
Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction


Abstract
:1. Introduction
2. Molecular Epidemiology
3. Molecular Classification
4. Molecular Diagnostics
5. Immunological Characteristics—The Tumor Immune Microenvironment
6. The Molecular Background of Melanoma Progression
7. Prognostic Markers: Gene Expression Pattern
8. Prognostic Markers: The Tumor Immune Microenvironment
9. Predictive Markers of Melanoma
10. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Tímár, J.; Vízkeleti, L.; Doma, V.; Barbai, T.; Rásó, E. Genetic progression of malignant melanoma. Cancer Metastasis Rev. 2016, 35, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; MacGregor, S.; Hayward, N.K. Melanoma genetics: Recent findings take us beyond well-travelled pathways. J. Investig. Dermatol. 2012, 132, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, A.J.; Hughes, M.C.; Kvaskoff, M.; Siskind, V.; Shekar, V.S.; Aitken, J.F.; Green, A.C.; Duffy, D.L.; Hayward, N.K.; Martin, N.G.; et al. The Queensland Study of Melanoma: Environmental and genetic associations (Q-MEGA): Study design, baseline characteristics and repeatability of phenotypes and sun exposure measures. Twin Res. Hum. Genet. 2008, 11, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos, C.I.; Wang, L.E.; Lee, J.E.; Gershenwald, J.E.; Chen, W.V.; Fang, S.; Kosoy, R.; Zhang, M.; Qureshi, A.A.; Vattathil, S.; et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 2011, 20, 5012–5023. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.H.; Iles, M.M.; Harland, M.; Taylor, J.C.; Aitken, J.F.; Andresen, P.A.; Akslen, L.A.; Armstrong, B.K.; Avril, M.F.; Azizi, E.; et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 2011, 43, 1108–1113. [Google Scholar] [CrossRef]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.S.; Stern, M.H.; Carvajal, R.D.; Bedfort, R.N.; Jia, R.; Shields, G.S.; et al. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 24. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Doma, V.; Barbai, T.; Beleaua, M.A.; Kovalszky, I.; Rásó, E.; Tímár, J. KIT mutation incidence and pattern of melanoma in central-east Europe. Pathol. Oncol. Res. 2020, 26, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [Green Version]
- Rabbie, R.; Ferguson, P.; Molina-Aguilar, C.; Adams, D.J. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019, 247, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Tímár, J.; Barbai, T.; Győrffy, B.; Rásó, E. Chapter 2. Understanding Melanoma Progression by Gene Expression Signatures. In Cancer Genomics; Pfeffer, U., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 47–79. [Google Scholar]
- Ordonez, N.G. Value of melanocytic-associated immunohistochemical markers in the diagnosis of malignant melanoma. Hum. Pathol. 2014, 45, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Deacon, D.C.; Smith, E.A.; Judson-Torres, R.L. Molecular biomarkers for melanoma screening, diagnosis and prognosis: Current state and future directions. Front. Med. 2021, 8, 642380. [Google Scholar] [CrossRef] [PubMed]
- Uguen, A.; Talagas, M.; Costa, S.; Duigou, S.; Bouvier, S.; De Braekeleer, M.; Marcorelles, P. A p16-ki-67-HMB45 immunohistochemistry score system as an ancillary diagnostic tool in the diagnosis of melanoma. Diagn. Pathol. 2015, 10, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, J.D.R.; Salim, S.; Velazquez, E.F.; Wang, L.; Williams, K.M.; Flejter, W.L.; Brooke, L.; Sunder, S.; Busam, K.J. Comparison of melanoma gene expression score with histopathology, FISH and SNP array for the classification of melanocytic neoplasms. Mod. Pathol. 2018, 31, 1733–1743. [Google Scholar] [CrossRef]
- Clarke, L.E.; Flake, D.D.; Busam, K.; Cockerell, C.; Helm, K.; McNiff, J.; Reed, J.; Tschen, J.; Kim, J.; Barnhill, K.; et al. An independent validation of a gene expression signature to differentiate malignant melanoma from benign melanocytic nevi. Cancer 2017, 123, 617–628. [Google Scholar] [CrossRef]
- Aivazian, K.; Ahmed, T.; El Sharouni, M.A.; Stretch, J.R.; Saw, R.P.M.; Spillane, A.J.; Shannon, K.F.; Ch’ng, S.; Nieweg, O.E.; Thompson, J.F.; et al. Histological regression in melanoma: Impact on sentinel lymph node status and survival. Mod. Pathol. 2021, 34, 1999–2006. [Google Scholar] [CrossRef]
- Ladányi, A.; Tímár, J. Immunologic and immunogenomic aspects of tumor progression. Semin. Cancer Biol. 2020, 60, 249–261. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef]
- Addeo, A.; Friedlander, A.; Banna, G.L.; Weiss, G.J. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol. Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas. J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelands, J.; Hendrickx, W.; Zoppoli, G.; Mall, R.; Saad, M.; Halliwill, K.; Curigliano, G.; Rinchai, D.; Decock, J.; Delogu, L.G.; et al. Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J. Immunother. Cancer 2020, 8, e000617. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Amon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Doma, V.; Kárpáti, S.; Rásó, E.; Barbai, T.; Tímár, J. Dynamic and unpredictable changes in mutant allele fractions of BRAF and NRAS during visceral progression of cutaneous malignant melanoma. BMC Cancer 2019, 19, 786. [Google Scholar]
- Papp, O.; Doma, V.; Gil, J.; Markó-Varga, G.; Kárpáti, S.; Tímár, J.; Vízkeleti, L. Organ specific copy number variations in visceral metastases of human melanoma. Cancers 2021, 13, 5984. [Google Scholar] [CrossRef]
- Alkaraki, A.; McArthur, G.A.; Sheppard, K.E.; Smith, L.K. Metabolic plasticity in melanoma progression and response to oncogene targeted therapies. Cancers 2021, 13, 5810. [Google Scholar] [CrossRef]
- Nath, A.; Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 2016, 6, 18669. [Google Scholar] [CrossRef] [Green Version]
- Safai, B.; Wu, A.G.; Hamby, C.Y. Prognostic biomarkers in melanoma: Tailoring treatments to patient. J. Clin. Aenest. Dermatol. 2021, 14, 44–48. [Google Scholar]
- Valenti, F.; Falcone, I.; Ungania, S.; Desiderio, F.; Giacomini, P.; Bazzichetto, C.; Conciatori, F.; Gallo, E.; Cognetti, F.; Ciliberto, G.; et al. Precision medicine and melanoma: Multi-omics approaches to monitoring the immunotherapy response. Int. J. Mol. Sci. 2021, 22, 3837. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, H.; Mizukami, Y.; Kaneko, A.; Kajihara, I.; Fukushima, S. Promising blood-based biomarkers for melanoma: Recent progress of liquid biopsy and its future perspectives. Curr. Treat. Options Oncol. 2022, 23, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, E.C.; DeBloom, J.R.; Lee, J.H.; Sussman, J.J.; Covington, K.R.; Caruso, H.G.; Quick, A.P.; Cook, R.W.; Slingluff, C.L.; McMaster, K.M. Long-term outcomes in a multicenter prospective cohort evaluating the prognostic 31-gene expression profile for cutaneous melanoma. JCO Precis. Oncol. 2021, 5, 589–601. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.P.; Zhang, Y.; Lv, J.W.; Li, Y.Q.; Wang, Y.Q.; He, Q.M.; Yang, X.J.; Sun, Y.; Mao, Y.P.; Yun, J.P.; et al. Genomic analysis of tumor microenvironment immune types across 14 solid cancer types: Immunotherapeutic implications. Theranostics 2017, 7, 3585–3594. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladányi, A. Prognostic and predictive significance of immune cell infiltrating cutaneous melanoma. Pigment Cell Melanoma Res. 2015, 28, 490–500. [Google Scholar] [CrossRef]
- Straker, R.J., III; Krupp, K.; Sharon, C.E.; Thaler, A.S.; Kelly, N.J.; Chu, E.Y.; Elder, D.E.; Xu, X.; Miura, J.T.; Karakousis, G.C. Prognostic significance of primary tumor-infiltrating lymphocytes in a contemporary melanoma cohort. Ann. Surg. Oncol. 2022. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Sivendran, S.; Chang, R.; Pham, L.; Phelps, R.G.; Harcharik, S.T.; Hall, L.D.; Bernardo, S.G.; Moskalenko, M.M.; Sivendran, E.; Fu, Y.; et al. Dissection of immune gene networks in primary melanoma tumors critical for antitumor surveillance of patients with stage II–III resectable disease. J. Investig. Dermatol. 2014, 134, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Schaafsma, E.; Gorlov, I.P.; Hernando, E.; Thomas, N.E.; Shen, R.; Turk, M.J.; Berwick, M.; Amos, C.I.; Cheng, C. A leukocyte infiltration score defined by a gene signature predicts melanoma patient prognosis. Mol. Cancer Res. 2019, 17, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poźniak, J.; Nsengimana, J.; Laye, J.P.; O’Shea, S.J.; Diaz, J.M.S.; Droop, A.P.; Filia, A.; Harland, M.; Davies, J.R.; Mell, T.; et al. Genetic and environmental determinants of immune response to cutaneous melanoma. Cancer Res. 2019, 79, 2684–2696. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Y.; Dai, X.; Lin, X.; Shan, Y.; Ye, J. The prognostic landscape of adaptive immune resistance signatures and infiltrating immune cells in the tumor microenvironment of uveal melanoma. Exp. Eye Res. 2020, 196, 108069. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Gu, X.; Wang, Y.; Yao, Z.; Zhou, C. Construction and validation of a novel immunosignature for overall survival in uveal melanoma. Front. Cell Dev. Biol. 2021, 9, 710558. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, I.H.G.; Jager, M.J. Uveal melanoma: The inflammatory microenvironment. J. Innate Immun. 2012, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Singh, M.K.; Kenney, M.C.; Jager, M.J.; Rizvi, M.A.; Meel, R.; Lomi, N.; Bakhshi, S.; Sen, S.; Kashyap, S. Prognostic significance of PD-1/PD-L1 expression in uveal melanoma: Correlation with tumor-infiltrating lymphocytes and clinicopathological parameters. Cancer Immunol. Immunother. 2021, 70, 1291–1303. [Google Scholar] [CrossRef]
- Seth, R.; Messerschmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQude, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic therapy of melanoma: ASCO guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef]
- Rajkumar, S.; Berry, D.; Heney, K.A.; Strong, C.; Ramsay, L.; Lajoie, M.; Alkallas, R.; Nguyen, T.T.; Thomson, C.; Ahanfeshar-Adams, M.; et al. Melanomas with concurrent BRAF non-p600 and NF1 loss-of-function mutations are targetable by BRAF/MEK inhibitor combination therapy. Cell Rep. 2022, 39, 110634. [Google Scholar] [CrossRef]
- Tímár, J.; Hegedűs, B.; Rásó, E. The role of lipid signaling in the progression of malignant melanoma. Cancer Metastasis Rev. 2018, 37, 245–255. [Google Scholar] [CrossRef]
- Larkin, J.L.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment—A review from the melanoma perspective. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatoni, T.; Mohos, A.; Papp, E.; Sebestyén, T.; Liszkay, G.; Oláh, J.; Varga, A.; Lengyel, Z.; Emri, G.; Gaudi, I.; et al. Tumor-infiltrating immune cells as potential biomarkers predicting response to treatment and survival in patients with metastatic melanoma receiving ipilimumab therapy. Cancer Immunol. Immunother. 2018, 67, 141–151. [Google Scholar] [CrossRef]
- Balatoni, T.; Ladányi, A.; Fröhlich, G.; Czirbesz, K.; Kovács, P.; Pánczél, G.; Bence, E.; Plótár, V.; Liszkay, G. Biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Pathol. Oncol. Res. 2020, 26, 317–325. [Google Scholar] [CrossRef]
- McCulloch, J.A.; Davar, D.; Rodrigues, R.R.; Badger, J.H.; Fang, J.R.; Cole, A.M.; Balaji, A.K.; Vetizou, M.; Prescott, S.M.; Fernandes, M.R.; et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 2022, 28, 545–556. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Ladányi, A.; Papp, E.; Mohos, A.; Balatoni, T.; Liszkay, G.; Oláh, J.; Varga, A.; Lengyel, Z.; Emri, G.; Ferrone, S. Role of the anatomic site in the association of HLA class I antigen expression level in metastases with clinical response to ipilimumab therapy in melanoma patients. J. Immunother. Cancer 2020, 8, e000209. [Google Scholar] [CrossRef]
- Ladányi, A.; Hegyi, B.; Balatoni, T.; Liszkay, G.; Rohregger, R.; Waldnig, C.; Dudás, J.; Ferrone, S. HLA class I downregulation in progressing metastases of melanoma patients treated with ipilimumab. Pathol. Oncol. Res. 2022, 28, 1610297. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torreyon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Michelakos, T.; Yamada, T.; Fan, S.; Wang, X.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol. Immunother. 2018, 67, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Sadagopan, A.; Moghaddam, A.S.; Ferrone, S. HLA class I antigen processing machinery defects in antitumor immunity and immunotherapy. Trends Cancer 2021, 7, 1089–1101. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T-cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Kakavand, H.; Jackett, L.A.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676. [Google Scholar] [CrossRef]
- Shukla, S.A.; Bachireddy, P.; Schilling, B.; Galonska, C.; Zhan, Q.; Bango, C.; Langer, R.; Lee, P.C.; Gusenleitner, D.; Keskin, D.B.; et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 2018, 173, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.J.; Kendall, S.M.; Makarov, V.; Walsh, L.A.; Desrichard, A.; Weinhold, N.; Chan, T.A. Recurrent SerpinB3 and SerpinB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016, 48, 1327–1329. [Google Scholar] [CrossRef]
- Ayers, M.A.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Ladányi, A.; Rásó, E.; Barbai, T.; Vízkeleti, L.; Puskás, L.G.; Kovács, S.; Győrffy, B.; Tímár, J. Identification of a tumor cell associated type I IFN resistance gene expression signature of human melanoma, the components of which have a predictive potential for immunotherapy. Int. J. Mol. Sci. 2022, 23, 2704. [Google Scholar] [CrossRef] [PubMed]
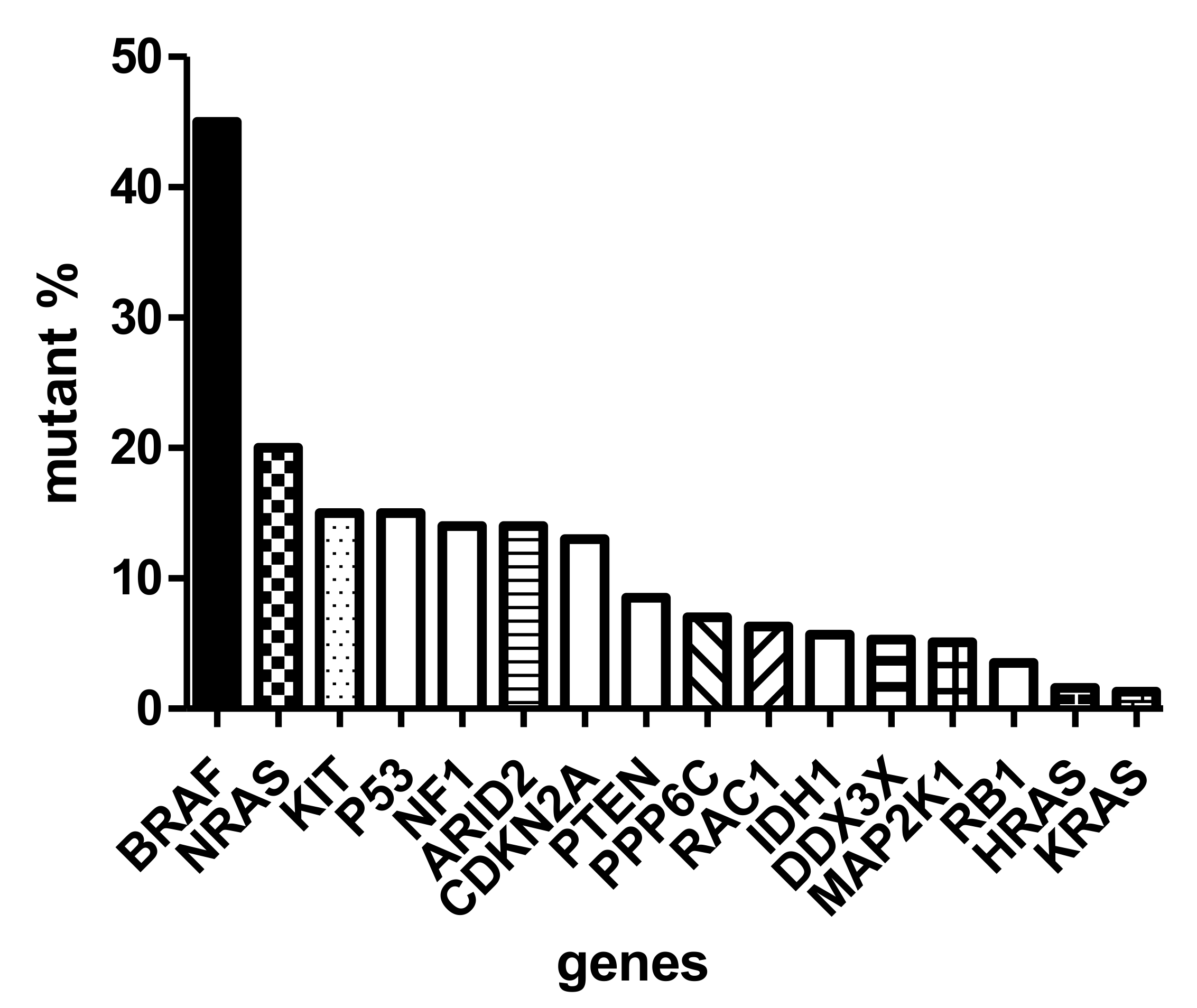

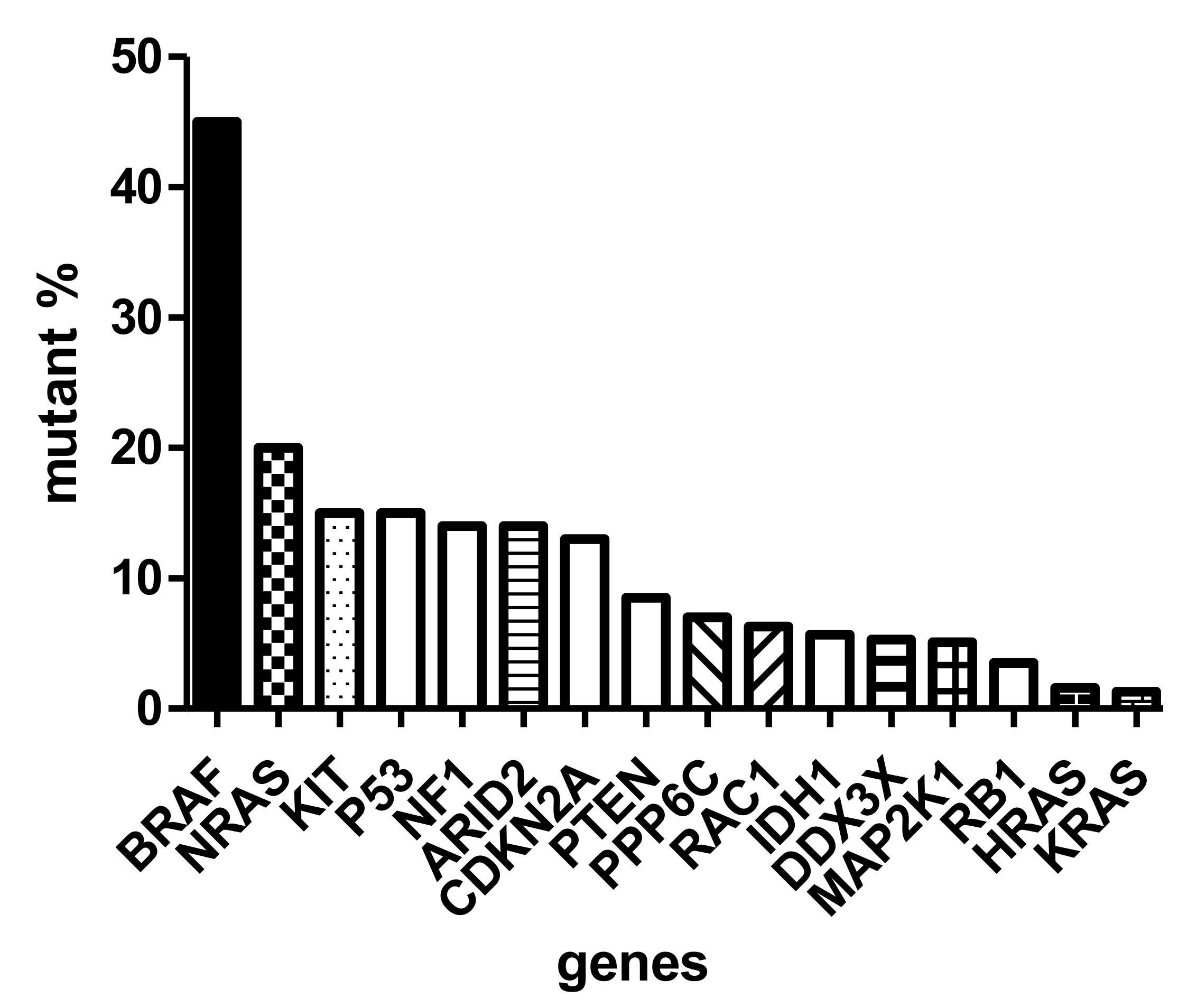{kind=link}
| BRAF-Mutant | RAS-Mutant | NF1-Mutant | Triple Wild-Type | |
|---|---|---|---|---|
| MAPK Signaling | + | + | + | − |
| Cell cycle | CDKN2Amut 60% CDK4mut rare | CDKN2Amut 70% CDK4mut rare CCND1amp 10% | CDKN2Amut 70% RB1mut 10% | CDKN2Amut 40% CDK4amp 15% CCND1amp 10% |
| DDR | TP53mut 10% | TP53mut 20% | TP53mut 30% | MDM2amp 15% |
| Epigenetics | ARID2mut 15% IDH1mut | ARID2mut 15% IDH1mut | ARID2mut 30% IDH1mut | IDH1mut |
| Others | PPP6Cmut 10% PD-L1amp MITFamp | PPP6Cmut 15% |
| Marker | Cellular Localization | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| S100B | cytoplasm | >93 | low |
| Pmel-17/gp100 | melanosome | >70 | >90 |
| MART-1/MelanA | melanosome | >85 | >95 |
| tyrosinase | melanosome | >80 | low |
| MITF | nuclear | >80 | low |
| SOX10 | nuclear | >95 | low |
| FISH [16] | myPath [17] | |
|---|---|---|
| TME | ||
| CCL5 | ||
| RREB1amp | Tumor | CXCL9/10 |
| CCND1amp | PRAME | CD38 |
| CDKN2A LOH | S100A7/8/9/12 | IRF1 |
| MYB LOH | PI3 | LCP2 |
| PTPRC | ||
| SEL1 | ||
| Gene Symbol | Gene Name | Regulation |
|---|---|---|
| BAP1 | BRCA1-associated protein 1 | down |
| MGP | Matrix G1a protein | down |
| SPP1 | Osteopontin | up |
| CXCL14 | Chemokine ligand 14 | down |
| CLCA2 | Chloride channel accessory 2 | down |
| S100A8 | S100 Ca-binding protein A8 | down |
| S100A9 | S100 Ca-binding protein A9 | down |
| BTG1 | B-cell translocation gene 1 | down |
| SAP130 | Sin3A-associated protein | down |
| ARG1 | Arginase 1 | down |
| KRT6B | Keratin 6B | up |
| KRT14 | Keratin 14 | down |
| GJA1 | Gap junction protein A1 | down |
| ID2 | Inhibitor of DNA binding 2 | down |
| EIF1B | Eukaryotic translocation initiator 1B | up |
| CRABP1 | Cellular retinoic acid binding protein 1 | down |
| ROBO1 | Roundabout guidance receptor 1 | down |
| RBM23 | RNA binding protein 23 | down |
| TACSTD2 | Tumor-associated Ca-signal transducer 2 | down |
| DSC1 | Desmocollin 1 | down |
| SPRR1B | Small proline-rich protein 1B | down |
| TRIM29 | Tripartite motif 29 | down |
| AQP3 | Aquaporin 3 | down |
| TYRP1 | Tyrosinase-related protein 1 | down |
| PPL | Periplakin | down |
| LTA4H | Leukotriene A4 hydrolase | down |
| CST6 | Cystatin E/M | down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tímár, J.; Ladányi, A. Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. Int. J. Mol. Sci. 2022, 23, 5384. https://doi.org/10.3390/ijms23105384
Tímár J, Ladányi A. Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. International Journal of Molecular Sciences. 2022; 23(10):5384. https://doi.org/10.3390/ijms23105384
Chicago/Turabian StyleTímár, József, and Andrea Ladányi. 2022. "Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction" International Journal of Molecular Sciences 23, no. 10: 5384. https://doi.org/10.3390/ijms23105384
APA StyleTímár, J., & Ladányi, A. (2022). Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. International Journal of Molecular Sciences, 23(10), 5384. https://doi.org/10.3390/ijms23105384