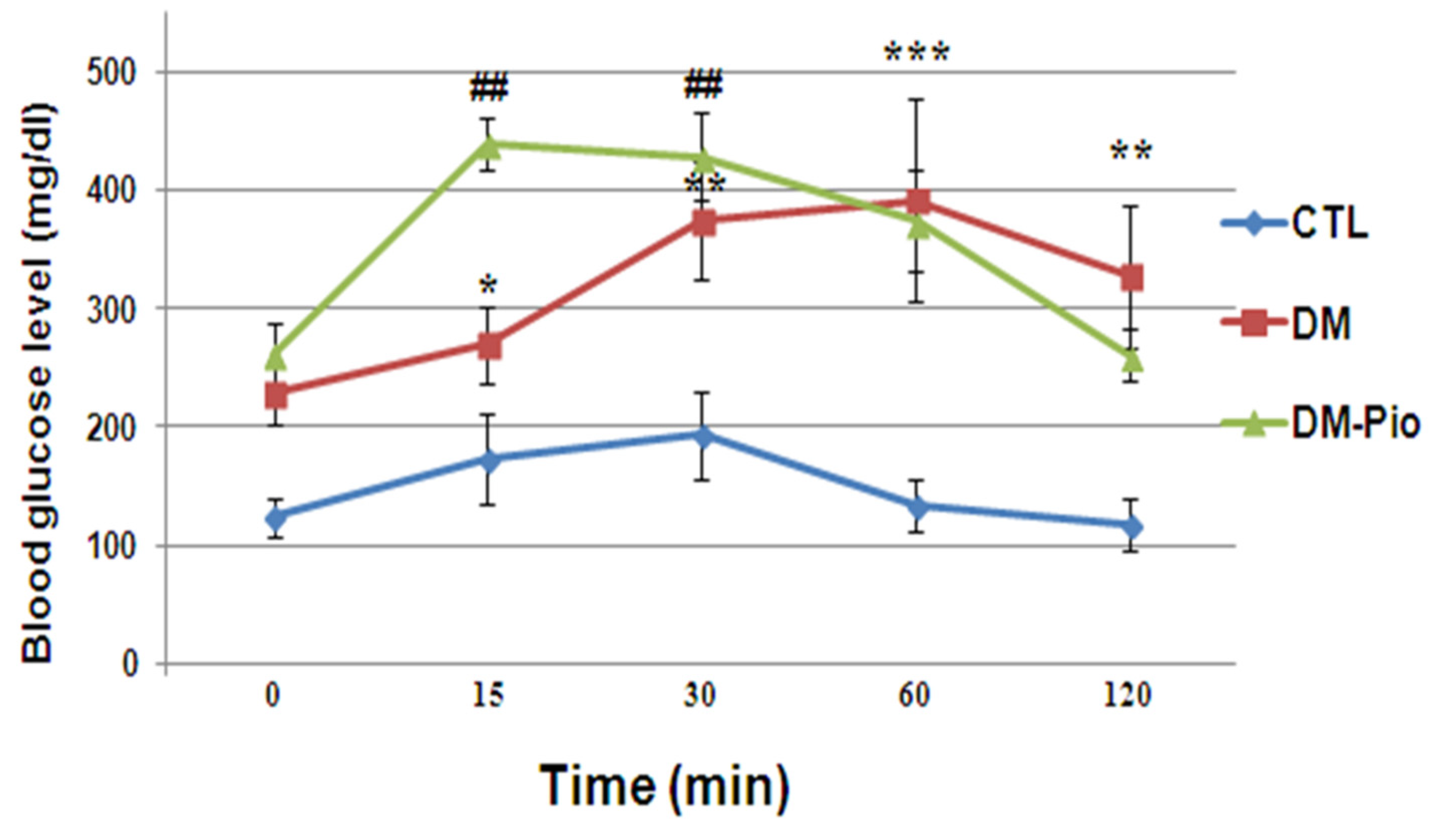
2.4. Effect of Pioglitazone on Level of Glucose during the Oral Glucose Tolerance Test (OGTT) in Diabetes Mice
2.5. Statistical Analysis
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Behavioral Tests
4.2.1. Open Field Test (OFT)
4.2.2. The Hole-Board Test (HBT)
4.2.3. The Novel Object Recognition Test (NOR)
4.3. Collection of Biological Materials for Research
4.3.1. Measurement of Cytokines in the Serum and Brain Supernatants
4.3.2. The Quantitative Real-Time PCR Analysis (qRT-PCR)
RNA Isolation from Hippocampus and Prefrontal Cortex of Mice
CDNA Synthesis
Real-Time PCR
4.3.3. Oral Glucose Tolerance Test (OGTT)
5. Conclusions and Prospects for the Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Wu, J.; Koc, S.; Lu, G. Genetic Imaging of Neuroinflammation in Parkinson’s Disease: Recent Advancements. Front. Cell Dev. Biol. 2021, 10, 1008. [Google Scholar] [CrossRef]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef]
- Comer, A.L.; Carrier, M.; Tremblay, M.E.; Cruz-Martín, A. The Inflamed Brain in Schizophrenia: The Convergence of Genetic and Environmental Risk Factors That Lead to Uncontrolled Neuroinflammation. Front. Cell Neurosci. 2020, 14, 274. [Google Scholar] [CrossRef]
- Benedetti, F.R.; Aggio, V.; Pratesi, M.L.; Greco, G.; Furlan, R. Neuroinflammation in Bipolar Depression. Front. Psychiatry 2020, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Giuliani, F. The Role of Inflammation in Depression and Fatigue. Front. Immunol. 2019, 10, 1696. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-H.; Zhang, G.-Z.; Li, B.; Li, M.; Woelfer, M.; Walter, M.; Wang, L. Role of inflammation in depression relapse. J. Neuroinflamm. 2019, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Asslih, S.; Damri, O.; Agam, G. Neuroinflammation as a Common Denominator of Complex Diseases. Int. J. Mol. Sci. 2021, 22, 6138. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Smith, C.D.; Abner, E.A.; Schmitt, F.A.; Scheff, S.W.; Davis, G.J.; Keller, J.N.; Jicha, G.A.; Davis, D.; Wang-Xia, W.; et al. Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim. Biophys. Acta 2009, 1792, 454–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderbom, G.; Zeng, B.Y. The NLRP3 inflammasome as a bridge between neuro-inflammation in metabolic and neurodegenerative; diseases. Int. Rev. Neurobiol. 2020, 154, 345–391. [Google Scholar] [PubMed]
- Xu, W.; Caracciolo, B.; Wang, H.X.; Winblad, B.; Backman, L.; Qiu, C.; Fratiglioni, L. Accelerated progression from mild cognitive impairment to dementia in people with diabetes. Diabetes 2010, 59, 2928–2935. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Bourgognon, J.M.; Cavanagh, J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci Adv. 2020, 4, 1–13. [Google Scholar] [CrossRef]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Zimmet, Z.P.; Magliano, J.D.; Herman, H.W.; Shaw, E.J. Diabetes: A 21st century challenge. Lancet 2014, 2, 56–64. [Google Scholar] [CrossRef]
- Wrighten, S.A.; Piroli, G.G.; Grillo, C.A.; Reagan, L.P. A look inside the diabetic brain:Contributors to diabetes induced brain aging. Biochim. Biophys Acta 2009, 1792, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Newgard, C.B. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef]
- Arner, P. Insulin resistance in type 2 diabetes-role of the adipokines. Curr. Mol. Med. 2005, 5, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Wang, Y.; Talaei, M.; Hu, F.B.; Wu, T. Relation of active, passive, and quitting smoking with incident type 2 diabetes: A systematic review and meta-analysis. Lancet Diab. Endocrinol. 2015, 3, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fu, A.K.Y.; Nancy, Y. Instructive roles of astrocytes in hippocampal synaptic plasticity: Neuronal activity-dependent regulatory mechanisms. FEBS 2021, 289, 2202–2218. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Kanaya, A.; Lindquist, K.; Simonsick, E.M.; Harris, T.; Shorr, R.I.; Tylavsky, F.A.; Newman, A.B. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 2004, 292, 2237–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skundric, D.S.; Lisak, R.P. Role of neuropoietic cytokines in development and progression of diabetic polyneuropathy: From glucose metabolism to neurodegeneration. J. Diabetes Res. 2003, 4, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Turrin, N.P.; Rivest, S. Tumor necrosis factor α but not interleukin 1β mediates neuroprotection in response to acute nitric oxide excitotoxicity. J. Neurosci. 2006, 26, 143–151. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M. Relationships between diabetes and cognitive impairment. Endocrinol. Metab. Clin. N. Am. 2014, 43, 245–267. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. GIOLA 2012, 61, 71–90. [Google Scholar] [CrossRef]
- Tan, Z.S.; Beiser, A.S.; Vasan, R.S.; Roubenoff, R.; Dinarello, C.A.; Harris, T.B.; Benjamin, E.J.; Au, R.; Kiel, D.P.; Wolf, P.A.; et al. Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology 2007, 68, 1902–1908. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Tesfaye, S.; Chaturvedi, N.; Eaton, S.E.M.; Ward, J.D.; Manes, C.; Ionescu-Tirgoviste, C.; Witte, D.R.; Fuller, J.H. Vascular risk factors and diabetic neuropathy. N. Eng. J. Med. 2005, 352, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.L.; Vincent, A.; Cheng, T.; Feldman, E.L. Diabetic neuropathy: Mechanisms to management. Pharmacol. Ther. 2008, 120, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.C.; Cheng, H.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Han, Q.; Lv, Y.; Sun, L.; Gang, X.; Wang, G. Biomarkers for cognitive decline in patients with diabetes mellitus: Evidence from clinical studies. Oncotarget 2017, 9, 7710–7726. [Google Scholar] [CrossRef]
- Yue, X.; Li, H.; Zhang, P.; Chang, L.; Li, T. Risk of Parkinson Disease in Diabetes Mellitus: An Updated Meta-Analysis of Population-Based Cohort Studies. Medicine 2016, 95, e3549. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes 1996, 45, 1661–1669. [Google Scholar] [CrossRef]
- Ji, S.; Kronenberg, G.; Balkaya, M.; Färber, K.; Gertz, K.; Kettenmann, H.; Endres, M. Acute neuroprotection by pioglitazone after mild brain ischemia without effect on long-term outcome. Exp. Neurol. 2009, 216, 321–328. [Google Scholar] [CrossRef]
- Jin, H.Y.; Lee, K.A.; Wu, J.Z.; Baek, H.S.; Park, T.S. The neuroprotective benefit from pioglitazone(PIO) addition on the alpha lipoic acid (ALA)-based treatment in experimental diabetic rats. Endocrine 2014, 47, 772–782. [Google Scholar] [CrossRef]
- Villapol, S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef]
- Bernardo, A.; Levi, G.; Minghetti, L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-Δ12, 14-prostaglandin J2 in the regulation of microglial functions. Eur. J. Neurosci. 2000, 12, 2215–2223. [Google Scholar] [CrossRef]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.J.; Quintanilla, R.A. Therapeutic Actions of the Thiazolidinediones in Alzheimer’s Disease. PPAR Res. 2015, 2015, 957248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, P.S.; Ho, B.L.; Yang, Y.H. Effects of pioglitazone on the incidence of dementia In patients with diabetes. J. Diabetes Its Complicat. 2017, 31, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Sauerbeck, A.; Gao, J.; Readnower, R.; Liu, M.; Pauly, J.R.; Bing, G.; Sullivan, P.G. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp. Neurol. 2011, 227, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegick, C.G.; Altheimer, M.D. Comparison of effects of thiazolidinediones on cardiovascular risk factors: Observations from a clinical practice. Endocr. Pract. 2001, 7, 162–169. [Google Scholar] [CrossRef]
- Xia, P.; Pan, Y.; Zhang, F.; Wang, N.; Wang, E.; Guo, Q.; Ye, Z. Pioglitazone Confers Neuroprotection Against Ischemia-Induced Pyroptosis due to its Inhibitory Effects on HMGB-1/RAGE and Rac1/ROS Pathway by Activating PPAR-ɤ. Cell Physiol. Biochem. 2018, 45, 2351–2368. [Google Scholar] [CrossRef]
- Yin, Q.Q.; Pei, J.J.; Xu, S.; Luo, D.Z.; Dong, S.Q.; Sun, M.H.; You, L.; Sun, Z.J.; Liu, X.P. Pioglitazone improves cognitive function via increasing insulin sensitivity and strengthening antioxidant defense system in fructose-drinking insulin resistance rats. PLoS ONE 2013, 8, e59313. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.; Ginty, M.; Kemp, K.; Scolding, N.; Wilkins, A. The PPAR-γ agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J. Neuroinflamm. 2012, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Inestrosa, N.C. Peroxisome proliferator-activated receptors and alzheimer’s disease: Hitting the blood-brain barrier. Mol. Neurobiol. 2013, 48, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cuzzocrea, S. Targeting the peroxisome proliferator-activated receptors (PPARs) in spinal cord injury. Expert Opin. Ther. Targets 2011, 15, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Sekulic-Jablanovic, M.; Petkovic, V.; Wright, M.B.; Kucharava, K.; Huerzeler, N.; Levano, S.; Brand, Y.; Leitmeyer, K.; Glutz, A.; Bausch, A.; et al. Effects of peroxisome proliferator activated receptors (PPAR)-γ and -α agonists on cochlear protection from oxidative stress. PLoS ONE 2017, 12, e0188596. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Blanchard, J.; Li, Y.; Iqbal, K.; Liu, F.; Gong, C.-X. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J. Neural. Transm. 2015, 122, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, G.; Ziouzenkova, O.; Devchand, P.R.; Nehr, V.; Hamdy, E.; Horton, O.S.; Plutzky, J. The Peroxisome Proliferator-Activated Receptor-γ Agonist Pioglitazone Represses Inflammation in a Peroxisome Proliferator-Activated Receptor-α–Dependent Manner In Vitro and In Vivo in Mice. J. Am. Coll. Cardiol. 2008, 52, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaf, N.E.; El-Shamarka, M.; Salemb, N.A.; Khadrawy, Y.A.; El Sayede, N.S. Neuroprotective effect of PPAR alpha and gamma agonists in a mouse model of amyloidogenesis through modulation of the Wnt/beta catenin pathway via targeting alpha- and beta-secretases. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109793. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajd, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Duran-Sandoval, D.; Thomas, A.C.; Bailleul, B.; Fruchart, J.C.; Staels, B. Pharmacology of PPARalpha, PPARgamma and dual PPARalpha/gamma agonists in clinical development. Med. Sci. 2003, 19, 819–825. [Google Scholar]
- Piatkowska-Chmiel, I.; Herbet, M.; Gawronska-Grzywacz, M.; Ostrowska-Lesko, M.; Dudka, J. The Role of Molecular and Inflammatory Indicators in the Assessment of Cognitive Dysfunction in a Mouse Model of Diabetes. Int. J. Mol. Sci. 2021, 22, 3878. [Google Scholar] [CrossRef]
- Barrientos, R.M.; Sprunger, D.B.; Campeau, S.; Higgins, E.A.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847–853. [Google Scholar] [CrossRef]
- Menachem-Zidon, O.B.; Goshen, I.; Kreisel, T.; Menahem, Y.B.; Reinhartz, E.; Hur, T.B.; Yirmiya, R. Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 2008, 33, 2251–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.W.; Chen, Y.C.; Yu, L.; Chen, H.I.; Jen, C.J.; Huang, A.M.; Tsai, H.J.; Chang, Y.T.; Kuo, Y.M. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J. Neurochem. 2007, 103, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Hara, M.; Nakamura, Y.; Kozaki, S.; Tsunoda, S.; Ihara, H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci. Lett. 2008, 432, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Gavillet, M.; Allaman, I.; Magistretti, P.J. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 2008, 56, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; Pinteaux, E.; Gibson, R.M.; Allan, S.M.; Rothwell, N.J. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J. Neurochem. 2006, 98, 258–266. [Google Scholar] [CrossRef]
- Reichenberg, A.; Yirmija, R.; Schulda, A.; Kraus, T.; Haack, M.; Morąg, A.; Polmacher, T. Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry 2001, 58, 445–452. [Google Scholar] [CrossRef]
- Weaver, J.D.; Huang, M.-H.; Albert, M.; Harris, T.; Rowe, J.W.; Seeman, T.E. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 2002, 59, 371–378. [Google Scholar] [CrossRef]
- Koo, J.W.; Duman, R.S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. USA 2008, 105, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.R.; Wang, M.; Ribeiro, D.; Cho, H.J.; Olmstead, R.; Breen, E.C.; Martinez-Maza, O.; Cole, S. Sleep loss activates cellular inflammatory signaling. Biol. Psychiatry 2008, 64, 538–540. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 5. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Kastin, A.J. TNFα transport across the blood-brain barrier is abolished in receptor knockout mice. Exp. Neurol. 2002, 174, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Kastin, A.J. Penetration of neurotrophins and cytokines across the blood-brain/blood-spinal cord barrier. Adv. Drug Deliv. Rev. 1999, 36, 291–298. [Google Scholar] [CrossRef]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, G.R.I.; Bird, F.; Alexander, V.; Warburton, E.C. Recognition Memory for Objects, Place, and Temporal Order: A Disconnection Analysis of the Role of the Medial Prefrontal Cortex and Perirhinal Cortex. J. Neurosci. 2007, 27, 2948–2957. [Google Scholar] [CrossRef] [Green Version]
- Aggleton, J.P.; Albasser, M.M.; Aggleton, D.J.; Poirier, G.L.; Pearce, J.M. Lesions of the rat perirhinal cortex spare the acquisition of a complex confgural visual discrimination yet impair object recognition. Behav. Neurosci. 2010, 124, 55–68. [Google Scholar] [CrossRef] [Green Version]
- DeVito, L.M.; Eichenbaum, H. Memory for the Order of Events in Specifc Sequences: Contributions of the Hippocampus and Medial Prefrontal Cortex. J. Neurosci. 2011, 31, 3169–3175. [Google Scholar] [CrossRef]
- Roberts, A.J.; Khom, S.; Bajo, M.; Vlkolinsky, R.; Polis, I.; Cates-Gatto, C.; Roberto, M.; Gruol, D.L. Increased IL-6 expression in astrocytes is associated with emotionality, alterations in central amygdala GABAergic transmission, and excitability during alcohol withdrawal. Brain Behav. Immun. 2019, 82, 188–202. [Google Scholar] [CrossRef]
- Eyre, H.; Papps, E.; Baune, B.T. Treating depression and depres-sion-like behavior with physical activity: An immune perspective. Front. Psychiatry 2013, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Rouen, S.; Barkus, K.M.; Dremina, Y.S.; Hui, D.; Christianson, J.A.; Wright, D.E.; Yoon, S.O.; Dobrowsky, R.T. Nerve growth factor blocks the glucose-induced down-regulation of caveolin-1 expression in Schwann cells via p75 neurotrophin receptor signaling. J. Biol. Chem. 2003, 278, 23151–23162. [Google Scholar] [CrossRef] [Green Version]
- Niesman, I.R.; Schilling, J.M.; Shapiro, L.A.; Kellerhals, S.E.; Bonds, J.A.; Kleschevnikov, A.M.; Cui, W.; Voong, A.; Krajewski, S.; Ali, S.S.; et al. Traumatic brain injury enhances neuroinflammation and lesion volume in caveolin deficient mice. J. Neuroinflamm. 2014, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS ONE 2010, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Charme, J.D.; Parisi, J.; McMurray, C.T. Neurological abnormalities in caveolin-1 knock out mice. Behav. Brain Res. 2006, 172, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.Q.; Liu, Z.J. Neuroinflammation and neuronal autophagic death were suppressed via Rosiglitazone treatment: New Brain injury via the PPARg/NF-kB/IL-6 signaling pathway evidence on neuroprotection in a rat model of global cerebral ischemia. J. Neurol. Sci. 2015, 349, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Woster, A.P.; Combs, C.K. Differential ability of thiazolidinedione PPARγ agonist to attenuate cytokine secretion in primary microglia and macrophage-like cells. J. Neurochem. 2007, 103, 67–76. [Google Scholar] [PubMed]
- Jung, H.J.; Kim, Y.J.; Eggert, S.; Chung, K.C.; Choi, K.S.; Park, S.A. Age-dependent increases in tau phosphorylation in the brains of type 2 diabetic rats correlate with a reduced expression of p62. Exp. Neurol. 2013, 248, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Phatak, H.M.; Yin, D.D. Factors associated with the effect-size of thiazolidinedione (TZD) therapy on HbA1c: A meta-analysis of published randomized clinical trials. Curr. Med. Res. Opin. 2006, 22, 2267–2278. [Google Scholar] [CrossRef]
- El-Sahar, A.E.; Safar, M.M.; Zaki, H.F.; Attia, A.S.; Ain-Shoka, A.A. Neuroprotective effects of pioglitazone against transient cerebral ischemic reperfusion injury in diabetic rats: Modulation of antioxidant, anti-inflammatory, and anti-apoptotic biomarkers. Pharmacol. Rep. 2015, 67, 901–906. [Google Scholar] [CrossRef]
- Barbiero, J.K.; Santiago, R.; Tonin, F.; Boschen, S.; da Silva, L.M.; Werner, M.F.; Da Cunha, C.; Lima, M.M.S.; Vital, M.A. PPAR-alpha agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 35–44. [Google Scholar] [CrossRef]
- Polak, P.E.; Kalinin, S.; Russo, C.D.; Gavrilyuk, V.; Sharp, A.; Peters, J.M.; Richardson, J.; Willson, T.M.; Weinberg, G.; Feinstein, D.L. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 168, 65–75. [Google Scholar] [CrossRef]
- Baghcheghi, Y.; Beheshti, F.; Salmani, H.; Soukhtanloo, M.; Hosseini, M. Protective Effect of PPARγ Agonists on Cerebellar Tissues Oxidative Damage in Hypothyroid Rats. Neurol. Res. Int. 2016, 2016, 1952561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masciopinto, F.; Di Pietro, N.; Corona, C.; Bomba, M.; Pip-ino, C.; Curcio, M.; Di Castelnuovo, A.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.T.; et al. Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 2012, 3, e448. [Google Scholar] [CrossRef] [PubMed]
- Maeshiba, Y.; Kiyota, Y.; Yamashita, K.; Yoshimura, Y.; Motohashi, M.; Tanayama, S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. ArzneimittelForschung 1997, 47, 29–35. [Google Scholar] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.-H.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.-W.; Kang, E.S.; et al. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef]
- Grommes, C.; Karlo, J.C.; Caprariello, A.; Blankenship, D.; Dechant, A.; Landreth, G.E. The PPARγ agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemother. Pharmacol. 2013, 71, 929–936. [Google Scholar] [CrossRef]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schäfers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-γ Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef]
- Wang, X.M.; Kim, H.P.; Song, R.; Choi, A.M.K. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am. J. Resp. Cell Mol. Biol. 2006, 34, 434–442. [Google Scholar] [CrossRef]
- Fazia, T.; Nova, A.; Gentilini, D.; Beecham, A.; Piras, M.; Saddi, V.; Ticca, A.; Bitti, P.; McCauley, J.L.; Berzuini, C.; et al. Investigating the Causal Effect of Brain Expression of CCL2, NFKB1, MAPK14, TNFRSF1A, CXCL10 Genes on Multiple Sclerosis: A Two-Sample Mendelian Randomization Approach. Front. Bioeng. Biotechnol. 2020, 8, 397. [Google Scholar] [CrossRef]
- Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. [Google Scholar] [CrossRef] [Green Version]
- Bettoni, I.; Comelli, F.; Rossini, C.; Granucci, F.; Giagnoni, G.; Peri, F.; Costa, B. Glial TLR4 receptor as new target to treat neuropathic pain: Efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 2008, 56, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Y.; Sun, Y.; Li, H.; Mi, W.; Jiang, Y. Analgesic effects of TLR4/NF-κB signaling pathway inhibition on chronic neuropathic pain in rats following chronic constriction injury of the sciatic nerve. Biomed. Pharmacother. 2018, 107, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Zhang, H.; Kosturakis, A.K.; Jawad, A.B.; Dougherty, P.M. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J. Pain 2014, 15, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Qiu, Y.; Wei, M.; Li, C.; Xie, Y.; Shen, L.; Huang, Y.; Ma, C. Suppression of MyD88-dependent signaling alleviates neuropathic pain induced by peripheral nerve injury in the rat. J. Neuroinflamm. 2017, 14, 70. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, X.; Deng, X.; Chen, H.; Xu, J.; Zhang, Z.; Liu, G.; Yong, Z.; Yuan, C.; Sun, X.; et al. Pioglitazone ameliorates neuronal damage after traumatic brain injury via the PPARg/NF-kB/ IL-6 signaling pathway. Genes Dis. 2019, 7, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Ogasawara, S.; Mizukami, H.; Yajima, N.; Wada, R.I.; Sugawara, A.; Yagihashi, S. Correction of protein kinase C activity and macrophage migration in peripheral nerve by pioglitazone, peroxisome proliferator activated-y-ligand, in insulin-deficient diabetic rats. J. Neurochem. 2008, 104, 491–499. [Google Scholar] [CrossRef]
- Hall, C.S.; Ballachey, E.L. A study of the rat’s behavior in a field: A contribution to method in comparative psychology. In University of California Publications in Psychology; University of California Press: Berkeley, CA, USA, 1932; pp. 1–12. [Google Scholar]
- Boissier, J.R.; Simon, P. Dissociation de deuxcomposantesdanslecomporte mnetd’investigation de la souris. Arch. Int. Pharmacodyn Ther. 1964, 147, 372–387. [Google Scholar]
- Boissier, J.R.; Simon, P. Automatisation du test de la planche a trous. Physiol. Behav. 1967, 2, 447–448. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid sensitive method for the quantification of microgram quantities of protein utilising the principle of protein-dye binding. Anal Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Assay ID | Gene Name | RefSeq | Amplicon Length (bp) |
|---|---|---|---|---|
| Tnfrsf1a | AB ID: Mm00441883_g1 | Tumor necrosis factor receptor superfamily, member 1a | NM_011609.4 | 82 |
| Cav1 | AB ID: Mm00483057_m1 | Caveolin 1 | NM_001243064.1 NM_007616.4 | 67 |
| Hprt | AB ID: Mm00446968_m1 | Hypoxanthine guanine phosphoribosyl transferase | NM_013556.2 | 65 |
| Tbp | AB ID: Mm00446974_m1 | TATA box binding protein | NM_013684.3 | 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piątkowska-Chmiel, I.; Herbet, M.; Gawrońska-Grzywacz, M.; Dudka, J. Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes. Int. J. Mol. Sci. 2022, 23, 5502. https://doi.org/10.3390/ijms23105502
Piątkowska-Chmiel I, Herbet M, Gawrońska-Grzywacz M, Dudka J. Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes. International Journal of Molecular Sciences. 2022; 23(10):5502. https://doi.org/10.3390/ijms23105502
Chicago/Turabian StylePiątkowska-Chmiel, Iwona, Mariola Herbet, Monika Gawrońska-Grzywacz, and Jarosław Dudka. 2022. "Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes" International Journal of Molecular Sciences 23, no. 10: 5502. https://doi.org/10.3390/ijms23105502
APA StylePiątkowska-Chmiel, I., Herbet, M., Gawrońska-Grzywacz, M., & Dudka, J. (2022). Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes. International Journal of Molecular Sciences, 23(10), 5502. https://doi.org/10.3390/ijms23105502