
Dopamine Photochemical Behaviour under UV Irradiation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples’ Preparation
2.2. Steady-State UV–Vis Absorption and Fluorescence Spectroscopy
2.3. Time-Resolved Fluorescence Spectroscopy
2.4. Theoretical Methods
3. Results and Discussion
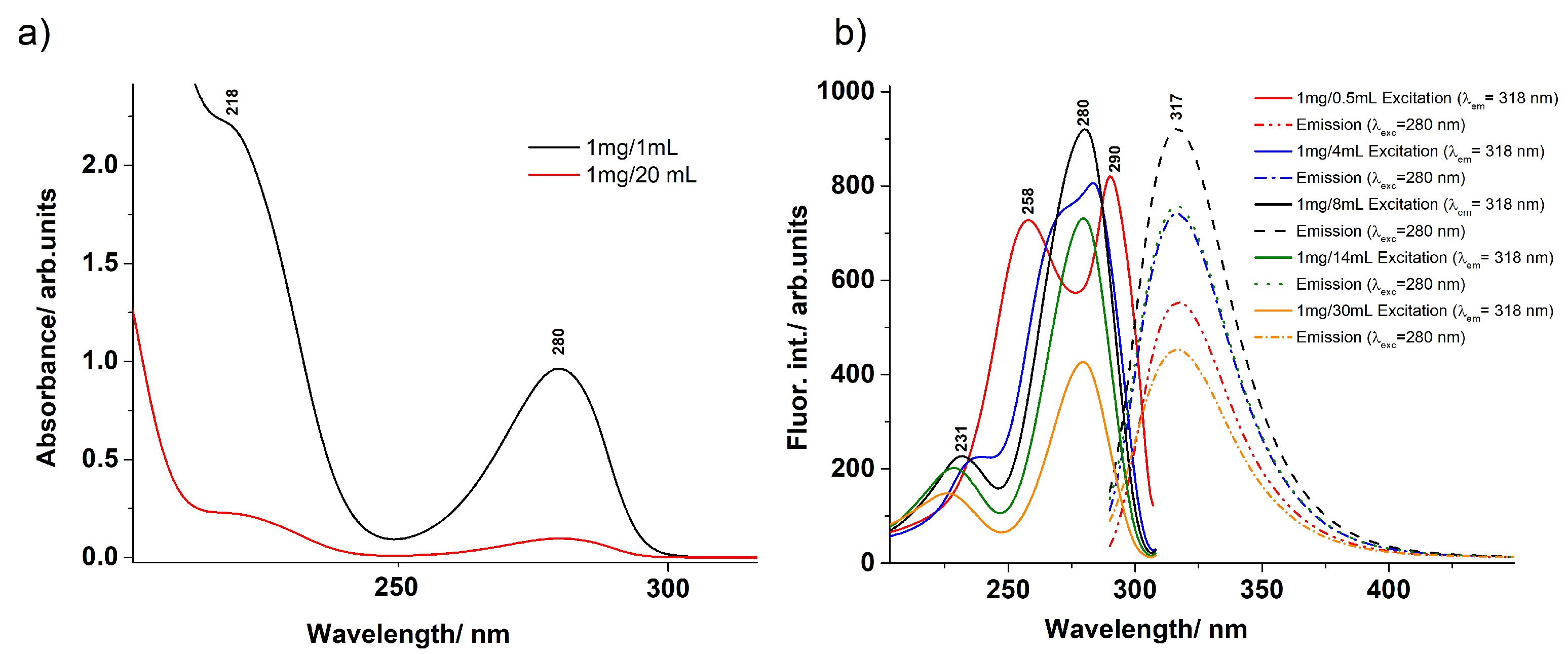
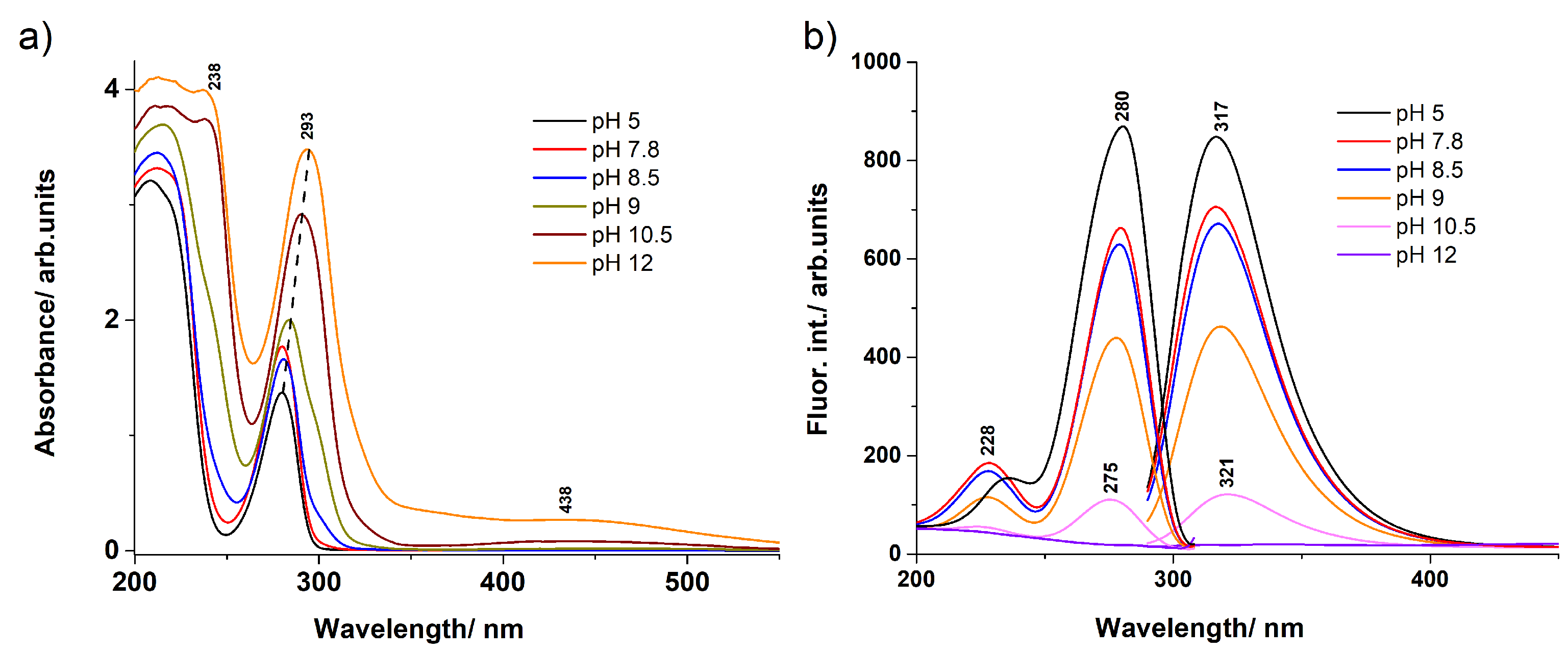
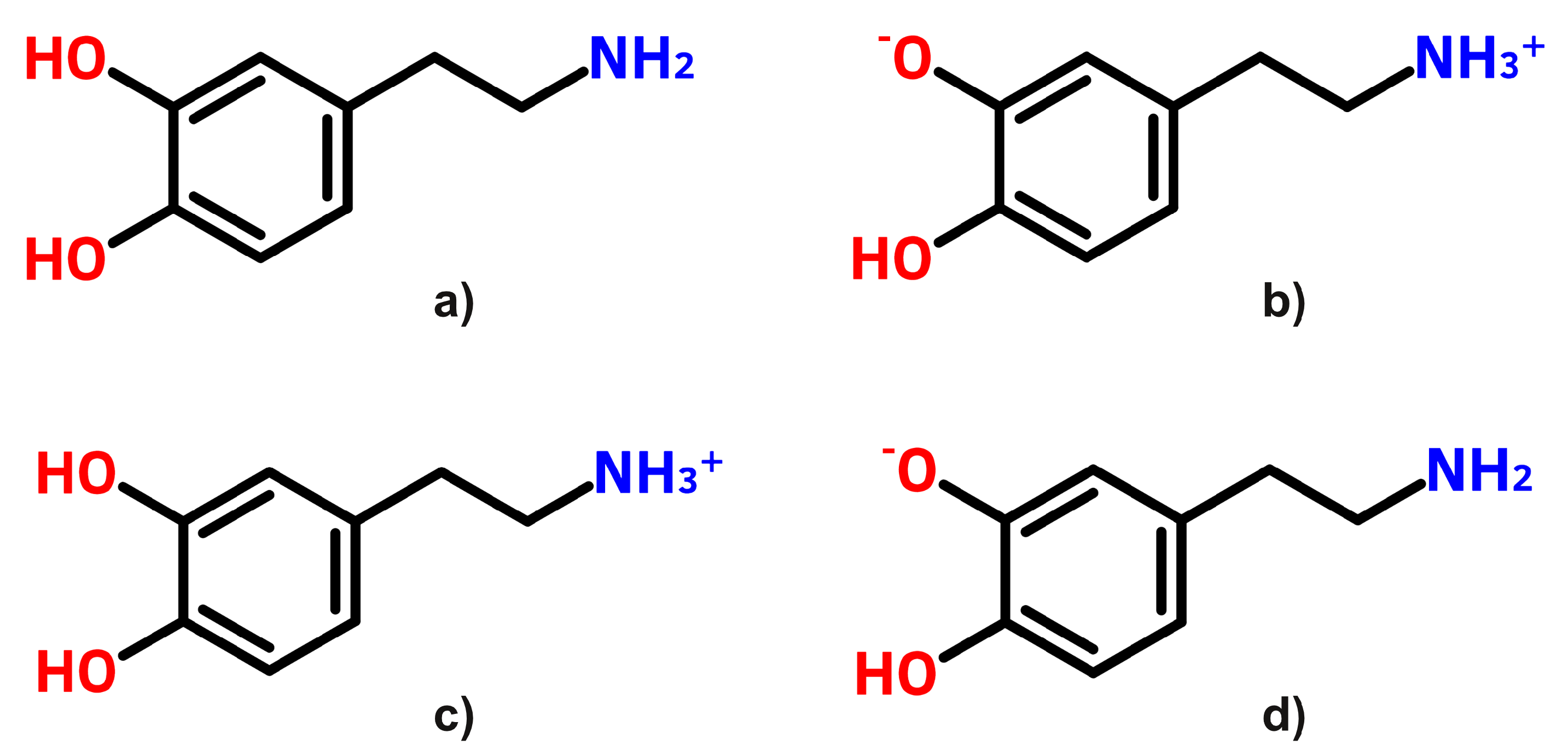
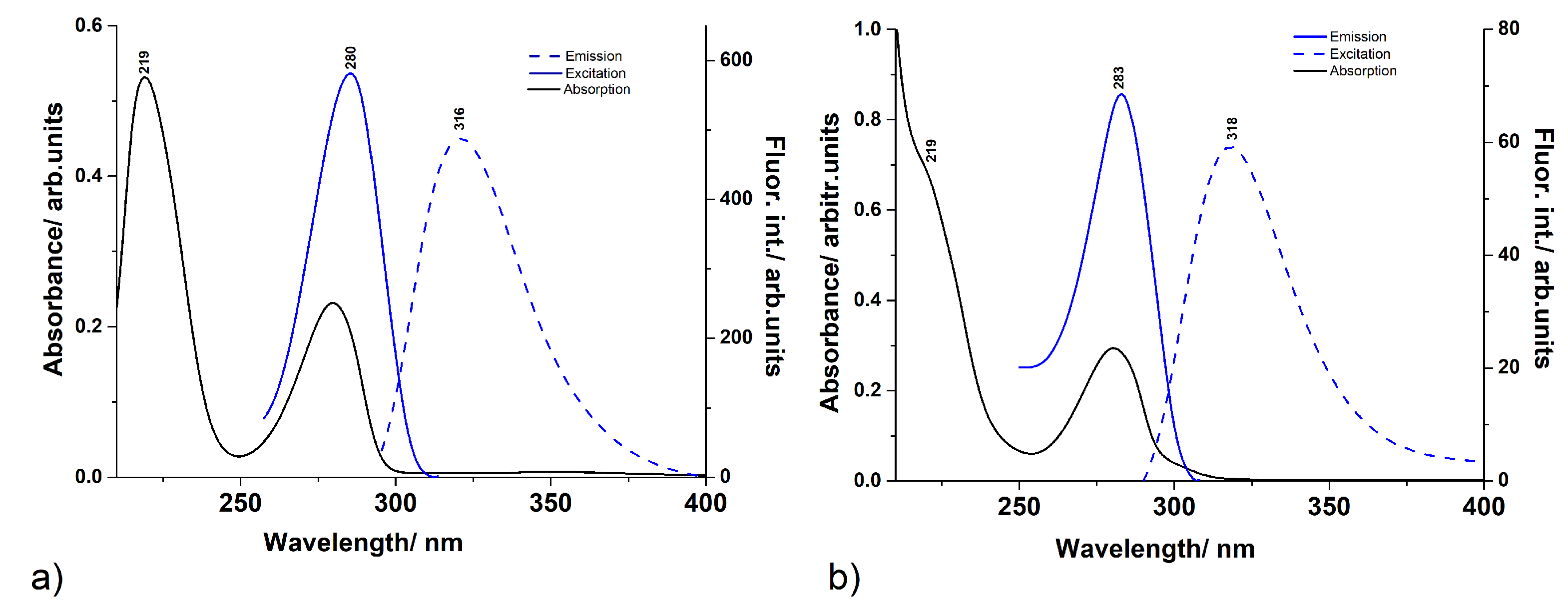
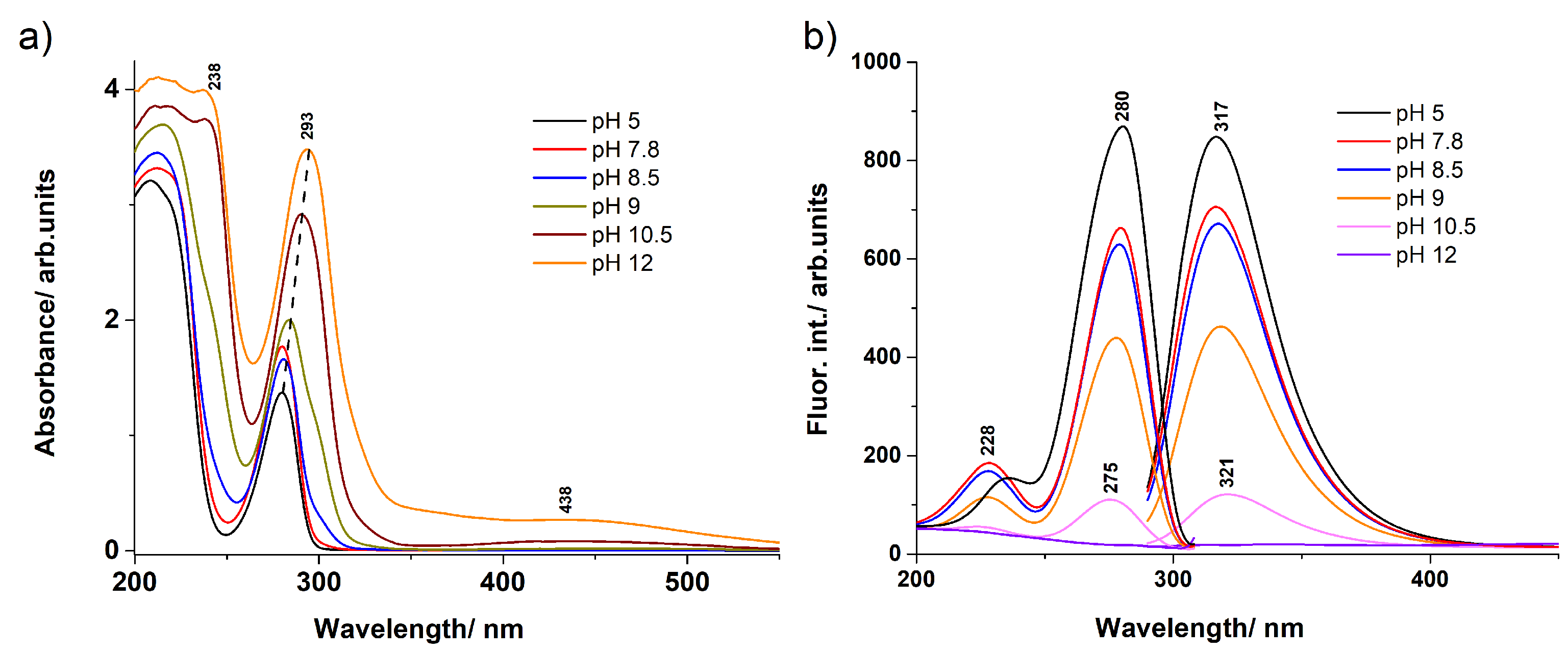
3.1. UV–Vis Absorption and Fluorescence Spectra
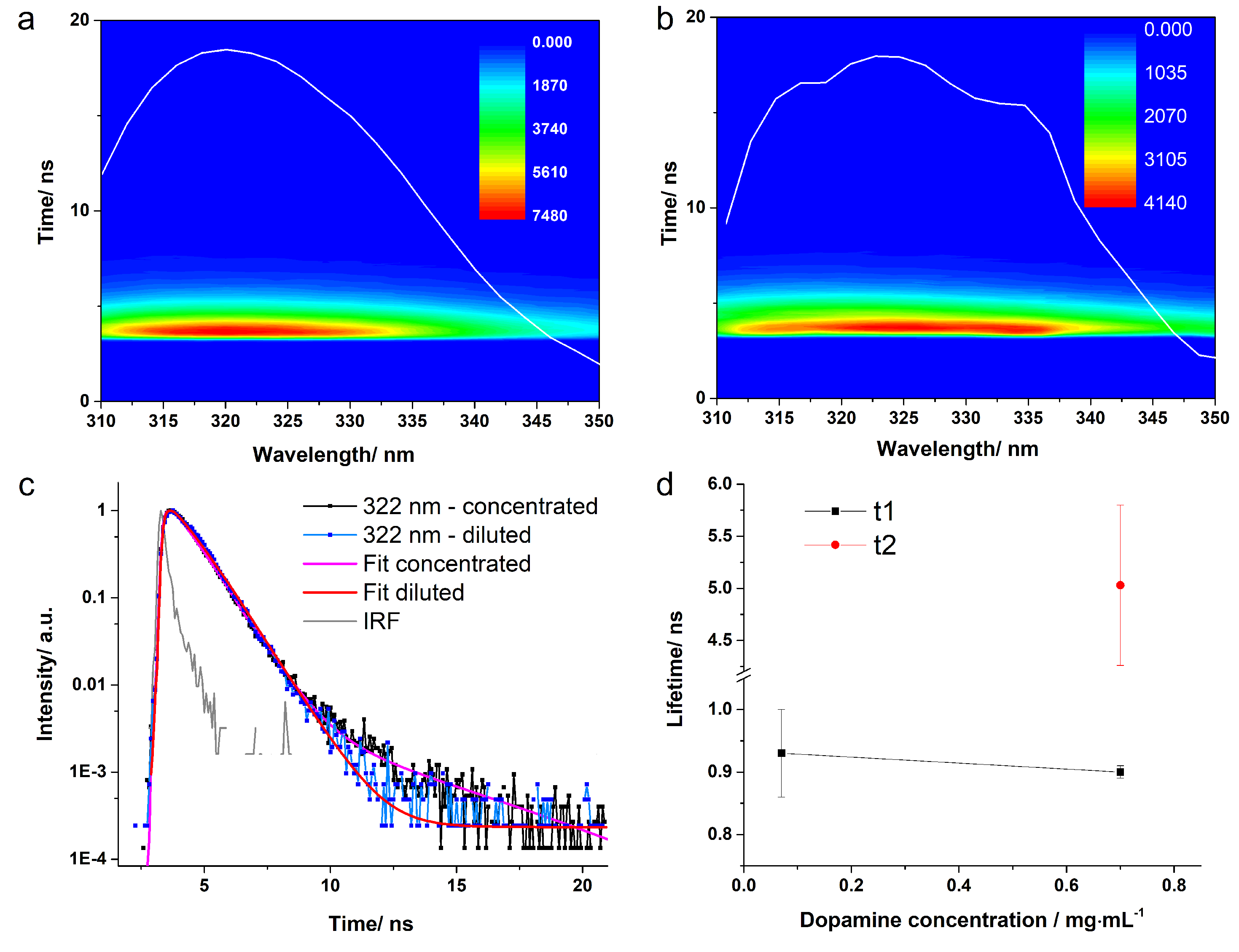
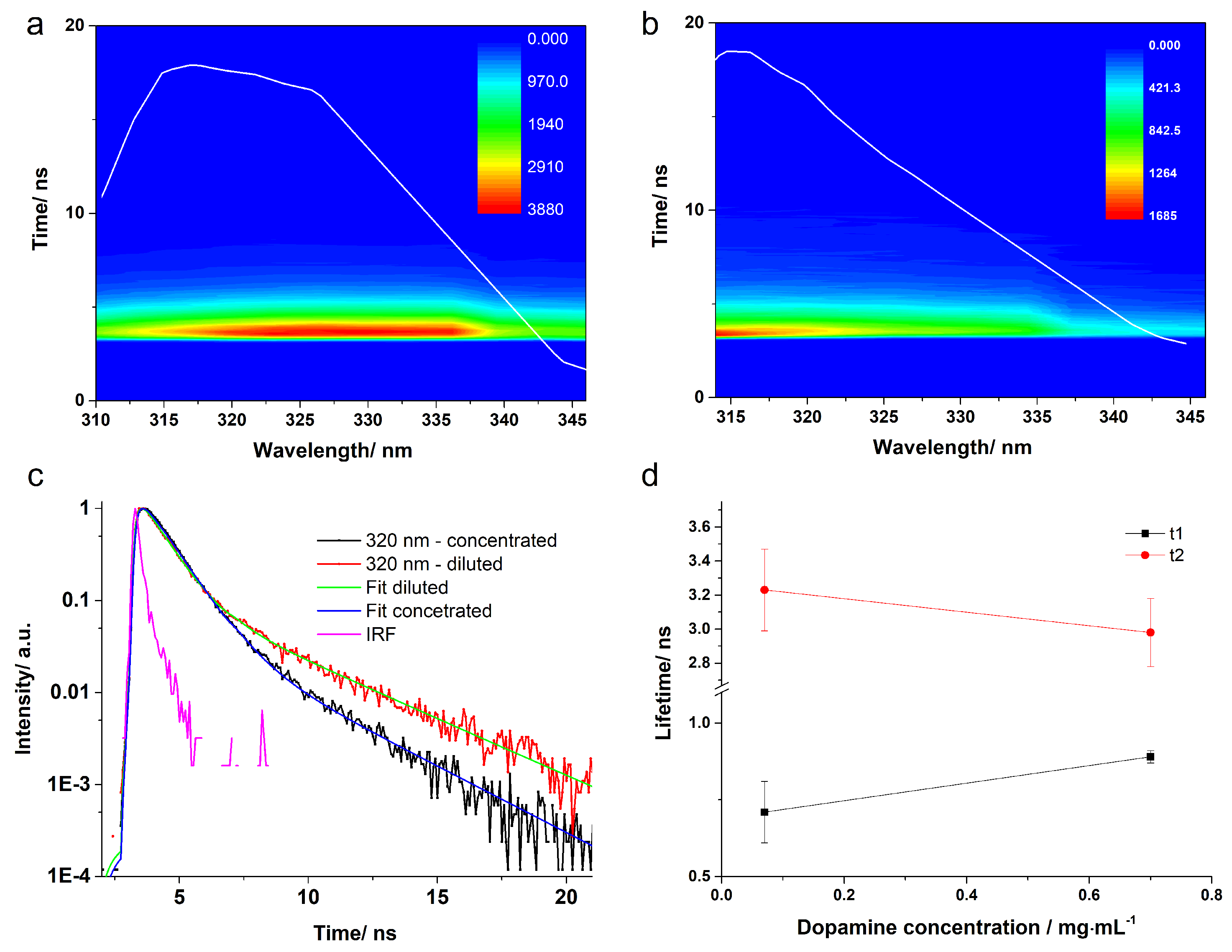
3.2. Fluorescence Lifetimes and Quantum Yield
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liebscher, J.; Mrówczynński, R.; Scheidt, H.A.; Filip, C.; Hădade, N.D.; Turcu, R.; Bende, A.; Beck, S. Structure of Polydopamine: A Never-Ending Story? Langmuir 2013, 29, 10539–10548. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, J. Chemistry of Polydopamine—Scope, Variation, and Limitation. Eur. J. Org. Chem. 2019, 31–32, 4976–4994. [Google Scholar] [CrossRef]
- Ball, V. Polydopamine Nanomaterials: Recent Advances in Synthesis Methods and Applications. Front. Bioeng. Biotechnol. 2018, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ai, K.; Lu, L. Polydopamine and Its Derivative Materials: Synthesis and Promising Applications in Energy, Environmental, and Biomedical Fields. Chem. Rev. 2014, 114, 5057–5115. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Park, E.; Lee, H. Polydopamine and Its Derivative Surface Chemistry in Material Science: A Focused Review for Studies at KAIST. Adv. Mater. 2020, 32, 1907505. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, J.; Liu, M.; Huang, H.; Zhang, X.; Wei, Y. Polydopamine-based functional materials and their applications in energy, environmental, and catalytic fields: State-of-the-art review. Chem. Eng. J. 2020, 387, 124019. [Google Scholar] [CrossRef]
- Yakhlifi, S.E.; Ball, V. Polydopamine as a stable and functional nanomaterial. Colloids Surf. B Biointerfaces 2020, 186, 110719. [Google Scholar] [CrossRef]
- Della Vecchia, N.F.; Avolio, R.; Alfè, M.; Errico, M.E.; Napolitano, A.; d’Ischia, M. Building-Block Diversity in Polydopamine Underpins a Multifunctional Eumelanin-Type Platform Tunable through a Quinone Control Point. Adv. Funct. Mater. 2013, 23, 1331–1340. [Google Scholar] [CrossRef]
- Cîrcu, M.; Filip, C. Closer to the polydopamine structure: New insights from a combined 13C/1H/2H solid-state NMR study on deuterated samples. Polym. Chem. 2018, 9, 3379–3387. [Google Scholar] [CrossRef]
- Mrówczyński, R.; Coy, L.E.; Scheibe, B.; Czechowski, T.; Augustyniak-Jabłokow, M.; Jurga, S.; Tadyszak, K. Electron Paramagnetic Resonance Imaging and Spectroscopy of Polydopamine Radicals. J. Phys. Chem. B 2015, 119, 10341–10347. [Google Scholar] [CrossRef]
- Ding, Y.; Weng, L.T.; Yang, M.; Yang, Z.; Lu, X.; Huang, N.; Leng, Y. Insights into the Aggregation/Deposition and Structure of a Polydopamine Film. Langmuir 2014, 30, 12258–12269. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Miller, D.J.; Freeman, B.D.; Paul, D.R.; Bielawski, C.W. Perspectives on poly(dopamine). Chem. Sci. 2013, 4, 3796–3802. [Google Scholar] [CrossRef]
- Chen, C.T.; Martin-Martinez, F.J.; Jung, G.S.; Buehler, M.J. Polydopamine and eumelanin molecular structures investigated with ab initio calculations. Chem. Sci. 2017, 8, 1631–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.C.; Ding, S.J. The pH-controlled nanoparticles size of polydopamine for anti-cancer drug delivery. J. Mater. Sci. Mater. Med. 2013, 24, 2381–2390. [Google Scholar] [CrossRef]
- Della Vecchia, N.F.; Luchini, A.; Napolitano, A.; D’Errico, G.; Vitiello, G.; Szekely, N.; d’Ischia, M.; Paduano, L. Tris Buffer Modulates Polydopamine Growth, Aggregation, and Paramagnetic Properties. Langmuir 2014, 30, 9811–9818. [Google Scholar] [CrossRef]
- Salomäki, M.; Marttila, L.; Kivelä, H.; Ouvinen, T.; Lukkari, J. Effects of pH and Oxidants on the First Steps of Polydopamine Formation: A Thermodynamic Approach. J. Phys. Chem. B 2018, 122, 6314–6327. [Google Scholar] [CrossRef] [PubMed]
- Salomäki, M.; Ouvinen, T.; Marttila, L.; Kivelä, H.; Leiro, J.; Mäkilä, E.; Lukkari, J. Polydopamine Nanoparticles Prepared Using Redox-Active Transition Metals. J. Phys. Chem. B 2019, 123, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Zhu, F.; Zhang, Z.; Cheng, Y.; Wang, Z.; Li, Y. Stimuli-responsive polydopamine-based smart materials. Chem. Soc. Rev. 2021, 50, 8319–8343. [Google Scholar] [CrossRef] [PubMed]
- Mostert, A.B.; Powell, B.J.; Pratt, F.L.; Hanson, G.R.; Sarna, T.; Gentle, I.R.; Meredith, P. Role of semiconductivity and ion transport in the electrical conduction of melanin. Proc. Natl. Acad. Sci. USA 2012, 109, 8943–8947. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Shawkey, M.D.; Dhinojwala, A. Bioinspired Melanin-Based Optically Active Materials. Adv. Opt. Mater. 2000, 8, 2000932. [Google Scholar] [CrossRef]
- Chen, X.; Yan, Y.; Mullner, M.; van Koeverden, M.P.; Noi, K.F.; Zhu, W.; Caruso, F. Engineering Fluorescent Poly(dopamine) Capsules. Langmuir 2014, 30, 2921–2925. [Google Scholar] [CrossRef] [PubMed]
- Son, E.J.; Kim, J.H.; Kim, K.; Park, C.B. Quinone and its derivatives for energy harvesting and storage materials. J. Mater. Chem. A 2016, 4, 11179–11202. [Google Scholar] [CrossRef]
- Dong, L.; Liu, X.; Xiong, Z.; Sheng, D.; Lin, C.; Zhou, Y.; Yang, Y. Fabrication of highly efficient ultraviolet absorbing PVDF membranes via surface polydopamine deposition. J. Appl. Polym. Sci. 2017, 134, 45746. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, S.; Chen, X.; Liu, X.; Wang, Z.; Li, Y. Recent developments in polydopamine fluorescent nanomaterials. Mater. Horiz. 2020, 7, 746–761. [Google Scholar] [CrossRef]
- Fedorenko, V.; Viter, R.; Mrówczyński, R.; Damberga, D.; Coy, E.; Iatsunskyi, I. Synthesis and photoluminescence properties of hybrid 1D core-shell structured nanocomposites based on ZnO/polydopamine. RSC Adv. 2020, 10, 29751–29758. [Google Scholar] [CrossRef]
- Zou, Y.; Chen, X.; Yang, P.; Liang, G.; Yang, Y.; Gu, Z.; Li, Y. Regulating the absorption spectrum of polydopamine. Sci. Adv. 2020, 6, eabb4696. [Google Scholar] [CrossRef]
- Aguilar-Ferrer, D.; Szewczyk, J.; Coy, E. Recent developments in polydopamine-based photocatalytic nanocomposites for energy production: Physico-chemical properties and perspectives. Catal. Today 2021, in press. [CrossRef]
- Mabuchi, M.; Shimada, J.; Okamoto, K.; Kawakami, Y.; Fujita, S.; Matsushige, K. Time-resolved fluorescence spectroscopy of dopamine in single cells. Proc. SPIE Adv. Fluoresc. Sens. Technol. V 2001, 4252, 140. [Google Scholar] [CrossRef]
- Wang, H.Y.; Sun, Y.; Tang, B. Study on fluorescence property of dopamine and determination of dopamine by fluorimetry. Talanta 2002, 57, 899–907. [Google Scholar] [CrossRef]
- Cruickshank, L.; Kennedy, A.R.; Shankland, N. Tautomeric and ionisation forms of dopamine and tyramine in the solid state. J. Mol. Struct. 2013, 1051, 132–136. [Google Scholar] [CrossRef]
- Nagy, P.I.; Takács-Novák, K. Tautomeric and conformational equilibria of biologically important (hydroxyphenyl)alkylamines in the gas phase and in aqueous solutiony. Phys. Chem. Chem. Phys. 2004, 6, 2838–2848. [Google Scholar] [CrossRef]
- Nagy, P.I.; Völgyi, G.; Takács-Novák, K. Tautomeric and conformational equilibria of tyramine and dopamine in aqueous solution. Mol. Phys. 2005, 103, 1589–1601. [Google Scholar] [CrossRef]
- Albarran, G.; Boggess, W.; Rassolov, V.; Schuler, R.H. Absorption Spectrum, Mass Spectrometric Properties, and Electronic Structure of 1,2-Benzoquinone. J. Phys. Chem. A 2010, 114, 7470–7478. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.S.; Li, G.D.; Mao, S.P.; Chai, J.D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theor. Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIRES Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIRES Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ’chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Nooijen, M.; Bartlett, R.J. Similarity transformed equation-of-motion coupled-cluster theory: Details, examples, and comparisons. J. Chem. Phys. 1997, 107, 6812–6830. [Google Scholar] [CrossRef]
- Nooijen, M.; Bartlett, R.J. A new method for excited states: Similarity transformed equation-of-motion coupled-cluster theory. J. Chem. Phys. 1997, 106, 6441–6448. [Google Scholar] [CrossRef]
- Berraud-Pache, R.; Neese, F.; Bistoni, G.; Izsák, R. Unveiling the Photophysical Properties of Boron-dipyrromethene Dyes Using a New Accurate Excited State Coupled Cluster Method. J. Chem. Theory Comput. 2020, 16, 564–575. [Google Scholar] [CrossRef]
- Sirohiwal, A.; Berraud-Pache, R.; Neese, F.; Izsák, R.; Pantazis, D.A. Accurate Computation of the Absorption Spectrum of Chlorophyll a with Pair Natural Orbital Coupled Cluster Methods. J. Phys. Chem. B 2020, 124, 8761–8771. [Google Scholar] [CrossRef]
- de Souza, B.; Neese, F.; Izsák, R. On the theoretical prediction of fluorescence rates from first principles using the path integral approach. J. Chem. Phys. 2018, 148, 034104. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Lewandowska-Andralojc, A.; Marciniak, B. Five Major Sins in Fluorescence Spectroscopy of Light-Harvesting Hybrid Materials. ACS Energy Lett. 2019, 4, 1898–1901. [Google Scholar] [CrossRef] [Green Version]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Rivera, A.E.; Corona-Avendaño, S.; Alarcón-Angeles, G.; Rojas-Hernández, A.; Ramírez-Silva, M.T.; Romero-Romo, M.A. Spectrophotometric study on the stability of dopamine and the determination of its acidity constants. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2003, 59, 3193–3203. [Google Scholar] [CrossRef]
- Corona-Avendaño, S.; Alarcón-Angeles, G.; Rosquete-Pina, G.A.; Rojas-Hernández, A.; Gutierrez, A.; Ramírez-Silva, M.T.; Romero-Romo, M.; Palomar-Pardavé, M. New Insights on the Nature of the Chemical Species Involved during the Process of Dopamine Deprotonation in Aqueous Solution: Theoretical and Experimental Study. J. Phys. Chem. B 2007, 111, 1640–1647. [Google Scholar] [CrossRef]
- Hirata, K.; Kasai, K.i.; Grégoire, G.; Ishiuchi, S.i.; Fujii, M. Hydration-controlled excited-state relaxation in protonated dopamine studied by cryogenic ion spectroscopy. J. Chem. Phys. 2021, 155, 151101. [Google Scholar] [CrossRef]
- Toure, O.; Dussap, C.G.; Lebert, A. Comparison of Predicted pKa Values for Some Amino-Acids, Dipeptides and Tripeptides, Using COSMO-RS, ChemAxon and ACD/Labs Methods. Oil & Gas Sci. Technol. Rev. IFP Energies Nouv. 2013, 68, 281–297. [Google Scholar] [CrossRef]
- Marvin 22.9 + Calculator Plugins. ChemAxon. Available online: http://www.chemaxon.com (accessed on 2 March 2022).
- Wang, J.; Durbeej, B. How accurate are TD-DFT excited-state geometries compared to DFT ground-state geometries? J. Comput. Chem. 2020, 41, 1718–1729. [Google Scholar] [CrossRef]
- Peng, Q.; Yi, Y.; Shuai, Z.; Shao, J. Excited state radiationless decay process with Duschinsky rotation effect: Formalism and implementation. J. Chem. Phys. 2007, 126, 114302. [Google Scholar] [CrossRef]
- Valiev, R.R.; Cherepanov, V.; Nasibullin, R.; Sundholm, D.; Kurtén, T. Calculating rate constants for intersystem crossing and internal conversion in the Franck-Condon and Herzberg-Teller approximations. Phys. Chem. Chem. Phys. 2019, 21, 18495–18500. [Google Scholar] [CrossRef]
- Huang, K.; Rhys, A.; Mott, N.F. Theory of light absorption and non-radiative transitions in F-centres. Proc. R. Soc. A: Math. Phys. Eng. Sci. 1950, 204, 406–423. [Google Scholar] [CrossRef]
- Rätsep, M.; Linnanto, J.M.; Muru, R.; Biczysko, M.; Reimers, J.R.; Freiberg, A. Absorption-emission symmetry breaking and the different origins of vibrational structures of the 1Qy and 1Qx electronic transitions of pheophytin. J. Chem. Phys. 2019, 151, 165102. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Geometries | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | DA | DAH | DAH | ||||||||
| S | 266 | 0.0275 | 292 | 0.0014 | 270 | 0.0379 | 302 | 0.0009 | |||
| S | 226 | 0.0024 | 286 | 0.0803 | 222 | 0.0011 | 287 | 0.0858 | |||
| S | 208 | 0.0637 | 249 | 0.0008 | 209 | 0.0378 | 255 | 0.0033 | |||
| S | 242 | 0.0033 | 250 | 0.0164 | |||||||
| S | 239 | 0.0088 | 230 | 0.0947 | |||||||
| S | 224 | 0.0108 | 226 | 0.1094 | |||||||
| S | 216 | 0.0119 | 219 | 0.0050 | |||||||
| S | 214 | 0.0034 | 216 | 0.0018 | |||||||
| S | 211 | 0.0045 | 216 | 0.0107 | |||||||
| S | 209 | 0.0351 | 211 | 0.0002 | |||||||
| pH | Solution | Reconv. Method | (ns) | Error | Ampl. (%) | (ns) | Error | Ampl. (%) | (ns) | Error |
|---|---|---|---|---|---|---|---|---|---|---|
| 5.5 | Concentrated | 1-exp | 0.89 | 0.01 | 100 | - | - | - | - | - |
| 2-exp | 0.91 | 0.02 | 99.58 | 4.86 | 0.38 | 0.42 | 0.99 | 0.03 | ||
| Diluted | 1-exp | 0.93 | 0.07 | 100 | - | - | - | - | - | |
| 8.0 | Concentrated | 2-exp | 0.89 | 0.01 | 95.83 | 2.98 | 0.20 | 4.17 | 0.97 | 0.05 |
| Diluted | 2-exp | 0.71 | 0.10 | 88.97 | 3.23 | 0.24 | 11.03 | 1.56 | 0.15 | |
| 9.0 | Concentrated | 2-exp | 0.69 | 0.02 | 92.61 | 2.41 | 0.20 | 7.40 | 0.82 | 0.01 |
| Diluted | 2-exp | 0.64 | 0.10 | 88.20 | 2.96 | 0.18 | 11.80 | 0.91 | 0.11 |
| DA | DA | DAH | DAH | |
|---|---|---|---|---|
| FC | 4.39 × 10 | 8.47 × 10 | 4.70 × 10 | 1.36 × 10 |
| (100%) | (100%) | (100%) | (100%) | |
| FC+HT | 3.79 × 10 | 2.75 × 10 | 4.51 × 10 | 1.14 × 10 |
| (86%, 14%) | (0%, 100%) | (96%, 4%) | (1%, 99%) | |
| FC+(HT+D ) | 3.16 × 10 | 1.02 × 10 | 2.95 × 10 | 7.10 × 10 |
| (81%, 19%) | (0%, 100%) | (78%, 22%) | (0%, 100%) |
| pH | Solution | QY (%) |
|---|---|---|
| 5.5 | Concentrated | 3.4 |
| Diluted | 3.4 | |
| 8.0 | Concentrated | 2.0 |
| Diluted | 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falamaş, A.; Petran, A.; Hada, A.-M.; Bende, A. Dopamine Photochemical Behaviour under UV Irradiation. Int. J. Mol. Sci. 2022, 23, 5483. https://doi.org/10.3390/ijms23105483
Falamaş A, Petran A, Hada A-M, Bende A. Dopamine Photochemical Behaviour under UV Irradiation. International Journal of Molecular Sciences. 2022; 23(10):5483. https://doi.org/10.3390/ijms23105483
Chicago/Turabian StyleFalamaş, Alexandra, Anca Petran, Alexandru-Milentie Hada, and Attila Bende. 2022. "Dopamine Photochemical Behaviour under UV Irradiation" International Journal of Molecular Sciences 23, no. 10: 5483. https://doi.org/10.3390/ijms23105483
APA StyleFalamaş, A., Petran, A., Hada, A.-M., & Bende, A. (2022). Dopamine Photochemical Behaviour under UV Irradiation. International Journal of Molecular Sciences, 23(10), 5483. https://doi.org/10.3390/ijms23105483