
GANAB and N-Glycans Substrates Are Relevant in Human Physiology, Polycystic Pathology and Multiple Sclerosis: A Review


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Glycobiology Overview
3. The Roles of Glycans
4. Recognition Patterns of Glycans
5. Glycosylation in Health and Disease
5.1. The Enzymatic Glycosylation and Its Modulation
5.1.1. N-Linked Glycosylation
5.1.2. O-Linked Glycosylation
5.1.3. C-Linked Glycosylation
5.1.4. Glypiation
5.1.5. Phosphoglycosylation
5.2. The Non-Enzymatic Glycosylation
6. Glycodrugs
6.1. The α-Glycosidases Inhibitors: Iminosugars
6.2. Glycodrugs in Diabetes Mellitus and Thesaurismosis
6.3. Viral Infections and Glycodrugs
6.4. Carbohydrate-Based Antibiotics
6.5. Carbohydrate-Based Cancer Drugs
6.6. Cardioactive Glycosides
6.7. Heparin
6.8. Carbohydrate-Based Vaccines
6.9. Carbohydrate-Based α-Glucosidases
6.10. Glycodrugs Miscellanea
7. Protein Folding and Folding Quality Control Machinery
The UPR
8. The GANAB
8.1. The Structure and Localization Glucosidase II
8.2. Enzyme Activity Assay and Interactions of Glucosidase II
9. UPR in Human Diseases: The Role of GII
9.1. Diabetes Mellitus
9.2. Neurodegeneration
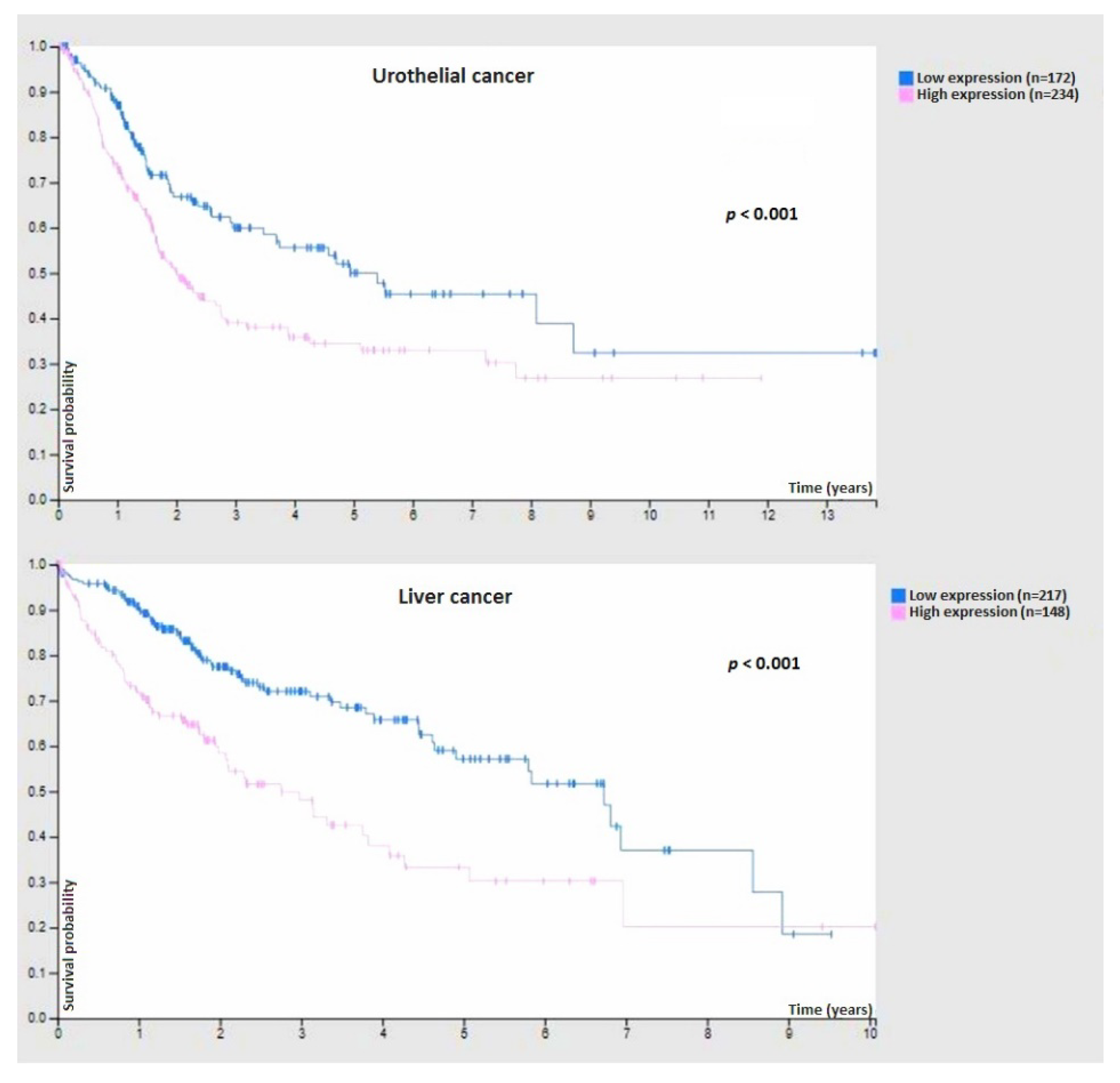
9.3. Cancer
9.4. Ischemia-Reperfusion Injury and Atherosclerotic
9.5. ADPLD and ADPKD
9.6. Epididymal Pathology and Male Infertility
9.7. Systemic Lupus Erythematosus
9.8. Multiple Sclerosis
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-Gal A | α-galactosidase A; |
| 18F-FDG | glucose-analogue, 2-deoxy-2(18F)fluoro-D-glucose; |
| 4-MU-Glc | 4-methylunbelliferyl-a-D-glucopyranoside; |
| AD | Alzheimer’s disease; |
| ADCC | antibody-dependent cell-mediated cytotoxicity; |
| ADCP | antibody-dependent cellular phagocytosis; |
| ADPKD | Autosomal Dominant Polycystic Kidney Disease; |
| ADPLD | Autosomal Dominant Polycystic Liver Disease; |
| AGEs | advanced glycation end-products; |
| ALS | Amyotrophic Lateral Sclerosis; |
| ARE | antioxidant response element; |
| Asn | asparagine; |
| ATF | activating transcription factor; |
| BBB | blood brain barrier; |
| BBE | Bickerstaff’s brainstem encephalitis; |
| BSA | bovine serum albumin; |
| C/EBP | CCAAT/enhancer binding protein; |
| CDC | complement-dependent cytotoxicity; |
| CDG | Congenital Disorders of Glycosylation; |
| CD-MPR | cation-dependent mannose-6-phosphate receptor; |
| CHOP | C/EBP homologous protein; |
| CI-MPR | cation-independent mannose-6-phosphate receptor; |
| CID | Chronic inflammatory disease polyneuropathy; |
| CML | N-carboxymethyllysine; |
| CNS | central nervous system; |
| CNX | Calnexin; |
| CRC | colorectal cancer; |
| CRM197 | diphtheria toxin; |
| CRT | Calreticulin; |
| CSGAGs | chondroitin sulfate/dermatan sulfate; |
| DAB | analog 1,4-dideoxy-1,4-imino-D-arabinitol; |
| DAMPs | damage-associated molecular patterns; |
| DD | disease duration; |
| DMF | Dimethyl Fumarate; |
| DMT | disease modifying therapy; |
| DNJ | 1-deoxynojirimycin; |
| EAE | experimental allergic encephalitis; |
| ER | endoplasmic reticulum; |
| ERAD | ER-associated degradation; |
| ERGIC | ER-Golgi intermediate compartment; |
| FAD | flavin adenine inucleotide; |
| GADD34 | growth arrest and DNA damage 34 complex; |
| GAGs | glycosaminoglycans; |
| GANC | GH31 neutral α-glycosidase C; |
| GBS | Guillain-Barré-Strohl syndrome; |
| GH31 | glycosyl hydrolase 31; |
| GIIα | α-subunit of α-glucosidase II; |
| GIIβ | β-subunit of α-glucosidase II; |
| GluI/GI | α-glucosidases I; |
| GluII/GII | α-glucosidases II; |
| GLUTs | glucose transporters; |
| GM-CSF | granulocyte-macrophage colony-stimulating factor; |
| GNS | N-acetylglucosamine-6-sulfatase; |
| GSD | glycogen storage diseases; |
| GSLs | glycosphingolipids; |
| HCAR2 | Hydroxycarboxylic Acid Receptor 2; |
| HCs | healthy controls; |
| HDEL | His-Asp-Glu-Leu; |
| HINCUT | noncoding ultra-conserved transcript; |
| HMGB1 | high mobility group box 1 protein; |
| HSGAGs | heparin/heparan sulfate; |
| IFN | interferon; |
| IRE1 | inositol requiring kinase 1; |
| KDEL | Lys-Asp-Glu-Leu; |
| KLH | keyhole limpet haemocyanin; |
| LABNAc | 2-acetamido-1,4-imino-1,2,4-tride-oxy-l-arabinitol; |
| LeY | Lewis Y; |
| LL | lesion load; |
| LPS | lipopolysaccharide; |
| MM | Multiple Myeloma; |
| MMP9 | matrix metalloproteinases type 9; |
| MOA | mechanisms of action; |
| MOGAD | Myelin oligodendrocyte glycoprotein antibody-associated disease; |
| MRH | mannose-6-phosphate receptor; |
| MS | Multiple Sclerosis; |
| NAD | nicotinamide adenine dinucleotide; |
| Neu5Gc | N-Glycolylneuraminic acid; |
| NLRs | NOD-like receptors; |
| NMO | Neuromyelitis Optica; |
| NMOSD | Neuromyelitis Optica Spectrum Disorders; |
| OGA | O-GlcNAcase; |
| O-GlcNAc | O-linked N-acetylglucosamine; |
| OGT | O-GlcNAc-transferase; |
| OST | oligosaccharyltransferase; |
| PAMPs | pathogen-associated molecular patterns; |
| PBMCs | peripheral blood mononuclear cells; |
| PC1 | polycystin-1; |
| PCKD | Polycystic Kidney Disease; |
| PCLD | Polycystic Liver Disease; |
| PD | Parkinson’s disease; |
| PDI | protein disulfide refolding isomerases; |
| PERK | pancreatic endoplasmic reticulum kinase; |
| PET | positron emission tomography; |
| PG | peptidoglycan; |
| pNP-Glc | p-nitrophenyl-a-D-glucopyranoside; |
| PP1 | protein phosphatase 1; |
| PQC | protein quality control; |
| PRKCSH | protein kinase C substrate 80 K-H; |
| Pro | proline; |
| PRR | pattern recognition receptors; |
| PSA | prostate specific antigen; |
| PSA1 | zwitterionic polysaccharide A1; |
| PSP | Progressive Supranuclear Palsy; |
| RAGE | receptor for advanced glycation end-products; |
| RRMS | relapsing remitting Multiple Sclerosis; |
| SAMPs | self-associated molecular patterns; |
| Ser | serine; |
| SGLT2 | sodium-dependent glucose cotransporter 2; |
| SLE | Systemic Lupus Erythematosus; |
| sLeA | sialyl Lewis A; |
| sLeX | sialyl Lewis X; |
| TACAs | tumor-associated carbohydrate antigens; |
| TAGE | toxic end-products of advanced glycation; |
| TCR | T-cell receptor; |
| Thr | threonine; |
| TLRs | Toll-like receptors; |
| TT | tetanus toxoid; |
| T-UCR | transcribed-ultra conserved regions; |
| UGGT | UDP-glucose glycoprotein glucosyltransferase; |
| UPR | unfolded protein response. |
References
- Wade, L.G. Organic Chemistry, 3rd ed.; Prentice-Hall, Inc.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Varki, A.; Schauer, R. Sialic Acids. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 14. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1920/ (accessed on 28 February 2022).
- Taylor, M.E.; Drickamer, K. Introduction to Glycobiology, 2nd ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Varki, A.; Cummings, R.; Esko, J.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1999; Chapter 16. Available online: https://www.ncbi.nlm.nih.gov/books/NBK20705/ (accessed on 28 February 2022).
- Zaia, J. Mass spectrometry and the emerging field of glycomics. Chem. Biol. 2008, 15, 881–892. [Google Scholar] [CrossRef]
- Laine, R.A. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide: The Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994, 4, 759–767. [Google Scholar] [CrossRef]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Drenth, J.P.; te Morsche, R.H.; Smink, R.; Bonifacino, J.S.; Jansen, J.B. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003, 33, 345–347. [Google Scholar] [CrossRef]
- Li, A.; Davila, S.; Furu, L.; Qian, Q.; Tian, X.; Kamath, P.S.; King, B.F.; Torres, V.E.; Somlo, S. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 2003, 72, 691–703. [Google Scholar] [CrossRef]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease; Harris, P.C. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef]
- De Masi, R.; Orlando, S. GANAB as a Novel Biomarker in Multiple Sclerosis: Correlation with Neuroinflammation and IFI35. Pharmaceuticals 2021, 14, 1195. [Google Scholar] [CrossRef]
- National Research Council. Transforming Glycoscience: A Roadmap for the Future; National Academies Press: Washington, WA, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK109958/ (accessed on 28 February 2022). [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef]
- Shi, D.; Sheng, A.; Chi, L. Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease. Front. Mol. Biosci. 2021, 8, 639666. [Google Scholar] [CrossRef]
- Mondal, S. UNIT—II: Carbohydrates Metabolism. Pharmacognosy and Phytochemistry, Lecture Notes for B Pharm VII Sem, PPH 401. 2019. Available online: https://doi.org/10.13140/RG.2.2.15056.25606 (accessed on 28 February 2022).
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Wong, N.; De Melo, J.; Tang, D. PKM2, a Central Point of Regulation in Cancer Metabolism. Int. J. Cell Biol. 2013, 2013, 242513. [Google Scholar] [CrossRef]
- Herrmann, K.; Benz, M.R.; Krause, B.J.; Pomykala, K.L.; Buck, A.K.; Czernin, J. (18)F-FDG-PET/CT in Evaluating Response to Therapy in Solid Tumors: Where We are and Where We Can Go. Q. J. Nucl. Med. Mol. Imaging 2011, 55, 620–632. [Google Scholar]
- Lim, J.A.; Li, L.; Raben, N. Pompe disease: From pathophysiology to therapy and back again. Front. Aging Neurosci. 2014, 6, 177. [Google Scholar] [CrossRef]
- Ozen, H. Glycogen storage diseases: New perspectives. World J. Gastroenterol. 2007, 13, 2541–2553. [Google Scholar] [CrossRef]
- Pyke, D.A. Diabetic ketosis and coma. J. Clin. Pathol. Suppl. 1969, 2, 57–65. [Google Scholar] [CrossRef]
- McFarlane, H.E.; Doring, A.; Persson, S. The cell biology of cellulose synthesis. Annu. Rev. Plant Biol. 2014, 65, 69–94. [Google Scholar] [CrossRef]
- Koch, B.E.; Stougaard, J.; Spaink, H.P. Keeping track of the growing number of biological functions of chitin and its interaction partners in biomedical research. Glycobiology 2015, 25, 469–482. [Google Scholar] [CrossRef]
- Bergstrom, K.S.; Xia, L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology 2013, 23, 1026–1037. [Google Scholar] [CrossRef]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergstrom, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.; Schutte, A.; van der Post, S.; Svensson, F.; Rodriguez-Pineiro, A.M.; Nystrom, E.E.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef]
- Bar Dolev, M.; Braslavsky, I.; Davies, P.L. Ice-binding proteins and their function. Annu. Rev. Biochem. 2016, 85, 515–542. [Google Scholar] [CrossRef]
- Duman, J.G. Animal ice-binding (antifreeze) proteins and glycolipids: An overview with emphasis on physiological function. J. Exp. Biol. 2015, 218, 1846–1855. [Google Scholar] [CrossRef]
- Iozzo, R.V. Basement membrane proteoglycans: From cellar to ceiling. Nat. Rev. Mol. Cell. Biol. 2005, 6, 646–656. [Google Scholar] [CrossRef]
- Farach-Carson, M.C.; Warren, C.R.; Harrington, D.A.; Carson, D.D. Border patrol: Insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 2014, 34, 64–79. [Google Scholar] [CrossRef]
- Aspberg, A. The different roles of aggrecan interaction domains. J. Histochem. Cytochem. 2012, 60, 987–996. [Google Scholar] [CrossRef]
- Mohammadi, H.; Mequanint, K.; Herzog, W. Computational aspects in mechanical modeling of the articular cartilage tissue. Proc. Inst. Mech. Eng. H 2013, 227, 402–420. [Google Scholar] [CrossRef]
- Pap, T.; Bertrand, J. Syndecans in cartilage breakdown and synovial inflammation. Nat. Rev. Rheumatol. 2013, 9, 43–55. [Google Scholar] [CrossRef]
- Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Molecules 2015, 20, 5286–5298. [Google Scholar] [CrossRef]
- Paszek, M.J.; DuFort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef]
- El Maarouf, A.; Petridis, A.K.; Rutishauser, U. Use of polysialic acid in repair of the central nervous system. Proc. Natl. Acad. Sci. USA 2006, 103, 16989–16994. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat. Rev. Neurosci. 2008, 9, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Sun, P.; Paller, A.S. Ganglioside GM3 blocks the activation of epidermal growth factor receptor induced by integrin at specific tyrosine sites. J. Biol. Chem. 2003, 278, 48770–48778. [Google Scholar] [CrossRef]
- Yoon, S.J.; Nakayama, K.; Hikita, T.; Handa, K.; Hakomori, S.I. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 18987–18991. [Google Scholar] [CrossRef]
- Coskun, U.; Grzybek, M.; Drechsel, D.; Simons, K. Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. USA 2011, 108, 9044–9048. [Google Scholar] [CrossRef]
- Garner, O.B.; Baum, L.G. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem. Soc. Trans. 2008, 36, 1472–1477. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef]
- Dennis, J.W.; Lau, K.S.; Demetriou, M.; Nabi, I.R. Adaptive regulation at the cell surface by N-glycosylation. Traffic 2009, 10, 1569–1578. [Google Scholar] [CrossRef]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; pp. 101–114. [Google Scholar]
- Partridge, E.A.; Le, R.C.; Di, G.G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Selleck, S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002, 71, 435–471. [Google Scholar] [CrossRef] [PubMed]
- Inatani, M.; Irie, F.; Plump, A.S.; Tessier-Lavigne, M.; Yamaguchi, Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science 2003, 302, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Stringer, S.E.; Rusch, M.A.; Selleck, S.B.; Ekker, S.C. A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol. 2005, 284, 364–376. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Domowicz, M.S. Chemistry and function of glycosaminoglycans in the nervous system. Adv. Neurobiol. 2014, 9, 89–115. [Google Scholar]
- Balasubramanian, R.; Zhang, X. Mechanisms of FGF gradient formation during embryogenesis. Semin Cell Dev Biol. 2016, 53, 94–100. [Google Scholar] [CrossRef]
- Springer, S.A.; Diaz, S.L.; Gagneux, P. Parallel evolution of a self-signal: Humans and new world monkeys independently lost the cell surface sugar Neu5Gc. Immunogenetics 2014, 66, 671–674. [Google Scholar] [CrossRef]
- Schauer, R. Sialic acids and their role as biological masks. Trends Biochem. Sci. 1985, 10, 357–360. [Google Scholar] [CrossRef]
- Muchmore, E.; Varki, A. Inactivation of influenza C esterase decreases infectivity without loss of binding; a probe for 9-O-acetylated sialic acids. Science 1987, 236, 1293–1295. [Google Scholar] [CrossRef]
- Uchimura, K.; Lemjabbar-Alaoui, H.; van Kuppevelt, T.H.; Rosen, S.D. Use of a phage display antibody to measure the enzymatic activity of the Sulfs. Methods Enzymol. 2010, 480, 51–64. [Google Scholar]
- Morimoto-Tomita, M.; Uchimura, K.; Werb, Z.; Hemmerich, S.; Rosen, S.D. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J. Biol. Chem. 2002, 277, 49175–49185. [Google Scholar] [CrossRef] [PubMed]
- Gorsi, B.; Stringer, S.E. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol. 2007, 17, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Vives, R.R.; Seffouh, A.; Lortat-Jacob, H. Post-synthetic regulation of HS structure: The Yin and Yang of the Sulfs in cancer. Front. Oncol. 2014, 3, 331. [Google Scholar] [CrossRef]
- Demetriou, M.; Granovsky, M.; Quaggin, S.; Dennis, J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 2001, 409, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.W.; Laferte, S.; Waghorne, C.; Breitman, M.L.; Kerbel, R.S. Beta 1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science 1987, 236, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Brazin, K.N.; Mallis, R.J.; Das, D.K.; Feng, Y.; Hwang, W.; Wang, J.H.; Wagner, G.; Lang, M.J.; Reinherz, E.L. Structural features of the αβTCR mechanotransduction apparatus that promote pMHC siscrimination. Front. Immunol. 2015, 6, 441. [Google Scholar] [CrossRef]
- Wang, C.C.; Chen, J.R.; Tseng, Y.C.; Hsu, C.H.; Hung, Y.F.; Chen, S.W.; Chen, C.M.; Khoo, K.H.; Cheng, T.J.; Cheng, Y.S.; et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 18137–18142. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; David, A.; Schmidt, L.; Gibbs, J.S.; Puigbo, P.; Ince, W.L.; Bennink, J.R.; Yewdell, J.W. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc. Natl. Acad. Sci. USA 2011, 108, E1417–E1422. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Dekan, G.; Gabel, C.; Farquhar, M.G. Sulfate contributes to the negative charge of podocalyxin, the major sialoglycoprotein of the glomerular filtration slits. Proc. Natl. Acad. Sci. USA 1991, 88, 5398–5402. [Google Scholar] [CrossRef]
- Ito, M.; Sugihara, K.; Asaka, T.; Toyama, T.; Yoshihara, T.; Furuichi, K.; Wada, T.; Asano, M. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS ONE 2012, 7, e29873. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B.; Sellmeier, M.; Schaper, W.; Blume, L.; Philippens, B.; Kats, E.; Bernard, U.; Galuska, S.P.; Geyer, H.; Geyer, R.; et al. Deficits in sialylation impair podocyte maturation. J. Am. Soc. Nephrol. 2012, 23, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Stow, J.L.; Soroka, C.J.; MacKay, K.; Striker, L.; Striker, G.; Farquhar, M.G. Basement membrane heparan sulfate proteoglycan is the main proteoglycan synthesized by glomerular epithelial cells in culture. Am. J. Pathol. 1989, 135, 637–646. [Google Scholar] [PubMed]
- Harvey, S.J.; Miner, J.H. Revisiting the glomerular charge barrier in the molecular era. Curr. Opin. Nephrol. Hypertens. 2008, 17, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.W.; Klein, R.S.; Ransohoff, R.M. The blood-brain barrier, chemokines and multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Engers, J.; Haque, M.; King, S.; Al-Omari, D.; Ma, T.Y. Matrix Metalloproteinase-9 (MMP-9) induced disruption of intestinal epithelial tight junction barrier is mediated by NF-κB activation. PLoS ONE 2021, 16, e0249544. [Google Scholar] [CrossRef]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcgammaRIIIa binding. Structure 2014, 22, 1478–1488. [Google Scholar] [CrossRef]
- Llop, E.; Gutierrez-Gallego, R.; Segura, J.; Mallorqui, J.; Pascual, J.A. Structural analysis of the glycosylation of gene-activated erythropoietin (epoetin delta, Dynepo). Anal. Biochem. 2008, 383, 243–254. [Google Scholar] [CrossRef]
- Kiss, Z.; Elliott, S.; Jedynasty, K.; Tesar, V.; Szegedi, J. Discovery and basic pharmacology of erythropoiesis-stimulating agents (ESAs), including the hyperglycosylated ESA, darbepoetin alfa: An update of the rationale and clinical impact. Eur. J. Clin. Pharmacol. 2010, 66, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhao, H.; Xia, H. Glycosylation-modified erythropoietin with improved half-life and biological activity. Int. J. Hematol. 2010, 91, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Cebon, J.; Nicola, N.; Ward, M.; Gardner, I.; Dempsey, P.; Layton, J.; Duhrsen, U.; Burgess, A.W.; Nice, E.; Morstyn, G. Granulocyte-macrophage colony stimulating factor from human lymphocytes. The effect of glycosylation on receptor binding and biological activity. J. Biol. Chem. 1990, 265, 4483–4491. [Google Scholar] [CrossRef]
- Niu, L.H.; Heaney, M.L.; Vera, J.C.; Golde, D.W. High-affinity binding to the GM-CSF receptor requires intact N-glycosylation sites in the extracellular domain of the β subunit. Blood 2000, 95, 3357–3362. [Google Scholar] [CrossRef]
- Rosen, L.B.; Freeman, A.F.; Yang, L.M.; Jutivorakool, K.; Olivier, K.N.; Angkasekwinai, N.; Suputtamongkol, Y.; Bennett, J.E.; Pyrgos, V.; Williamson, P.R.; et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J. Immunol. 2013, 190, 3959–3966. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Parekh, R.B.; Dwek, R.A.; Sutton, B.J.; Fernandes, D.L.; Leung, A.; Stanworth, D.; Rademacher, T.W.; Mizuochi, T.; Taniguchi, T.; Matsuta, K.; et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985, 316, 452–457. [Google Scholar] [CrossRef]
- Rademacher, T.W.; Williams, P.; Dwek, R.A. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc. Natl. Acad. Sci. USA 1994, 91, 6123–6127. [Google Scholar] [CrossRef]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef]
- Lewis, B.A.; Hanover, J.A. O-GlcNAc and the epigenetic regulation of gene expression. J. Biol. Chem. 2014, 289, 34440–34448. [Google Scholar] [CrossRef]
- Olivier-Van Stichelen, S.; Hanover, J.A. You are what you eat: O-linked N-acetylglucosamine in disease, development and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell. Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Boren, T.; Falk, P.; Roth, K.A.; Larson, G.; Normark, S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993, 262, 1892–1895. [Google Scholar] [CrossRef]
- Walz, A.; Odenbreit, S.; Mahdavi, J.; Boren, T.; Ruhl, S. Identifcation and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology 2005, 15, 700–708. [Google Scholar] [CrossRef]
- Kobayashi, M.; Lee, H.; Nakayama, J.; Fukuda, M. Roles of gastric mucin-type O-glycans in the pathogenesis of Helicobacter pylori infection. Glycobiology 2009, 19, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Duraisingh, M.T.; Maier, A.G.; Triglia, T.; Cowman, A.F. Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 4796–4801. [Google Scholar] [CrossRef]
- Malpede, B.M.; Lin, D.H.; Tolia, N.H. Molecular basis for sialic acid dependent receptor recognition by Plasmodium falciparum erythrocyte binding antigen 140/BAEBL. J. Biol. Chem. 2013, 288, 12406–12415. [Google Scholar] [CrossRef]
- Schultze, B.; Gross, H.J.; Klenk, H.D.; Brossmer, R.; Herrler, G. Differential reactivity of bovine coronavirus (BCV) and influenza C virus with N-acetyl-9-O-acetylneuraminic acid (NEU5,9AC2)-containing receptors. Adv. Exp. Med. Biol. 1990, 276, 115–119. [Google Scholar]
- Song, H.; Qi, J.; Khedri, Z.; Diaz, S.; Yu, H.; Chen, X.; Varki, A.; Shi, Y.; Gao, G.F. An open receptor-binding cavity of hemagglutinin-esterase-fusion glycoprotein from newly-identified influenza D virus: Basis for its broad cell tropism. PLoS Pathog. 2016, 12, e1005411. [Google Scholar]
- Geno, K.A.; Gilbert, G.L.; Song, J.Y.; Skovsted, I.C.; Klugman, K.P.; Jones, C.; Konradsen, H.B.; Nahm, M.H. Pneumococcal capsules and their types: Past, present, and future. Clin. Microbiol. Rev. 2015, 28, 871–899. [Google Scholar] [CrossRef]
- Hutson, A.M.; Atmar, R.L.; Graham, D.Y.; Estes, M.K. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 2002, 185, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thornton, S.; Morrow, A.L.; Jiang, X. Norovirus and histo-blood group antigens: Demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Obojes, K.; Vlasak, R. The sialate-4-O-acetylesterases of coronaviruses related to mouse hepatitis virus: A proposal to reorganize group 2 Coronaviridae. J. Gen. Virol. 2002, 83, 395–402. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 2004, 78, 12665–12667. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Zhang, X.Q.; Senaati, H.P.; Chen, H.W.; Varki, N.M.; Schooley, R.T.; Gagneux, P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol. J. 2013, 10, 321. [Google Scholar] [CrossRef]
- von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Hayden, F.G.; Osterhaus, A.D.M.E.; Treanor, J.J.; Fleming, D.M.; Aoki, F.Y.; Nicholson, K.G.; Bohnen, A.M.; Hirst, H.M.; Keene, O.; Wightman, K. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenza virus infections. N. Engl. J. Med. 1997, 337, 874–880. [Google Scholar] [CrossRef]
- Meindl, P.; Bodo, G.; Palese, P.; Schulman, J.; Tuppy, H. Inhibition of neuraminidase activity by derivatives of 2-deoxy-2,3-dehydro-N-acetylneuraminic acid. Virology 1974, 58, 457–463. [Google Scholar] [CrossRef]
- Moustafa, I.; Connaris, H.; Taylor, M.; Zaitsev, V.; Wilson, J.C.; Kiefel, M.J.; von Itzstein, M.; Taylor, G. Sialic acid recognition by Vibrio cholerae neuraminidase. J. Biol. Chem. 2004, 279, 40819–40826. [Google Scholar] [CrossRef]
- Janeway, C.A.J.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Moresco, E.M.; LaVine, D.; Beutler, B. Toll-like receptors. Curr. Biol. 2011, 21, R488–R493. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef]
- Marshall, A.S.; Gordon, S. Commentary: C-type lectins on the macrophage cell surface—Recent findings. Eur. J. Immunol. 2004, 34, 18–24. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Croci, D.O. Regulatory circuits mediated by lectin–glycan interactions in autoimmunity and cancer. Immunity 2012, 36, 322–335. [Google Scholar] [CrossRef]
- Mahla, R.S.; Reddy, M.C.; Prasad, D.V.; Kumar, H. Sweeten PAMPs: Role of sugar complexed PAMPs in innate immunity and vaccine biology. Front. Immunol. 2013, 4, 248. [Google Scholar] [CrossRef]
- Varki, A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology 2011, 21, 1121–1124. [Google Scholar] [CrossRef]
- Chen, G.Y.; Brown, N.K.; Zheng, P.; Liu, Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology 2014, 24, 800–806. [Google Scholar] [CrossRef]
- Libbey, J.E.; McCoy, L.L.; Fujinami, R.S. Molecular mimicry in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 127–147. [Google Scholar] [CrossRef]
- Vaishnav, R.A.; Liu, R.; Chapman, J.; Roberts, A.M.; Ye, H.; Rebolledo-Mendez, J.D.; Tabira, T.; Fitzpatrick, A.H.; Achiron, A.; Running, M.P.; et al. Aquaporin 4 molecular mimicry and implications for neuromyelitis optica. J. Neuroimmunol. 2013, 15, 260, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Hemmer, B.; Muhlau, M. The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J. Autoimmun. 2010, 34, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Shahrizaila, N.; Yuki, N. Bickerstaff brainstem encephalitis and Fisher syndrome: Anti-GQ1b antibody syndrome. J. Neurol. Neurosurg. Psychiatry 2013, 84, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Vatti, N.; Ramirez-Santana, C.; Chang, C.; Mancera-Paez, O.; Gershwin, M.E.; Anaya, J.M. Chronic inflammatory demyelinating polyneuropathy as an autoimmune disease. J. Autoimmun. 2019, 102, 8–37. [Google Scholar] [CrossRef]
- van der Meché, F.G.; van Doorn, P.A. Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy: Immune mechanisms and update on current therapies. Ann. Neurol. 1995, 37, S14–S31. [Google Scholar] [CrossRef]
- Spadaro, M.; Gerdes, L.A.; Krumbholz, M.; Ertl-Wagner, B.; Thaler, F.S.; Schuh, E.; Metz, I.; Blaschek, A.; Dick, A.; Bruck, W.; et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e257. [Google Scholar] [CrossRef]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; de Haidar Jorge, F.M.; Takahashi, T.; Nakashima, I.; Apostolos-Pereira, S.L.; Talim, N.; Simm, R.F.; et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Y.; Fu, Y.; Yang, L.; Su, L.; Shi, K.; Li, M.; Liu, Q.; Borazanci, A.; Liu, Y.; et al. Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD. Sci. China Life Sci. 2016, 59, 1270–1281. [Google Scholar] [CrossRef]
- Latov, N. Antibody testing in neuropathy associated with anti-Myelin-Associated Glycoprotein antibodies: Where we are after 40 years. Curr. Opin. Neurol. 2021, 34, 625–630. [Google Scholar] [CrossRef]
- Yuki, N. Guillain-Barré syndrome and anti-ganglioside antibodies: A clinician-scientist’s journey. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 299–326. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Kuwahara, M.; Morikawa, M.; Kusunoki, S. Bickerstaff brainstem encephalitis with or without anti-GQ1b antibody. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e889. [Google Scholar] [CrossRef] [PubMed]
- Pianta, A.; Arvikar, S.L.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Investig. 2017, 127, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; van Vliet, S.J.; Engering, A.; Hart, B.A.T.; van Kooyk, Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu. Rev. Immunol. 2004, 22, 33–54. [Google Scholar] [CrossRef] [PubMed]
- McGreal, E.P.; Miller, J.L.; Gordon, S. Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 2005, 17, 18–24. [Google Scholar] [CrossRef]
- Li, E.; Tabas, I.; Kornfeld, S. The synthesis of complex-type oligosaccharides. I. Structure of the lipid-linked oligosaccharide precursor of the complex-type oligosaccharides of the vesicular stomatitis virus G protein. J. Biol. Chem. 1978, 253, 7762–7770. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Parodi, A.J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kamiya, Y.; Kato, K. The role of MRH domain-containing lectins in ERAD. Glycobiology 2010, 20, 651–660. [Google Scholar] [CrossRef]
- Zhang, B.; Cunningham, M.A.; Nichols, W.C.; Bernat, J.A.; Seligsohn, U.; Pipe, S.W.; McVey, J.H.; Schulte-Overberg, U.; de Bosch, N.B.; Ruiz-Saez, A.; et al. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat. Genet. 2003, 34, 220–225. [Google Scholar] [CrossRef]
- Stavenhagen, K.; Hinneburg, H.; Thaysen-Andersen, M.; Hartmann, L.; Varón Silva, D.; Fuchser, J.; Kaspar, S.; Rapp, E.; Seeberger, P.H.; Kolarich, D. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: An evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass. Spectrom. 2013, 48, 627–639. [Google Scholar] [CrossRef]
- Zacchi, L.F.; Schulz, B.L. N-glycoprotein macroheterogeneity: Biological implications and proteomic characterization. Glycoconj. J. 2016, 33, 359–376. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.J.; Andre, S.; Jimenez-Barbero, J.; Romero, A.; Solís, D. From lectin structure to functional glycomics: Principles of the sugar code. Trends Biochem. Sci. 2011, 36, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.J.; Kayser, K. Introduction to glycopathology: The concept, the tools and the perspectives. Diagn. Pathol. 2014, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Moehler, T.M.; Seckinger, A.; Hose, D.; Andrulis, M.; Moreaux, J.; Hielscher, T.; Willhauck-Fleckenstein, M.; Merling, A.; Bertsch, U.; Jauch, A.; et al. The glycome of normal and malignant plasma cells. PLoS ONE 2013, 8, e83719. [Google Scholar] [CrossRef]
- Barfeld, S.J.; East, P.; Zuber, V.; Mills, I.G. Metaanalysis of prostate cancer gene expression data identifies a novel discriminatory signature enriched for glycosylating enzymes. BMC Med. Genomics 2014, 7, 513. [Google Scholar] [CrossRef]
- Venturi, G.; Gomes Ferreira, I.; Pucci, M.; Ferracin, M.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Impact of sialyltransferase ST6GAL1 overexpression on different colon cancer cell types. Glycobiology 2019, 29, 684–695. [Google Scholar] [CrossRef]
- Norton, P.A.; Mehta, A.S. Expression of genes that control core fucosylation in hepatocellular carcinoma: Systematic review. World J. Gastroenterol. 2019, 25, 2947–2960. [Google Scholar] [CrossRef]
- Azevedo, R.; Peixoto, A.; Gaiteiro, C.; Fernandes, E.; Neves, M.; Lima, L.; Santos, L.L.; Ferreira, J.A. Over forty years of bladder cancer glycobiology: Where do glycans stand facing precision oncology? Oncotarget 2017, 8, 91734–91764. [Google Scholar] [CrossRef]
- Christiansen, M.N.; Chik, J.; Lee, L.; Anugraham, M.; Abrahams, J.L.; Packer, N.H. Cell surface protein glycosylation in cancer. Proteomics 2014, 14, 525–546. [Google Scholar] [CrossRef]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF Nglycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteomics 2016, 131, 29–37. [Google Scholar] [CrossRef]
- Lauc, G.; Pezer, M.; Rudan, I.; Campbell, H. Mechanisms of disease: The human N-glycome. Biochim. Biophys. Acta 2016, 1860, 1574–1582. [Google Scholar] [CrossRef]
- Kizuka, Y. Epigenetic regulation of and by glycosylation. In Glycoscience: Biology and Medicine; Taniguchi, N., Endo, T., Hart, G.W., Seeberger, P.H., Wong, C.H., Eds.; Springer: Tokyo, Japan, 2015; pp. 1129–1134. [Google Scholar]
- Agrawal, P.; Kurcon, T.; Pilobello, K.T.; Rakus, J.F.; Koppolu, S.; Liu, Z.; Batista, B.S.; Eng, W.S.; Hsu, K.L.; Liang, Y.; et al. Mapping posttranscriptional regulation of the human glycome uncovers microRNA defining the glycocode. Proc. Natl. Acad. Sci. USA 2014, 111, 4338–4343. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, B.; Jin, G.; Zhang, J.; Wang, X.; Feng, Y.; Bian, Z.; Fei, B.; Yin, Y.; Huang, Z. An Integrated Three-Long Non-coding RNA Signature Predicts Prognosis in Colorectal Cancer Patients. Front. Oncol. 2019, 9, 1269. [Google Scholar] [CrossRef] [PubMed]
- Ferdin, J.; Nishida, N.; Wu, X.; Nicoloso, M.S.; Shah, M.Y.; Devlin, C.; Ling, H.; Shimizu, M.; Kumar, K.; Cortez, M.A.; et al. HINCUTs in cancer: Hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ. 2013, 20, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Dehelean, L.; Sarbu, M.; Petrut, A.; Zamfir, A.D. Trends in glycolipid biomarker discovery in neurodegenerative disorders by mass spectrometry. Adv. Exp. Med. Biol. 2019, 1140, 703–729. [Google Scholar]
- Ghazarian, H.; Idoni, B.; Oppenheimer, S.B. A glycobiology review: Carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem. 2011, 113, 236–247. [Google Scholar] [CrossRef]
- Bohnsack, R.N.; Song, X.; Olson, L.J.; Kudo, M.; Gotschall, R.R.; Canfield, W.M.; Cummings, R.D.; Smith, D.F.; Dahms, N.M. Cation-independent mannose 6-phosphate receptor: A composite of distinct phosphomannosyl binding sites. J. Biol. Chem. 2009, 284, 35215–35226. [Google Scholar] [CrossRef]
- Kolb-Bachofen, V.; Schlepper-Schäfer, J.; Vogell, W.; Kolb, H. Electron microscopic evidence for an asialoglycoprotein receptor on Kupffer cells: Localization of lectin-mediated endocytosis. Cell 1982, 29, 859–866. [Google Scholar] [CrossRef]
- Skorepa, O.; Pazicky, S.; Kalousková, B.; Bláha, J.; Abreu, C.; Ječmen, T.; Rosůlek, M.; Fish, A.; Sedivy, A.; Harlos, K.; et al. Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers 2020, 12, 1998. [Google Scholar] [CrossRef]
- Rottger, S.; White, J.; Wandall, H.H.; Olivo, J.C.; Stark, A.; Bennett, E.P.; Whitehouse, C.; Berger, E.G.; Clausen, H.; Nilsson, T. Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J. Cell Sci. 1998, 111, 45–60. [Google Scholar] [CrossRef]
- Patsos, G.; Hebbe-Viton, V.; Robbe-Masselot, C.; Masselot, D.; San Martin, R.; Greenwood, R.; Paraskeva, C.; Klein, A.; Graessmann, M.; Michalski, J.C.; et al. O-Glycan inhibitors generate aryl-glycans, induce apoptosis and lead to growth inhibition in colorectal cancer cell lines. Glycobiology 2009, 19, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Schachter, H.; Brockhausen, I. The Biosynthesis of Serine (Threonine)-N-acetylgalactosamine-Linked Carbohydrate Moieties. In Glycoconjugates Composition, Structure and Function; Allen, H.J., Kisailus, E.C., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1992; pp. 262–332. [Google Scholar]
- Mitra, N.; Sinha, S.; Ramya, T.N.; Surolia, A. N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem. Sci. 2006, 31, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A. Glycan-dependent signaling: O-linked N-acetylglucosamine. FASEB J. 2001, 15, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc. Biochim. Biophys. Acta 2006, 1761, 599–617. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G.; Pratt, C.W. Fundamentals of Biochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 9. [Google Scholar]
- Zachara, N.; Akimoto, Y.; Hart, G.W. The O-GlcNAc Modification. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; Chapter 19. Available online: https://www.ncbi.nlm.nih.gov/books/NBK453063/ (accessed on 28 February 2022). [CrossRef]
- Gardiner, E.E.; De Luca, M.; McNally, T.; Michelson, A.D.; Andrews, R.K.; Berndt, M.C. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood 2001, 98, 1440–1447. [Google Scholar] [CrossRef]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Sugahara, K.; Mikami, T.; Uyama, T.; Mizuguchi, S.; Nomura, K.; Kitagawa, H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. [Google Scholar] [CrossRef]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell. Sci. 2000, 113, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, J.L. Keratan sulfate: Structure, biosynthesis, and function. Glycobiology 2000, 10, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed]
- Julenius, K. NetCGlyc 1.0: Prediction of mammalian C-mannosylation sites. Glycobiology 2007, 17, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Manabe, S.; Ikezaki, M.; Inai, Y.; Matsui, I.S.; Ohta, Y.; Muroi, E.; Ito, Y. C-Mannosylated peptides derived from the thrombospondin type 1 repeat interact with Hsc70 to modulate its signaling in RAW264.7 cells. Glycobiology 2010, 20, 1298–1310. [Google Scholar] [CrossRef]
- Ihara, Y.; Inai, Y.; Ikezaki, M.; Matsui, I.S.; Manabe, S.; Ito, Y. C-Mannosylation: A modification on tryptophan in cellular proteins. In Glycoscience: Biology and Medicine; Endo, T., Seeberger, P., Hart, G., Wong, C.H., Taniguchi, N., Eds.; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Shcherbakova, A.; Preller, M.; Taft, M.H.; Pujols, J.; Ventura, S.; Tiemann, B.; Buettner, F.F.; Bakker, H. C-mannosylation supports folding and enhances stability of thrombospondin repeats. eLife 2019, 8, e52978. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zimmers, T.; Neumann, D.; Longmore, G.; Yoshimura, Y.; Lodish, H.F. Mutations in the Trp-Ser-X-Trp-Ser motif of the erythropoietin receptor abolish processing, ligand binding, and activation of the receptor. J. Biol. Chem. 1992, 267, 11619–11625. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Hart, G.W.; Kinoshita, T. Glycosylphosphatidylinositol Anchors. In Essentials of Glycobiology [Internet], 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015–2017; Chapter 12. [Google Scholar] [CrossRef]
- Haynes, P.A. Phosphoglycosylation: A new structural class of glycosylation? Glycobiology 1998, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.J.; Allen, A.K.; Snary, D. Studies on the structure of a phosphoglycoprotein from the parasite protozoan Trypanosoma cruzi. Biochem. J. 1983, 213, 313–319. [Google Scholar] [CrossRef]
- Ilg, T.; Harbecke, D.; Wiese, M.; Overath, P. Monoclonal antibodies directed against Leishmania secreted acid phosphatase and lipophosphoglycan. Eur. J. Biochem. 1993, 217, 603–615. [Google Scholar] [CrossRef]
- Yoshida-Moriguchi, T.; Yu, L.; Stalnaker, S.H.; Davis, S.; Kunz, S.; Madson, M.; Oldstone, M.B.; Schachter, H.; Wells, L.; Campbell, K.P. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science 2010, 327, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C.; Brancaccio, A. The evolution of the dystroglycan complex, a major mediator of muscle integrity. Biol. Open 2015, 4, 1163–1179. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Stehouwer, C.D.; Schalkwijk, C.G. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr. Opin. Lipidol. 2013, 24, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef]
- Miroliaei, M.; Aminjafari, A.; Ślusarczyk, S.; Nawrot-Hadzik, I.; Rahimmalek, M.; Matkowski, A. Inhibition of glycation-induced cytotoxicity, protein glycation, and activity of proteolytic enzymes by extract from Perovskia atriplicifolia roots. Pharmacogn. Mag. 2017, 13, S676–S683. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med. Hypotheses. 2004, 63, 449–452. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Ahmad, S.; Choi, I.; Ahmad, N.; Farhan, M.; Tatyana, G.; Shahab, U. Recent advances in detection of AGEs: Immunochemical, bioanalytical and biochemical approaches. IUBMB Life 2015, 67, 897–913. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. Involvement of Toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of advanced glycation end products (Review). Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef]
- Sampath, C.; Zhu, Y.; Sang, S.; Ahmedna, M. Bioactive compounds isolated from apple, tea, and ginger protect against dicarbonyl induced stress in cultured human retinal epithelial cells. Phytomedicine 2016, 23, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Federico, G.; Gori, M.; Randazzo, E.; Vierucci, F. Skin advanced glycation end-products evaluation in infants according to the type of feeding and mother’s smoking habits. SAGE Open Med. 2016, 4, 2050312116682126. [Google Scholar] [CrossRef] [PubMed]
- Schroter, D.; Hohn, A. Role of advanced glycation end products in carcinogenesis and their therapeutic implications. Curr. Pharm. Des. 2019, 24, 5245–5251. [Google Scholar] [CrossRef]
- Tessier, F.J.; Boulanger, E.; Howsam, M. Metabolic transit of dietary advanced glycation end-products—the case of NƐ-carboxymethyllysine. Glycoconj. J. 2021, 38, 311–317. [Google Scholar] [CrossRef]
- Hashemzaei, M.; Tabrizian, K.; Alizadeh, Z.; Pasandideh, S.; Rezaee, R.; Mamoulakis, C.; Tsatsakis, A.; Skaperda, Z.; Kouretas, D.; Shahraki, J. Resveratrol, curcumin and gallic acid attenuate glyoxal-induced damage to rat renal cells. Toxicol. Rep. 2020, 7, 1571–1577. [Google Scholar] [CrossRef]
- Kuzan, A.; Chwiłkowska, A.; Kobielarz, M.; Pezowicz, C.; Gamian, A. Glycation of extracellular matrix proteins and its role in atherosclerosis. Postepy. Hig. Med. Dosw. 2012, 66, 804–809. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox. Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef]
- Dhananjayan, K.; Gunawardena, D.; Hearn, N.; Sonntag, T.; Moran, C.; Gyengesi, E.; Srikanth, V.; Münch, G. Activation of Macrophages and Microglia by Interferon-γ and Lipopolysaccharide Increases Methylglyoxal Production: A New Mechanism in the Development of Vascular Complications and Cognitive Decline in Type 2 Diabetes Mellitus? J. Alzheimers Dis. 2017, 59, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Role of ligands of receptor for advanced glycation end products (RAGE) in peripheral artery disease. Rejuvenation Res. 2018, 21, 456–463. [Google Scholar] [CrossRef] [PubMed]
- van Eupen, M.G.; Schram, M.T.; Colhoun, H.M.; Hanssen, N.M.; Niessen, H.W.; Tarnow, L.; Parving, H.H.; Rossing, P.; Stehouwer, C.D.; Schalkwijk, C.G. The methylglyoxal-derived AGE tetrahydropyrimidine is increased in plasma of individuals with type 1 diabetes mellitus and in atherosclerotic lesions and is associated with sVCAM-1. Diabetologia 2013, 56, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Drury, S.; Hudson, B.I.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. Rage and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef]
- Cho, H.J.; Xie, C.; Cai, H. AGE-induced neuronal cell death is enhanced in G2019S LRRK2 mutation with increased RAGE expression. Transl. Neurodegener. 2018, 7, 1. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Sternberg, Z.; Chiotti, A.; Tario, J.; Chichelli, T.; Patel, N.; Chadha, K.; Yu, J.; Karmon, Y. Reduced expression of membrane-bound (m)RAGE is a biomarker of multiple sclerosis disease progression. Immunobiology 2016, 221, 193–198. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef]
- Dalfó, E.; Portero-Otín, M.; Ayala, V.; Martínez, A.; Pamplona, R.; Ferrer, I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J. Neuropathol. Exp. Neurol. 2005, 64, 816–830. [Google Scholar] [CrossRef]
- Kren, V.; Martínková, L. Glycosides in medicine: The role of glycosidic residue in biological activity. Curr. Med. Chem. 2001, 8, 1303–1328. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I.; Green, M.D. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin. Pharmacokinet. 2005, 44, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Luo, S.; Zhang, B. Glycan analysis of therapeutic glycoproteins. MAbs 2016, 8, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.; Wilson, F.X. Therapeutic applications of iminosugars: Current perspectives and future opportunities. Prog. Med. Chem. 2011, 50, 135–176. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Mellor, H.R.; Neville, D.C.A.; Harvey, D.J.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Cellular effects of deoxynojirimycin analogues: Inhibition of N-linked oligosaccharide processing and generation of free glucosylated oligosaccharides. Biochem. J. 2004, 381, 867–875. [Google Scholar] [CrossRef]
- Chang, J.; Guo, J.T.; Du, Y.; Block, T. Imino sugar glucosidase inhibitors as broadly active anti-filovirus agents. Emerg. Microbes Infect. 2013, 2, e77. [Google Scholar] [CrossRef]
- Yagi, M.; Kouno, T.; Aoyagi, Y.; Murai, H. The structure of moranoline, a piperidine alkaloid from Morus species. J. Agric. Chem. Soc. Jpn. 1976, 50, 571–572. [Google Scholar]
- Nash, R.J.; Kato, A.; Yu, C.Y.; Fleet, G.W. Iminosugars as Therapeutic Agents: Recent Advances and Promising Trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Zheng, C.; Wang, T.; Zhao, H.; Wang, J.; Wang, Z.; Zhai, X.; Jia, Z.; Chen, J.; Zhou, Y.; et al. 1-Deoxynojirimycin: Occurrence, Extraction, Chemistry, Oral Pharmacokinetics, Biological Activities and In Silico Target Fishing. Molecules 2016, 21, 1600. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.R.; Mansfield, K.; You, J.E.; Tennant, B.C.; Kim, Y.H. Natural iminosugar derivatives of 1-deoxynojirimycin inhibit glycosylation of hepatitis viral envelope proteins. J. Microbiol. 2007, 45, 431–440. [Google Scholar]
- Ashraf, G.M.; Greig, N.H.; Khan, T.A.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.A.; Zaidi, S.K.; Akram, M.; Jabir, N.R.; et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Somsak, L.; Nagy, V.; Hadady, Z.; Docsa, T.; Gergely, P. Glucose Analog Inhibitors of Glycogen Phosphorylases as Potential Antidiabetic Agents: Recent Developments. Curr. Pharm. Des. 2003, 9, 1177–1189. [Google Scholar] [CrossRef]
- Ferhati, X.; Matassini, C.; Fabbrini, M.G.; Goti, A.; Morrone, A.; Cardona, F.; Moreno-Vargas, A.J.; Paoli, P. Dual targeting of PTP1B and glucosidases with new bifunctional iminosugar inhibitors to address type 2 diabetes. Bioorg. Chem. 2019, 87, 534–549. [Google Scholar] [CrossRef]
- Durantel, D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr. Opin. Investig. Drugs 2009, 10, 860–870. [Google Scholar]
- Fischl, M.A.; Resnick, L.; Coombs, R.; Kremer, A.B.; Pottage, J.C., Jr.; Fass, R.J.; Fife, K.H.; Powderly, W.G.; Collier, A.C.; Aspinall, R.L.; et al. The safety and efficacy of combination N-butyl-deoxynojirimycin (SC-48334) and zidovudine in patients with HIV-1 infection and 200–500 CD4 cells/mm3. J. Acquir. Immune Defic. Syndr. 1994, 7, 139–147. [Google Scholar]
- Voss, A.A.; Díez-Sampedro, A.; Hirayama, B.A.; Loo, D.D.; Wright, E.M. Imino sugars are potent agonists of the human glucose sensor SGLT3. Mol. Pharmacol. 2007, 71, 628–634. [Google Scholar] [CrossRef]
- McCormack, P.L.; Goa, K.L. Miglustat. Drugs 2003, 63, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef]
- Tropak, M.B.; Blanchard, J.E.; Withers, S.G.; Brown, E.D.; Mahuran, D. High-throughput screening for human lysosomal beta-N-Acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem. Biol. 2007, 14, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Lee, C.J.; Liao, C.L.; Dwek, R.A.; Zitzmann, N.; Lin, Y.L. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 2002, 76, 3596–35604. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Collin, M.; Karlsson, G.; James, W.; Butters, T.; Davis, S.; Gordon, S.; Dwek, R.; Platt, F. The glucosidase inhibitor N-butyldeoxynojirimycin inhibits human immunodeficiency virus entry at the level of post-CD4 binding. J. Virol. 1995, 69, 5791–5797. [Google Scholar] [CrossRef]
- Taylor, D.L.; Sunkara, P.; Liu, P.; Kang, M.S.; Bowlin, T.L.; Tyms, A.S. 6-O-butanoylcastanospermine (MDL 28,574) inhibits glycoprotein processing and the growth of HIVs. AIDS 1991, 5, 693–698. [Google Scholar] [CrossRef]
- Sorbera, L.A.; Castaner, J.; Garcia-Capdevila, L. Celgosivir: A-glucosidase inhibitor. Antihepatitis-C virus drug. Drugs Future 2005, 30, 545–552. [Google Scholar] [CrossRef]
- Pavlovic, D.; Neville, D.C.; Argaud, O.; Blumberg, B.; Dwek, R.A.; Fischer, W.B.; Zitzmann, N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. USA 2003, 100, 6104–6108. [Google Scholar] [CrossRef]
- Cabezas, J.A.; Reglero, A.; Calvo, P. Glycosidases. (Fucosidases, galactosidases, glucosidases, hexosaminidases and glucuronidase from some molluscs and vertebrates, and neuraminidase from virus). Int. J. Biochem. 1983, 15, 243–259. [Google Scholar] [CrossRef]
- Glanz, V.Y.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Inhibition of sialidase activity as a therapeutic approach. Drug Des. Devel. Ther. 2018, 12, 3431–3437. [Google Scholar] [CrossRef]
- Mishra, S.; Upadhaya, K.; Mishra, K.B.; Shukla, A.K.; Tripathi, R.P.; Tiwari, V.K. Carbohydrate-Based Therapeutics: A Frontier in Drug Discovery and Development. Stud. Nat. Prod. Chem. 2016, 49, 307–361. [Google Scholar] [CrossRef]
- Ritter, T.K.; Wong, C.H. Carbohydrate-Based Antibiotics: A New Approach to Tackling the Problem of Resistance. Angew. Chem. Int. Ed. Engl. 2001, 40, 3508–3533. [Google Scholar] [CrossRef]
- Bugg, T.D.; Rodolis, M.T.; Mihalyi, A.; Jamshidi, S. Inhibition of phospho-MurNAc-pentapeptide translocase (MraY) by nucleoside natural product antibiotics, bacteriophage ϕX174 lysis protein E, and cationic antibacterial peptides. Bioorg. Med. Chem. 2016, 24, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.A.; Fleet, G.W.J.; Asano, N.; Molyneux, R.J.; Nash, R.J. Polyhydroxylated alkaloids—Natural occurrence and therapeutic applications. Phytochemistry 2001, 56, 265–295. [Google Scholar] [CrossRef]
- Hamaguchi, J.; Nakagawa, H.; Takahashi, M.; Kudo, T.; Kamiyama, N.; Sun, B.; Oshima, T.; Sato, Y.; Deguchi, K.; Todo, S.; et al. Swainsonine reduces 5-fluorouracil tolerance in the multistage resistance of colorectal cancer cell lines. Mol. Cancer 2007, 6, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J.; Matsumoto, K.; White, S.L.; Olden, K. Inhibition of experimental metastasis by castanospermine in mice: Blockage of two distinct stages of tumor colonization by oligosaccharide processing inhibitors. Cancer Res. 1986, 46, 5215–5222. [Google Scholar]
- Taniguchi, N.; Kizuka, Y. Glycans and cancer: Role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 2015, 126, 11–51. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Shaikh, A.S.; Wang, F. Recent Advance in Tumor-Associated Carbohydrate Antigens (TACAs)-based Antitumor Vaccines. ACS Chem. Biol. 2016, 11, 850–863. [Google Scholar] [CrossRef]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, Q. Recent Development in Carbohydrate-Based Cancer Vaccines. Curr. Opin. Chem. Biol. 2009, 13, 608–617. [Google Scholar] [CrossRef]
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune Recognition of Tumor-Associated Mucin MUC1 is Achieved by a Fully Synthetic Aberrantly Glycosylated MUC1 Tripartite Vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Wu, D.; Ragupathi, G.; Lu, S.; Williams, L.; Hwu, W.J.; Johnson, D.; Livingston, P.O. Sequential Immunization of Melanoma Patients with GD3 Ganglioside Vaccine and Anti-Idiotypic Monoclonal Antibody That Mimics GD3 Ganglioside. Clin. Cancer Res. 2004, 10, 4717. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Suciu, S.; Rutkowski, P.; Marsden, J.; Santinami, M.; Corrie, P.; Aamdal, S.; Ascierto, P.A.; Patel, P.M.; Kruit, W.H.; et al. Adjuvant ganglioside GM2–KLH/QS-21 vaccination versus observation after resection of primary tumor > 1.5 mm in patients with stage II melanoma: Results of the EORTC 18961 randomized phase III trial. J. Clin. Oncol. 2013, 31, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Theratope®® Study Group. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef]
- Thakkar, S.G.; Heeger, P.; Wacker, B.; Roddy, M.; Waxenecker, G.; Himmler, H.G.; Loibner, G.; Elson, P.; Bukowski, R. A phase Ib trial of an anti-idiotypic vaccine for Lewis Y (IGN 301) administered subcutaneously in patients with refractory solid tumors. J. Clin. Oncol. 2004, 22, 2586. [Google Scholar] [CrossRef]
- Hossain, M.K.; Wall, K.A. Immunological Evaluation of Recent MUC1 Glycopeptide Cancer Vaccines. Vaccines 2016, 4, 25. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose Conjugation for the Specific Targeting and Treatment of Cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef]
- Smith, B.A.H.; Bertozzi, C.R. The clinical impact of glycobiology: Targeting selectins, Siglecs and mammalian glycans. Nat. Rev. Drug Discov. 2021, 20, 217–243. [Google Scholar] [CrossRef]
- Wrodnigg, T.M.; Steiner, A.J.; Ueberbacher, B.J. Natural and Synthetic Iminosugars as Carbohydrate Processing Enzyme Inhibitors for Cancer Therapy. Anticancer. Agents Med. Chem. 2008, 8, 77–85. [Google Scholar] [CrossRef]
- Vallee, F.; Karaveg, K.; Herscovics, A.; Moremen, K.W.; Howell, P.L. Structural Basis for Catalysis and Inhibition of N-glycan Processing Class I alpha 1,2-mannosidases. J. Biol. Chem. 2000, 275, 41287–41298. [Google Scholar] [CrossRef]
- Allan, G.; Ouadid-Ahidouch, H.; Sanchez-Fernandez, E.M.; Risquez-Cuadro, R.; Fernandez, J.M.; Ortiz-Mellet, C.; Ahidouch, A. New Castanospermine Glycoside Analogues Inhibit Breast Cancer Cell Proliferation and Induce Apoptosis Without Affecting Normal Cells. PLoS ONE 2013, 8, e76411. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Tiwari, V.K.; Schmidt, R.R. Recent trends and challenges on carbohydrate-based molecular scaffolding: General consideration toward impact of carbohydrates in drug discovery and development. In Carbohydrates in Drug Discovery and Development; Tiwari, V.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Chapter 1; pp. 1–69. [Google Scholar] [CrossRef]
- Whayne, T.F., Jr. Clinical Use of Digitalis: A State of the Art Review. Am. J. Cardiovasc. Drugs 2018, 18, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Rudd, T.; Yates, E. New Applications of Heparin and Other Glycosaminoglycans. Molecules 2017, 22, 749. [Google Scholar] [CrossRef] [PubMed]
- Bashiri, S.; Koirala, P.; Toth, I.; Skwarczynski, M. Carbohydrate Immune Adjuvants in Subunit Vaccines. Pharmaceutics 2020, 12, 965. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.C.; Rogers, P.D.; Shelton, C.M. A Review of Pneumococcal Vaccines: Current Polysaccharide Vaccine Recommendations and Future Protein Antigens. J. Pediatr. Pharmacol. Ther. 2016, 21, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.; Lepenies, B. Carbohydrate-Based Vaccines: An Overview. Methods Mol. Biol. 2015, 1331, 1–10. [Google Scholar] [CrossRef]
- Feng, L.S.; Zheng, M.J.; Zhao, F.; Liu, D. 1,2,3-Triazole hybrids with anti-HIV-1 activity. Arch. Pharm. 2021, 354, e2000163. [Google Scholar] [CrossRef]
- Sun, L.; Huang, T.; Dick, A.; Meuser, M.E.; Zalloum, W.A.; Chen, C.H.; Ding, X.; Gao, P.; Cocklin, S.; Lee, K.H.; et al. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur. J. Med. Chem. 2020, 190, 112085. [Google Scholar] [CrossRef]
- Ramprasad, J.; Sthalam, V.K.; Thampunuri, R.L.M.; Bhuky, S.; Ummanni, R.; Balasubramanian, S.; Pabbaraja, S. Synthesis and evaluation of a novel quinoline-triazole analogs for antitubercular properties via molecular hybridization approach. Bioorg. Med. Chem. Lett. 2019, 29, 126671. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Ali, A.A. 1,2,3-Triazoles: Synthesis and Biological Application. In Azoles—Synthesis, Properties, Applications and Perspectives; Kuznetsov, A., Ed.; IntechOpen: London, UK, 2020; Available online: https://www.intechopen.com/chapters/72394 (accessed on 28 February 2022).
- Da Silva, P.N.; da Conceição, R.A.; do Couto Maia, R.; de Castro Barbosa, M.L. Sodium-glucose cotransporter 2 (SGLT-2) inhibitors: A new antidiabetic drug class. MedChemComm 2018, 9, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Aracri, P.; Colombo, E.; Scalmani, P.; Mantegazza, M.; Avanzini, G.; Franceschetti, S. Phosphorylation of sodium channels mediated by protein kinase-C modulates inhibition by topiramate of tetrodotoxin-sensitive transient sodium current. Br. J. Pharmacol. 2007, 150, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Gryder, D.S.; Rogawski, M.A. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J. Neurosci. 2003, 23, 7069–7074. [Google Scholar] [CrossRef] [PubMed]
- Meurs, A.; Clinckers, R.; Ebinger, G.; Michotte, Y.; Smolders, I. Substantia nigra is an anticonvulsant site of action of topiramate in the focal pilocarpine model of limbic seizures. Epilepsia 2006, 47, 1519–1535. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.M.; Shipman, C., Jr.; Drach, J.C. Antiviral activity of arabinosyladenine and arabinosylhypoxanthine in herpes simplex virus-infected KB cells: Selective inhibition of viral deoxyribonucleic acid synthesis in the presence of an adenosine deaminase inhibitor. Antimicrob. Agents Chemother. 1976, 10, 64–74. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holzer, S.; Rzechorzek, N.J.; Short, I.R.; Jenkyn-Bedford, M.; Pellegrini, L.; Kilkenny, M.L. Structural Basis for Inhibition of Human Primase by Arabinofuranosyl Nucleoside Analogues Fludarabine and Vidarabine. ACS Chem. Biol. 2019, 14, 1904–1912. [Google Scholar] [CrossRef]
- Huchzermeyer, H.; Schumann, C. Lactulose-a multifaceted substance. Z. Gastroenterol. 1997, 35, 945–955. [Google Scholar]
- Mukherjee, S.; John, S. Lactulose. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kudaravalli, P.; John, S. Sucralfate. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lee, E.S.; Kim, J.S.; Lee, H. Auranofin, an Anti-rheumatic Gold Drug, Aggravates the Radiation-Induced Acute Intestinal Injury in Mice. Front. Pharmacol. 2019, 10, 417. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef]
- Breitling, J.; Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef]
- Wang, F.; Chan, C.; Weir, N.R.; Denic, V. The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 2014, 512, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.J. Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 2000, 69, 69–93. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta 2010, 1803, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Friedlander, R.; Jarosch, E.; Urban, J.; Volkwein, C.; Sommer, T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000, 2, 379–384. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef]
- Naidoo, N. Er and aging-protein folding and the er stress response. Ageing Res. Rev. 2009, 8, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. Er stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Onda, M.; Nagai, H.; Nagahata, T.; Ogawa, K.; Emi, M. Upregulation and overexpression of human x-box binding protein 1 (hxbp-1) gene in primary breast cancers. Breast Cancer 2003, 10, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of tumor progression by suppression of stress protein grp78/ bip induction in fibrosarcoma b/c10me. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. Xbp1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the atf6, xbp1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the er stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Araki, E.; Oyadomari, S.; Mori, M. Endoplasmic reticulum stress and diabetes mellitus. Intern. Med. 2003, 42, 7–14. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Burcin, M.; Gromada, J.; Urano, F. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr. Opin. Pharmacol. 2009, 9, 763–770. [Google Scholar] [CrossRef]
- Ishihara, H.; Takeda, S.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Yamada, T.; Inoue, H.; Soga, H.; Katagiri, H.; et al. Disruption of the wfs1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum. Mol. Genet. 2004, 13, 1159–1170. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. Wfs1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S. A model of the unfolded protein response: Pancreatic beta-cell as a case study. Cell Physiol. Biochem. 2009, 23, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin- proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology 2006, 66, S102–S109. [Google Scholar] [CrossRef]
- Ugalde, R.A.; Staneloni, R.J.; Leloir, L.F. Action of glycosidases on the saccharide moiety of the glucose-containing dolichyl diphosphate oligosaccharide. FEBS Lett. 1978, 91, 209–212. [Google Scholar] [CrossRef]
- Ugalde, R.A.; Staneloni, R.J.; Leloir, L.F. Microsomal glucosidases of rat liver. Partial purification and inhibition by disaccharides. Eur. J. Biochem. 1980, 113, 97–103. [Google Scholar] [CrossRef]
- Grinna, L.S.; Robbins, P.W. Glycoprotein biosynthesis. Rat liver microsomal glucosidases, which process oligosaccharides. J. Biol. Chem. 1979, 254, 8814–8818. [Google Scholar] [CrossRef]
- Scher, M.G.; Waechter, C.J. A glucosylated oligosaccharide lipid intermediate in calf brain. Evidence for the transfer of the oligosaccharide into membrane glycoprotein and subsequent removal of glucosyl residues. J. Biol. Chem. 1979, 254, 2630–2637. [Google Scholar] [CrossRef]
- D’Alessio, C.; Caramelo, J.J.; Parodi, A.J. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell Dev. Biol. 2010, 21, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Caramelo, J.J.; Castro, O.A.; Alonso, L.G.; De Prat-Gay, G.; Parodi, A.J. UDP-Glc: Glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. USA 2003, 100, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wong, H.N.; Schauerte, J.A.; Kaufman, R.J. The protein kinase/endoribonuclease IRE1α that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J. Biol. Chem. 2002, 277, 18346–18356. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Protein folding in the endoplasmic reticulum and the unfolded protein response. In Molecular Chaperones in Health and Disease. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 69–91. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Choy, M.S.; Yusoff, P.; Lee, I.C.; Newton, J.C.; Goh, C.W.; Page, R.; Shenolikar, S.; Peti, W. Structural and Functional Analysis of the GADD34:PP1 eIF2α Phosphatase. Cell Rep. 2015, 11, 1885–1891. [Google Scholar] [CrossRef]
- Lin, W.; Stone, S. Unfolded protein response in myelin disorders. Neural Regen. Res. 2020, 15, 636–645. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Lei, Z.; Yue, Y.; Stone, S.; Wu, S.; Lin, W. NF-κB Activation Accounts for the Cytoprotective Effects of PERK Activation on Oligodendrocytes during EAE. J. Neurosci. 2020, 40, 6444–6456. [Google Scholar] [CrossRef] [PubMed]
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Keum, Y.S.; Han, Y.H.; Liew, C.; Kim, J.H.; Xu, C.; Yuan, X.; Shakarjian, M.P.; Chong, S.; Kong, A.N. Induction of heme oxygenase-1 (HO-1) and NAD[P]H: Quinone oxidoreductase 1 (NQO1) by a phenolic antioxidant, butylated hydroxyanisole (BHA) and its metabolite, tert-butylhydroquinone (tBHQ) in primary-cultured human and rat hepatocytes. Pharm. Res. 2006, 23, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. 2017, 23, 1875–1883. [Google Scholar] [CrossRef]
- Getts, M.T.; Getts, D.R.; Kohm, A.P.; Miller, S.D. Endoplasmic reticulum stress response as a potential therapeutic target in multiple sclerosis. Therapy 2008, 5, 631–640. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef]
- Goedert, M. Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef]
- Hu, Y.; Benedict, M.A.; Ding, L.; Nunez, G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999, 18, 3586–3595. [Google Scholar] [CrossRef]
- Pollard, M.G.; Travers, K.J.; Weissman, J.S. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1998, 1, 171–182. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Prols, F. Dual topology of co-chaperones at the membrane of the endoplasmic reticulum. Cell Death Discov. 2021, 7, 203. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Soto, M.M.; McCullagh, E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: A role in neurodegeneration. Front. Aging Neurosci. 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Tempio, T.; Orsi, A.; Sicari, D.; Valetti, C.; Yoboue, E.D.; Anelli, T.; Sitia, R. A virtuous cycle operated by ERp44 and ERGIC-53 guarantees proteostasis in the early secretory compartment. iScience 2021, 24, 102244. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef]
- Helenius, A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol. Biol. Cell 1994, 5, 253–265. [Google Scholar] [CrossRef]
- Van der Goot, A.T.; Pearce, M.M.P.; Leto, D.E.; Shaler, T.A.; Kopito, R.R. Redundant and Antagonistic Roles of XTP3B and OS9 in Decoding Glycan and Non-glycan Degrons in ER-Associated Degradation. Mol. Cell 2018, 70, 516–530.e6. [Google Scholar] [CrossRef]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999, 1426, 239–257. [Google Scholar] [CrossRef]
- Satoh, T.; Toshimori, T.; Yan, G.; Yamaguchi, T.; Kato, K. Structural basis for two-step glucose trimming by glucosidase II involved in ER glycoprotein quality control. Sci. Rep. 2016, 6, 20575. [Google Scholar] [CrossRef]
- Alonso, J.M.; Santa-Cecilia, A.; Calvo, P. Effect of bromoconduritol on glucosidase II from rat liver. A new kinetic model for the binding and hydrolysis of the substrate. Eur. J. Biochem. 1993, 215, 37–42. [Google Scholar] [CrossRef]
- Hirai, M.; Shimizu, N. Purification of two distinct proteins of approximate Mr 80,000 from human epithelial cells and identification as proper substrates for protein kinase C. Biochem. J. 1990, 270, 583–589. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Simons, J.F.; Helenius, A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J. Biol. Chem. 1996, 271, 27509–27516. [Google Scholar] [CrossRef] [PubMed]
- Herscovics, A. Importance of glycosidases in mammalian glycoprotein biosynthesis. Biochim. Biophys. Acta 1999, 1473, 96–107. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Parodi, A.J. N-glycan processing and glycoprotein folding. Adv. Protein Chem. 2002, 59, 303. [Google Scholar]
- Treml, K.; Meimaroglou, D.; Hentges, A.; Bause, E. The a- and b-subunits are required for expression of catalytic activity in the hetero-dimeric glucosidase II complex from human liver. Glycobiology 2000, 10, 493–502. [Google Scholar] [CrossRef]
- Arendt, C.W.; Ostergaard, H.L. Two distinct domains of the b-subunit of glucosidase II interact with the catalytic a-subunit. Glycobiology 2000, 10, 487–492. [Google Scholar] [CrossRef]
- Feng, J.; Romaniouk, A.V.; Samal, S.K.; Vijay, I.K. Processing enzyme glucosidase II: Proposed catalytic residues and developmental regulation during the ontogeny of the mouse mammary gland. Glycobiology 2004, 14, 909–921. [Google Scholar] [CrossRef]
- Henrissat, B.; Bairoch, A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1993, 293, 781–788. [Google Scholar] [CrossRef]
- Munro, S. The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr. Biol. 2001, 11, R499–R501. [Google Scholar] [CrossRef]
- D’Alessio, C.; Dahms, N.M. Glucosidase II and MRH-domain containing proteins in the secretory pathway. Curr. Protein Pept. Sci. 2015, 16, 31–48. [Google Scholar] [CrossRef]
- Olson, L.J.; Orsi, R.; Alculumbre, S.G.; Peterson, F.C.; Stigliano, I.D.; Parodi, A.; D’Alessio, C.; Dahms, N.M. Structure of the lectin MRH domain of Glucosidase II, an enzyme that regulates glycoprotein folding quality control in the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 16460–16475. [Google Scholar] [CrossRef]
- Nairn, A.V.; Moremen, K.W. Glucosidase, Alpha Neutral AB.; Glucosidase II Subunit Beta (GANAB, PRKCSH, α-Glucosidase II). In Handbook of Glycosyltransferases and Related Genes; Taniguchi, N., Honke, K., Fukuda, M., Narimatsu, H., Yamaguchi, Y., Angata, T., Eds.; Springer: Tokyo, Japan, 2014. [Google Scholar] [CrossRef]
- Brada, D.; Dubach, U.C. Isolation of a homogeneous glucosidase II from pig kidney microsomes. Eur. J. Biochem. 1984, 141, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M.; Touster, O. Purification and characterization of glucosidase II, an endoplasmic reticulum hydrolase involved in glycoprotein biosynthesis. J. Biol. Chem. 1982, 257, 9990–10000. [Google Scholar] [CrossRef]
- Hettkamp, H.; Bause, E.; Legler, G. Inhibition by nojirimycin and 1-deoxynojirimycin of microsomal glucosidases from calf liver acting on the glycoprotein oligosaccharides Glc1-3Man9GlcNAc2. Biosci. Rep. 1982, 2, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Martiniuk, F.; Ellenbogen, A.; Hirschhorn, R. Identity of neutral a-glucosidase AB and the glycoprotein processing enzyme glucosidase II. Biochemical and genetic studies. J. Biol. Chem. 1985, 260, 1238–1242. [Google Scholar] [CrossRef]
- Alonso, J.M.; Santa-Cecilia, A.; Calvo, P. Glucosidase II from rat liver microsomes. Kinetic model for binding and hydrolysis. Biochem. J. 1991, 278, 721–727. [Google Scholar] [CrossRef]
- Hentges, A.; Bause, E. Affinity purification and characterization of glucosidase II from pig liver. Biol. Chem. 1997, 378, 1031–1038. [Google Scholar] [CrossRef]
- Saunier, B.; Kilker, R.D., Jr.; Tkacz, J.S.; Quaroni, A.; Herscovics, A. Inhibition of N-linked complex oligosaccharide formation by 1-deoxynojirimycin, an inhibitor of processing glucosidases. J. Biol. Chem. 1982, 257, 14155–14161. [Google Scholar] [CrossRef]
- Datema, R.; Romero, P.A.; Legler, G.; Schwarz, R.T. Inhibition of formation of complex oligosaccharides by the glucosidase inhibitor bromoconduritol. Proc. Natl. Acad. Sci. USA 1982, 79, 6787–6791. [Google Scholar] [CrossRef]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.; Thomas, D.Y. The heterodimeric structure of glucosidase II is required for its activity, solubility, and localization in vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef]
- Reitman, M.L.; Trowbridge, I.S.; Kornfeld, S. A lectin-resistant mouse lymphoma cell line is deficient in glucosidase II, a glycoprotein-processing enzyme. J. Biol. Chem. 1982, 257, 10357–10363. [Google Scholar] [CrossRef]
- Flura, T.; Brada, D.; Ziak, M.; Roth, J. Expression of a cDNA encoding the glucose trimming enzyme glucosidase II in CHO cells and molecular characterization of the enzyme deficiency in a mutant mouse lymphoma cell line. Glycobiology 1997, 7, 617–624. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fedeles, S.V.; Gallagher, A.R.; Somlo, S. Polycystin-1: A master regulator of intersecting cystic pathways. Trends Mol. Med. 2014, 20, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.E.; Hurley, U.A.; Birch, R.J.; Arioli, T.; Cork, A.; Williamson, R.E. The cellulose-deficient Arabidopsis mutant rsw3 is defective in a gene encoding a putative glucosidase II, an enzyme processing N-glycans during ER quality control. Plant. J. 2002, 32, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Galli, C.; Vanoni, O.; Arnold, S.M.; Kaufman, R.J. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol. Cell 2005, 20, 503–512. [Google Scholar] [CrossRef]
- Li, J.; Zhao-Hui, C.; Batoux, M.; Nekrasov, V.; Roux, M.; Chinchilla, D.; Zipfel, C.; Jones, J.D.G. Specific ER quality control components required for biogenesis of the plant innate immune receptor EFR. Proc. Natl. Acad. Sci. USA 2009, 106, 15973–15978. [Google Scholar] [CrossRef]
- Arendt, C.W.; Ostergaard, H.L. Identification of the CD45-associated 116-kDa and 80-kDa proteins as the a- and b-subunits of a-glucosidase II. J. Biol. Chem. 1997, 272, 13117–13125. [Google Scholar] [CrossRef][Green Version]
- Roach, T.; Slater, S.; Koval, M.; White, L.; Cahir McFarland, E.D.; Okumura, M.; Thomas, M.; Brown, E. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr. Biol. 1997, 7, 408–417. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Walport, M.; Shlomchik, M.J. Antigen receptor structure and signaling pathways. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK27130/ (accessed on 28 February 2022).
- D’Alessio, D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 126–132. [Google Scholar] [CrossRef]
- Liu, M.; Wright, J.; Guo, H.; Xiong, Y.; Arvan, P. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitam. Horm. 2014, 95, 35–62. [Google Scholar] [CrossRef]
- Ron, D. Proteotoxicity in the endoplasmic reticulum: Lessons from the Akita diabetic mouse. J. Clin. Investig. 2002, 109, 443–445. [Google Scholar] [CrossRef]
- Stoy, J.; Edghill, E.L.; Flanagan, S.E.; Ye, H.; Paz, V.P.; Pluzhnikov, A.; Below, J.E.; Hayes, M.G.; Cox, N.J.; Lipkind, G.M.; et al. Neonatal Diabetes International Collaborative Group. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. 2007, 104, 15040–15044. [Google Scholar] [CrossRef] [PubMed]
- Delepine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; PSP Genetics Study Group; Lee, V.M.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef]
- Unterberger, U.; Hoftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtlander, T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. NeuroBiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek-Kaminska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Park, Y.K.; Lee, J.H.; Park, K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-ε/ERK/AP-1 signaling cascade. Cancer Res. 2001, 61, 8322–8330. [Google Scholar]
- Chen, X.; Ding, Y.; Liu, C.G.; Mikhail, S.; Yang, C.S. Overexpression of glucose-regulated protein 94 (Grp94) in esophageal adenocarcinomas of a rat surgical model and humans. Carcinogenesis 2002, 23, 123–130. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Orlowski, R.Z. The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 189–213. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.L.; Li, J.; Zhou, Z.H.; Liu, J.R.; Huang, B.H.; Zheng, D.; Su, C. Differentiation induction enhances bortezomib efficacy and overcomes drug resistance in multiple myeloma. Biochem. Biophys. Res. Commun. 2012, 420, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Lau, E.K.; Al-Shabeeb, A.; Nikolic, A.; Catalano, A.; Iland, H.; Horvath, N.; Ho, P.J.; Harrison, S.; Fleming, S.; et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012, 97, 64–72. [Google Scholar] [CrossRef]
- Nilsson, B.; Rehncrona, S.; Siesjo, B.K. Coupling of cerebral metabolism and blood flow in epileptic seizures, hypoxia and hypoglycaemia. Ciba Found. Symp. 1978, 56, 199–218. [Google Scholar] [CrossRef]
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: Unfolded protein response (upr)-dependent and upr-independent pathways. Mol. Cancer Res. 2006, 4, 423–436. [Google Scholar] [CrossRef]
- Kunz, A.; Iadecola, C. Cerebral vascular dysregulation in the ischemic brain. Handb. Clin. Neurol. 2009, 92, 283–305. [Google Scholar] [CrossRef]
- Traystman, R.J.; Kirsch, J.R.; Koehler, R.C. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J. Appl. Physiol. 1991, 71, 1185–1195. [Google Scholar] [CrossRef]
- Zhou, A.X.; Tabas, I. The UPR in atherosclerosis. Semin. Immunopathol. 2013, 35, 321–332. [Google Scholar] [CrossRef]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, M.; Guo, Y.S.; Shen, X.Y.; Gao, Z.K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef]
- Jaeken, J.; Peanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.J.; Waanders, E.; Woudenberg, J.; Lefeber, D.J.; Drenth, J.P.H. Congenital disorders of glycosylation in hepatology: The example of polycystic liver disease. J. Hepatol. 2010, 52, 432–440. [Google Scholar] [CrossRef]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Cnossen, W.R.; te Morsche, R.H.; Hoischen, A.; Gilissen, C.; Chrispijn, M.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Veltman, J.A.; Drenth, J.P. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5343–5348. [Google Scholar] [CrossRef] [PubMed]
- Waanders, E.; te Morsche, R.H.; de Man, R.A.; Jansen, J.B.; Drenth, J.P. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum. Mutat. 2006, 27, 830. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; Yao, Q.; Li, W.; Huang, Q.; Outeda, P.; Cebotaru, V.; Chiaravalli, M.; Boletta, A.; Piontek, K.; et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat. Commun. 2014, 5, 5482. [Google Scholar] [CrossRef]
- Wilson, E.M.; Choi, J.; Torres, V.E.; Somlo, S.; Besse, W. Large Deletions in GANAB and SEC63 explain 2 cases of polycystic kidney and liver disease. Kidney Int. Rep. 2020, 5, 727–731. [Google Scholar] [CrossRef]
- Van Keimpema, L.; De Koning, D.B.; van Hoek, B.; van den Berg, A.P.; van Oijen, M.G.; De Man, R.A.; Nevens, F.; Drenth, J.P. Patients with isolated polycystic liver disease referred to liver centres: Clinical characterization of 137 cases. Liver Int. 2011, 31, 92–98. [Google Scholar] [CrossRef]
- Audrezet, M.P.; Cornec-Le Gall, E.; Chen, J.M.; Redon, S.; Quere, I.; Creff, J.; Benech, C.; Maestri, S.; Le Meur, Y.; Ferec, C. Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum. Mutat. 2012, 33, 1239–1250. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Wegierski, T.; Skouloudaki, K.; Putz, M.; Fu, X.; Engel, C.; Boehlke, C.; Peng, H.; Kuehn, E.W.; et al. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum. Mol. Genet. 2010, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Gainullin, V.G.; Hopp, K.; Ward, C.J.; Hommerding, C.J.; Harris, P.C. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Investig. 2015, 125, 607–620. [Google Scholar] [CrossRef]
- Grunblatt, E.; Nemoda, Z.; Werling, A.M.; Roth, A.; Angyal, N.; Tarnok, Z.; Thomsen, H.; Peters, T.; Hinney, A.; Hebebrand, J.; et al. The involvement of the canonical Wnt-signaling receptor LRP5 and LRP6 gene variants with ADHD and sexual dimorphism: Association study and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 365–376. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, Q.H.; Lu, Y.J.; Ren, K.; Yi, G.H. Involvement of the IRE1α-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol. 2015, 34, 6–18. [Google Scholar] [CrossRef]
- Vivas-Acevedo, G.; Lozano-Hernández, R.; Camejo, M.I. Varicocele decreases epididymal neutral α-glucosidase and is associated with alteration of nuclear DNA and plasma membrane in spermatozoa. BJU Int. 2014, 113, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Krause, W.; Bohring, C. Why do we determine alpha-glucosidase activity in human semen during infertility work-up? Andrologia 1999, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Cheon, Y.P.; Kim, C.H. Impact of glycosylation on the unimpaired functions of the sperm. Clin. Exp. Reprod. Med. 2015, 42, 77–85. [Google Scholar] [CrossRef]
- Cooper, T.G.; Yeung, C.H.; Nashan, D.; Jockenhövel, F.; Nieschlag, E. Improvement in the assessment of human epididymal function by the use of inhibitors in the assay of alpha-glucosidase in seminal plasma. Int. J. Androl. 1990, 13, 297–305. [Google Scholar] [CrossRef]
- Dohle, G.R. Inflammatory-associated obstructions of the male reproductive tract. Andrologia 2003, 35, 321–324. [Google Scholar] [CrossRef]
- Kret, B.; Milad, M.; Jeyendran, R.S. New discriminatory level for glucosidase activity to diagnose epididymal obstruction or dysfunction. Arch. Androl. 1995, 35, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Decheux, J.L.; Gatti, J.L.; Dacheux, F. Contribution of epididymal secretory proteins for spermatozoa maturation. Microsc. Res. Tech. 2003, 61, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Ben Ali, H.; Guerin, J.F.; Pinatel, M.C.; Mathieu, C.; Boulieu, D.; Tritar, B. Relationship between semen characteristics, alpha glucosidase and the capacity of spermatozoa to bind to the human zona pellucida. Int. J. Androl. 1994, 17, 121–126. [Google Scholar]
- Levrant, S.; Watanabe, M.; Land, S.; Sauer, R.; Jeyendran, R.S. The relevance of neutral alpha-glucosidase activity in andrology. Syst. Biol. Reprod. Med. 2009, 55, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.J.; Huang, Z.X.; Zhou, C.J.; Wang, J.W.; You, Y.; Song, Z.Q.; Xiang, M.M.; Zhong, B.Y.; Hao, F. Gene profiling involved in immature CD4+ T lymphocyte responsible for systemic lupus erythematosus. Mol. Immunol. 2006, 43, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- De Masi, R.; Vergara, D.; Pasca, S.; Acierno, R.; Greco, M.; Spagnolo, L.; Blasi, E.; Sanapo, F.; Trianni, G.; Maffia, M. PBMCs protein expression profile in relapsing IFN-treated multiple sclerosis: A pilot study on relation to clinical findings and brain atrophy. J. Neuroimmunol. 2009, 210, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.W.; Kim, S.H.; Jeong, I.H.; Ahn, S.W.; Huh, S.Y.; Park, M.S.; Eom, Y.I.; Joo, I.S.; Cho, J.Y.; Cho, E.B.; et al. Utility of the rio score and modified rio score in korean patients with multiple sclerosis. PLoS ONE 2015, 10, e0129243. [Google Scholar] [CrossRef]
- Ostolaza Ibanez, A.; Corroza Laviñeta, J.; Ayuso Blanco, T. Immunosenescence: The role of age in multiple sclerosis. Neurologia 2020, 20, S0213-485330226-7. [Google Scholar] [CrossRef]
- De Masi, R.; Orlando, S.; De Donno, A. The Age-Related Efficacy of Dimethyl Fumarate and Natalizumab in the Real-World Management of Multiple Sclerosis. Pharmaceuticals 2021, 14, 81. [Google Scholar] [CrossRef]
- Li, Q.; Ye, X.S. Iminosugars as Immunomodulating Agents: Synthesis and Biological Activities of 1-Deoxynojirimycin and Related Compounds. Isr. J. Chem. 2015, 55, 336–346. [Google Scholar] [CrossRef]
- Walter, S.; Fassbender, K.; Gulbins, E.; Liu, Y.; Rieschel, M.; Herten, M.; Bertsch, T.; Engelhardt, B. Glycosylation processing inhibition by castanospermine prevents experimental autoimmune encephalomyelitis by interference with IL-2 receptor signal transduction. J. Neuroimmunol. 2002, 132, 1–10. [Google Scholar] [CrossRef]
- Willenborg, D.O.; Parish, C.R.; Cowden, W.B. Inhibition of experimental allergic encephalomyelitis by the alpha-glucosidase inhibitor castanospermine. J. Neurol. Sci. 1989, 90, 77–85. [Google Scholar] [CrossRef]
- Parodi, B.; Sanna, A.; Cedola, A.; Uccelli, A.; Kerlero de Rosbo, N. Hydroxycarboxylic Acid Receptor 2, a Pleiotropically Linked Receptor for the Multiple Sclerosis Drug, Monomethyl Fumarate. Possible Implications for the Inflammatory Response. Front. Immunol. 2021, 12, 655212. [Google Scholar] [CrossRef] [PubMed]
- Gkalpakiotis, S.; Arenberger, P.; Gkalpakioti, P.; Meluzinova, E.; Chandran, D.; Arenbergerova, M. Management of psoriasis vulgaris and multiple sclerosis with fumaric acid. J. Am. Acad Dermatol. 2014, 70, e60–e61. [Google Scholar] [CrossRef]
- Zhao, M.; Luo, J.; Xiao, B.; Tang, H.; Song, F.; Ding, X.; Yang, G. Endoplasmic reticulum stress links psoriasis vulgaris with keratinocyte inflammation. Postepy Dermatol. Alergol. 2020, 37, 34–40. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Masi, R.; Orlando, S. GANAB and N-Glycans Substrates Are Relevant in Human Physiology, Polycystic Pathology and Multiple Sclerosis: A Review. Int. J. Mol. Sci. 2022, 23, 7373. https://doi.org/10.3390/ijms23137373
De Masi R, Orlando S. GANAB and N-Glycans Substrates Are Relevant in Human Physiology, Polycystic Pathology and Multiple Sclerosis: A Review. International Journal of Molecular Sciences. 2022; 23(13):7373. https://doi.org/10.3390/ijms23137373
Chicago/Turabian StyleDe Masi, Roberto, and Stefania Orlando. 2022. "GANAB and N-Glycans Substrates Are Relevant in Human Physiology, Polycystic Pathology and Multiple Sclerosis: A Review" International Journal of Molecular Sciences 23, no. 13: 7373. https://doi.org/10.3390/ijms23137373
APA StyleDe Masi, R., & Orlando, S. (2022). GANAB and N-Glycans Substrates Are Relevant in Human Physiology, Polycystic Pathology and Multiple Sclerosis: A Review. International Journal of Molecular Sciences, 23(13), 7373. https://doi.org/10.3390/ijms23137373