
Antitumoral Activity of a CDK9 PROTAC Compound in HER2-Positive Breast Cancer

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
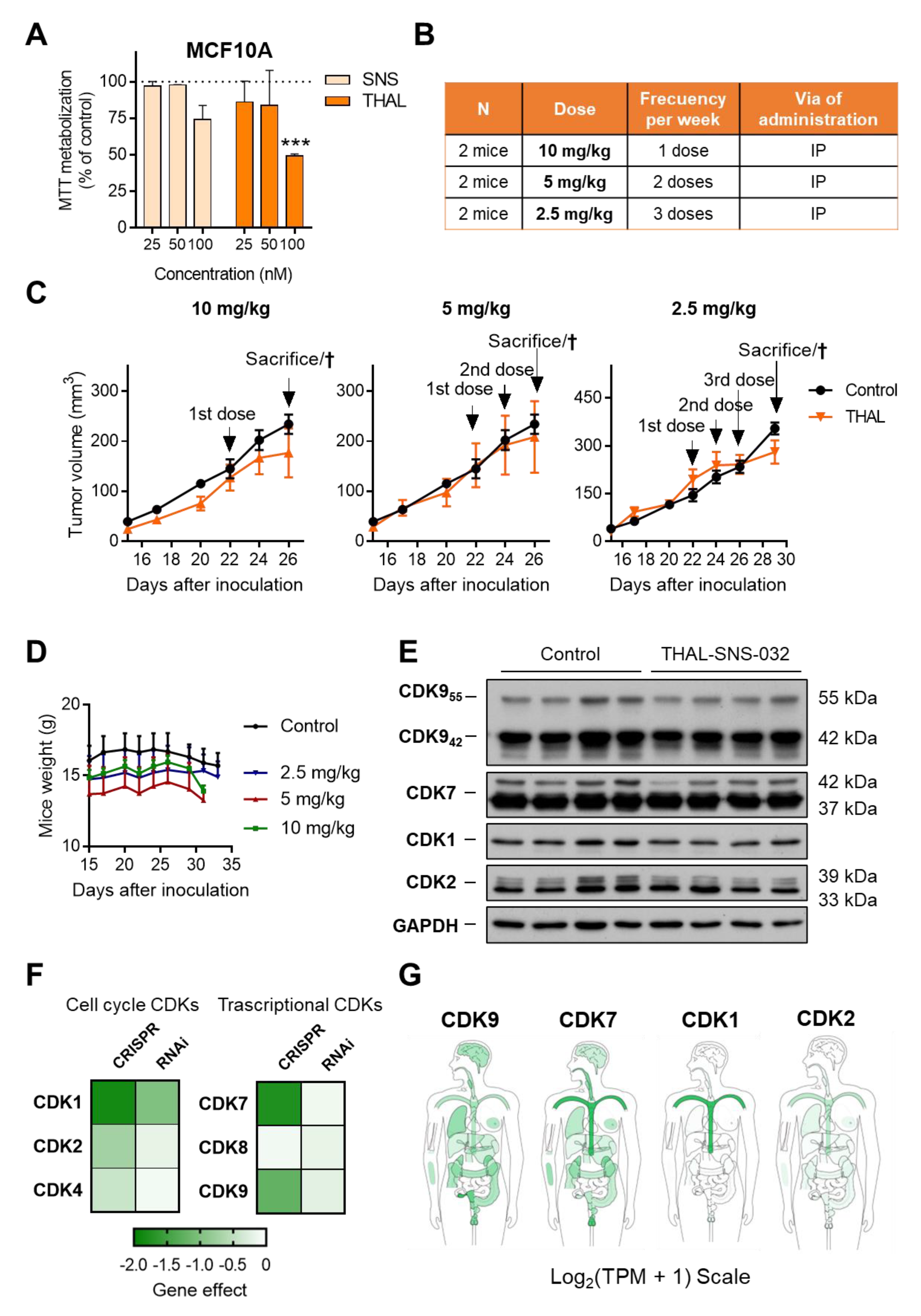
2.1. In Silico Analysis of CDKs in Breast Cancer
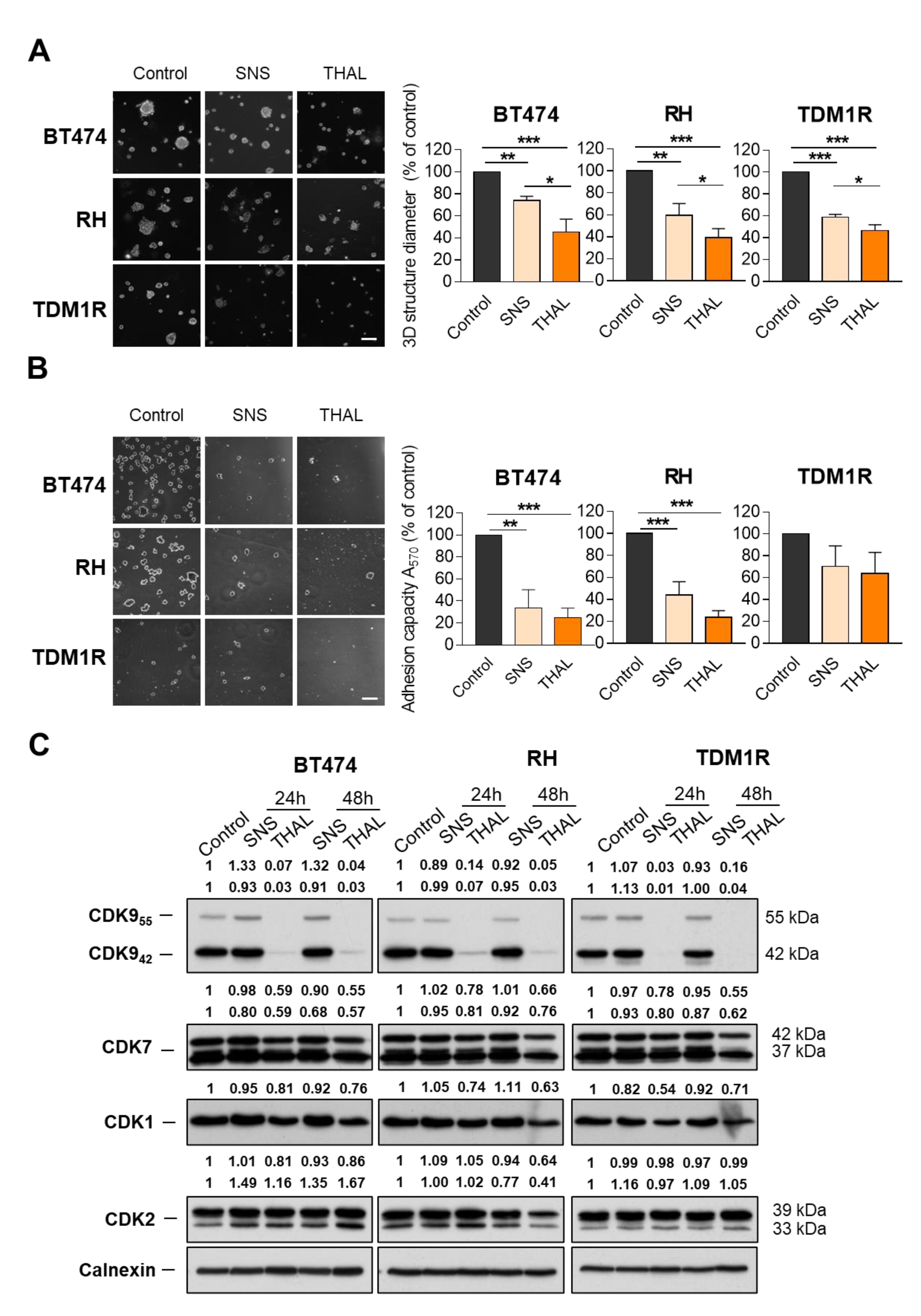
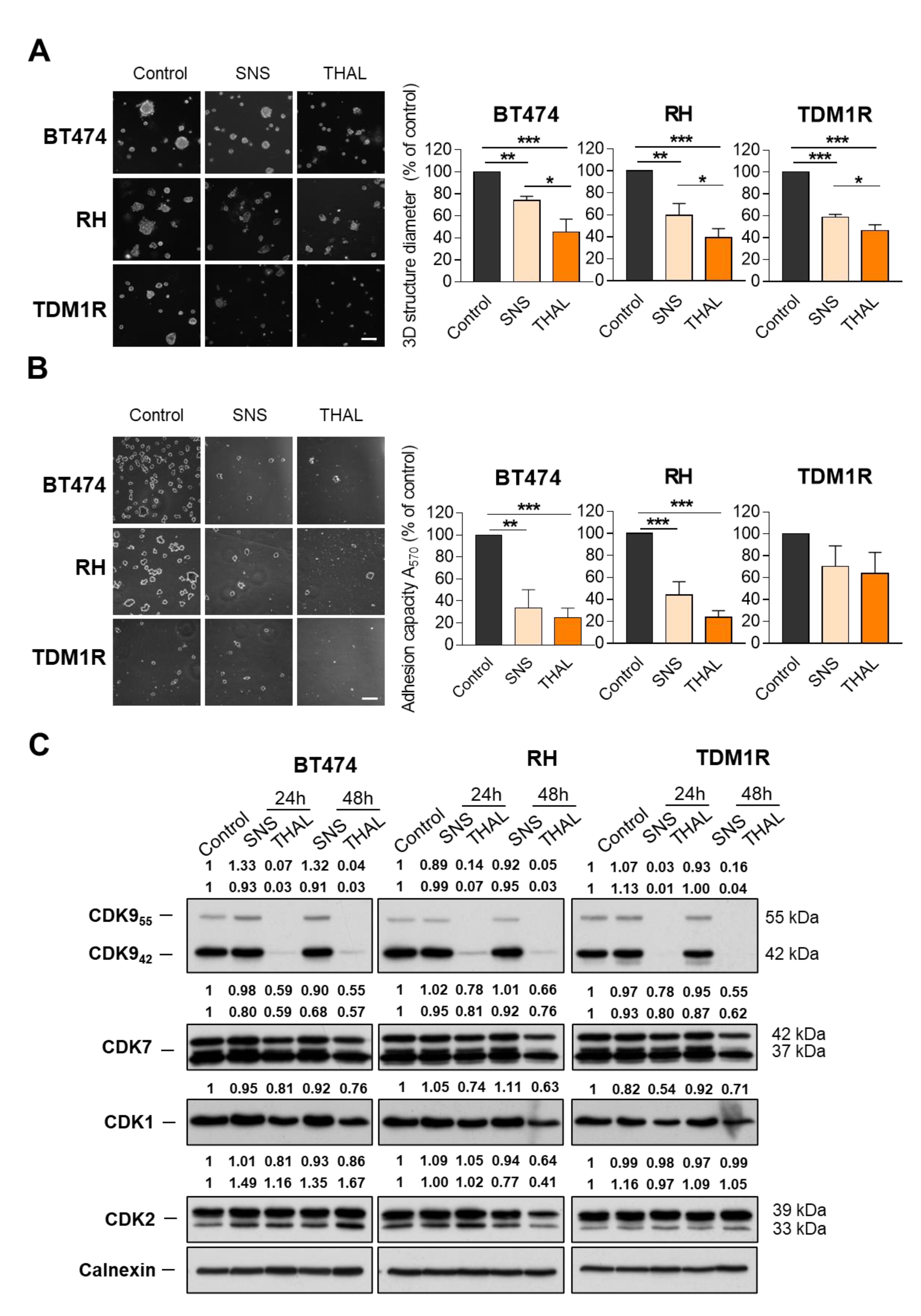
2.2. THAL-SNS-032 Is Active in HER2-Positive Cell Lines
2.3. THAL-SNS-032 Is More Potent Than the Parental CDK9 Kinase Inhibitor
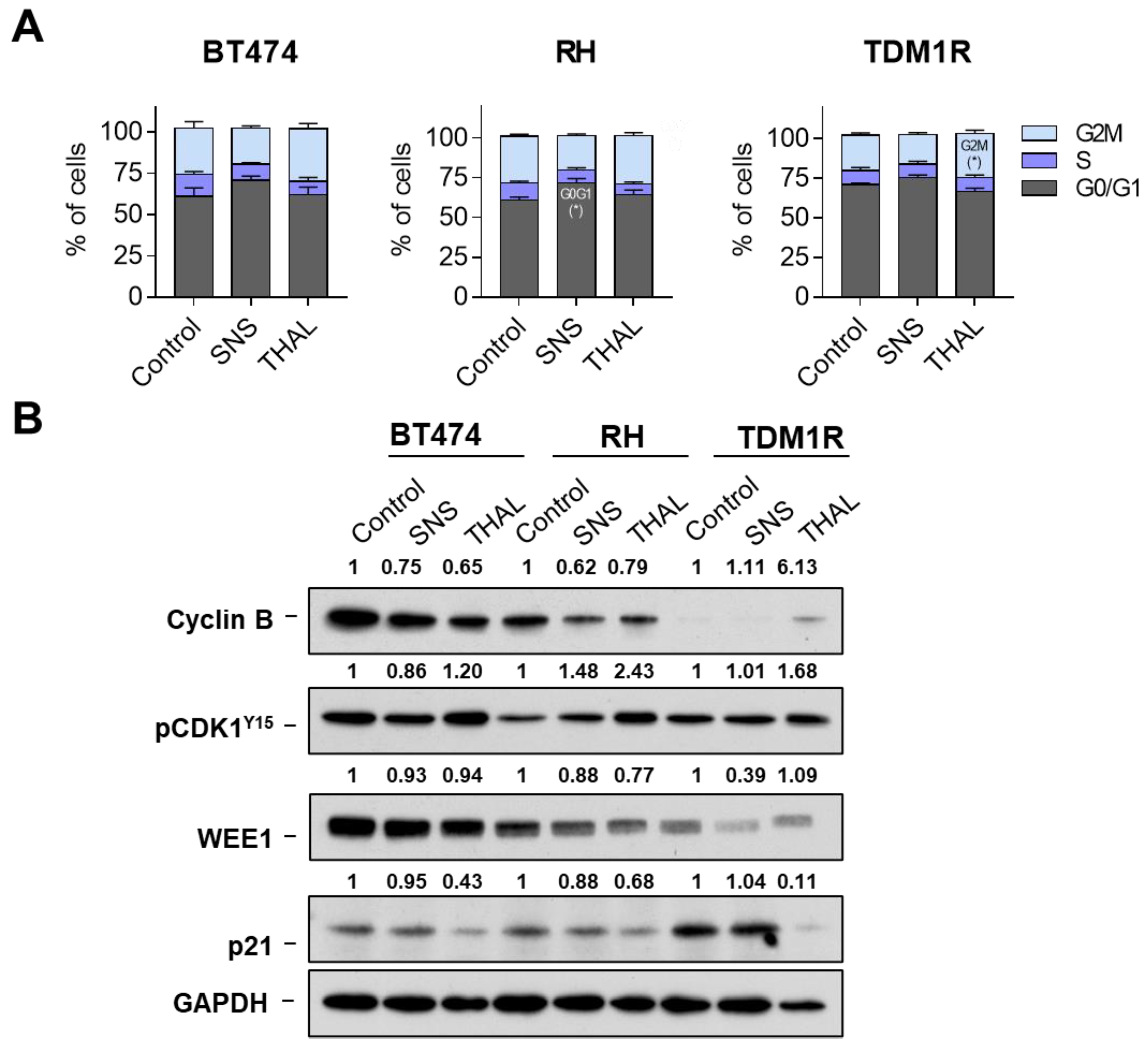
2.4. Effect of THAL-SNS-032 on the Cell Cycle
2.5. THAL-SNS-032 Induces Cell Death
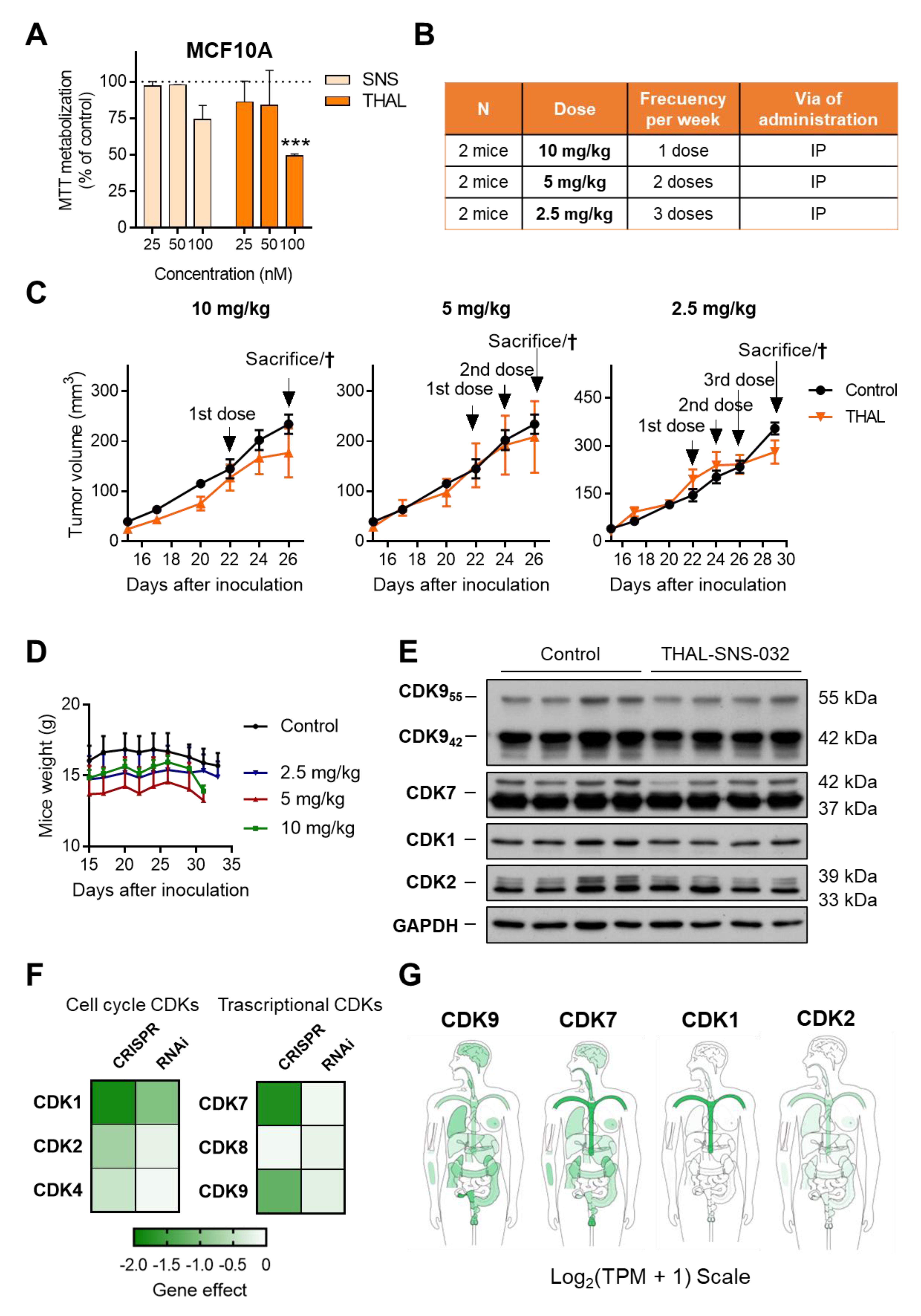
2.6. THAL-SNS-032 Has an Inverse Therapeutic Index
3. Discussion
4. Materials and Methods
4.1. Clinical Outcome Evaluation
4.2. Cell Culture and Drugs
4.3. Proliferation Assay (MTT) and EC50 Value
4.4. Matrigel-3D Tumor Sphere-Forming Assay
4.5. Fibronectin-Adhesion Assay
4.6. Protein Expression Analysis: Western Blot
4.7. Flow Cytometry Experiments
4.8. Xenograft Mice (In Vivo)
4.9. Dependency Study and Cell Line Expression Analyses
4.10. Small Interfering RNA CDK9
4.11. Quantitative Reverse-Transcription PCR
4.12. CDKs Expression in Normal Tissues
4.13. Caspase 3 Activity
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.; Endicott, J.A. Structural insights into the functional diversity of the CDK–cyclin family. Open Biol. 2018, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Senderowicz, A.M. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Investig. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, C.; Sun, X.; Shi, X.; Jin, B.; Ding, K.; Yeung, S.C.J.; Pan, J. Cyclin-dependent kinase 7/9 inhibitor SNS-032 abrogates FIP1-like-1 platelet-derived growth factor receptor α and Bcr-Abl oncogene addiction in malignant hematologic cells. Clin. Cancer Res. 2012, 18, 1966–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, J.J.; Von Karstedt, S.; El Hay, M.A.; Conti, A.M.F.; Arce, F.T.; Montinaro, A.; Papenfuss, K.; El-Bahrawy, M.A.; Walczak, H. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014, 21, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; Kim, W.; Jo, H.R.; Shin, Y.J.; Kim, M.H.; Jeong, J.H. Anticancer and radiosensitizing effects of the cyclin-dependent kinase inhibitors, AT7519 and SNS-032, on cervical cancer. Int. J. Oncol. 2018, 53, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Settleman, J.; Neto, J.M.F.; Bernards, R. Thinking Differently about Cancer Treatment Regimens. Cancer Discov. 2021, 11, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, A.; Pandiella, A. Proteolysis targeting chimeras (PROTACs) in cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Baliu-Piqué, M.; Pandiella, A.; Ocaña, A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers 2020, 12, 3271. [Google Scholar] [CrossRef]
- Noblejas-López, M.D.M.; Nieto-Jiménez, C.; Galán-Moya, E.M.; Tebar-García, D.; Montero, J.C.; Pandiella, A.; Burgos, M.; Ocaña, A. MZ1 co-operates with trastuzumab in HER2 positive breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–12. [Google Scholar] [CrossRef]
- Gandullo-Sánchez, L.; Capone, E.; Ocaña, A.; Iacobelli, S.; Sala, G.; Pandiella, A. HER3 targeting with an antibody-drug conjugate bypasses resistance to anti-HER2 therapies. EMBO Mol. Med. 2020, 12, e11498. [Google Scholar] [CrossRef]
- Mallareddy, J.R.; Singh, S.; Boghean, L.; Natarajan, A. Selective CDK9 degradation using a proteolysis-targeting chimera (PROTAC) strategy. Future Med. Chem. 2022, 14, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Lei, W.; Ma, Y.; Wang, Y.; Wei, B.; Chen, X.; Ru, G.; He, X.; Mou, X.; Wang, S. Synergistic antitumor effects of CDK inhibitor SNS-032 and an oncolytic adenovirus co-expressing TRAIL and Smac in pancreatic cancer. Mol. Med. Rep. 2017, 15, 3521–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.B.; Burris, H.A.; et al. Trastuzumab emtansine with or without pertuzumab versus trastuzumab with taxane for human epidermal growth factor receptor 2–positive advanced breast cancer: Final results from MARIANNE. Cancer 2019, 125, 3974–3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Chan, A.; Moy, B.; Mansi, J.; Ejlertsen, B.; Holmes, F.A.; Chia, S.; Iwata, H.; Gnant, M.; Loibl, S.; Barrios, C.H.; et al. Final Efficacy Results of Neratinib in HER2-positive Hormone Receptor-positive Early-stage Breast Cancer from the Phase III ExteNET Trial. Clin. Breast Cancer 2021, 21, 80–91.e7. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Anshabo, A.T.; Milne, R.; Wang, S.; Albrecht, H. CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front. Oncol. 2021, 11, 1573. [Google Scholar] [CrossRef]
- Xie, G.; Tang, H.; Wu, S.; Chen, J.; Liu, J.; Liao, C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int. J. Oncol. 2014, 45, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P.; Pereira, L.A.; Drabsch, Y.; Ramsay, R.G.; Gonda, T.J. Estrogen receptor-α recruits P-TEFb to overcome transcriptional pausing in intron 1 of the MYB gene. Nucleic Acids Res. 2012, 40, 5988–6000. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P.; Yang, R.-M.; Sutton, J.; Ramsay, R.G.; Gonda, T. CDK9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget 2016, 7, 9069–9083. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Li, Y.; Yu, B.; Ren, J.; Huang, H.; Wang, M.; Ding, H.; Li, Z.; Wang, J.; Bian, J. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur. J. Med. Chem. 2021, 211, 113091. [Google Scholar] [CrossRef] [PubMed]
- Cimas, F.J.; Niza, E.; Juan, A.; Noblejas-López, M.D.M.; Bravo, I.; Lara-Sanchez, A.; Alonso-Moreno, C.; Ocaña, A. Controlled delivery of bet-protacs: In vitro evaluation of MZ1-loaded polymeric antibody conjugated nanoparticles in breast cancer. Pharmaceutics 2020, 12, 986. [Google Scholar] [CrossRef]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Luci, C.; Díaz-Rodríguez, E.; Gandullo-Sánchez, L.; Díaz-Gil, L.; Ocaña, A.; Pandiella, A. Adaptive resistance to trastuzumab impairs response to neratinib and lapatinib through deregulation of cell death mechanisms. Cancer Lett. 2020, 470, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Luci, C.R.; García-Alonso, S.; Díaz-Rodriguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocaña, A.; Pandiella, A. Resistance to the antibody–drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 2017, 77, 4639–4651. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Rodriguez, E.; Pérez-Peña, J.; Ríos-Luci, C.; Arribas, J.; Ocaña, A.; Pandiella, A. TRAIL receptor activation overcomes resistance to trastuzumab in HER2 positive breast cancer cells. Cancer Lett. 2019, 453, 34–44. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noblejas-López, M.d.M.; Gandullo-Sánchez, L.; Galán-Moya, E.M.; López-Rosa, R.; Tébar-García, D.; Nieto-Jiménez, C.; Gómez-Juárez, M.; Burgos, M.; Pandiella, A.; Ocaña, A. Antitumoral Activity of a CDK9 PROTAC Compound in HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2022, 23, 5476. https://doi.org/10.3390/ijms23105476
Noblejas-López MdM, Gandullo-Sánchez L, Galán-Moya EM, López-Rosa R, Tébar-García D, Nieto-Jiménez C, Gómez-Juárez M, Burgos M, Pandiella A, Ocaña A. Antitumoral Activity of a CDK9 PROTAC Compound in HER2-Positive Breast Cancer. International Journal of Molecular Sciences. 2022; 23(10):5476. https://doi.org/10.3390/ijms23105476
Chicago/Turabian StyleNoblejas-López, María del Mar, Lucía Gandullo-Sánchez, Eva M. Galán-Moya, Raquel López-Rosa, David Tébar-García, Cristina Nieto-Jiménez, Mónica Gómez-Juárez, Miguel Burgos, Atanasio Pandiella, and Alberto Ocaña. 2022. "Antitumoral Activity of a CDK9 PROTAC Compound in HER2-Positive Breast Cancer" International Journal of Molecular Sciences 23, no. 10: 5476. https://doi.org/10.3390/ijms23105476
APA StyleNoblejas-López, M. d. M., Gandullo-Sánchez, L., Galán-Moya, E. M., López-Rosa, R., Tébar-García, D., Nieto-Jiménez, C., Gómez-Juárez, M., Burgos, M., Pandiella, A., & Ocaña, A. (2022). Antitumoral Activity of a CDK9 PROTAC Compound in HER2-Positive Breast Cancer. International Journal of Molecular Sciences, 23(10), 5476. https://doi.org/10.3390/ijms23105476