
Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation

 ,
, 
Abstract
1. Introduction
2. Short-Chain Fatty Acids (SCFAs)
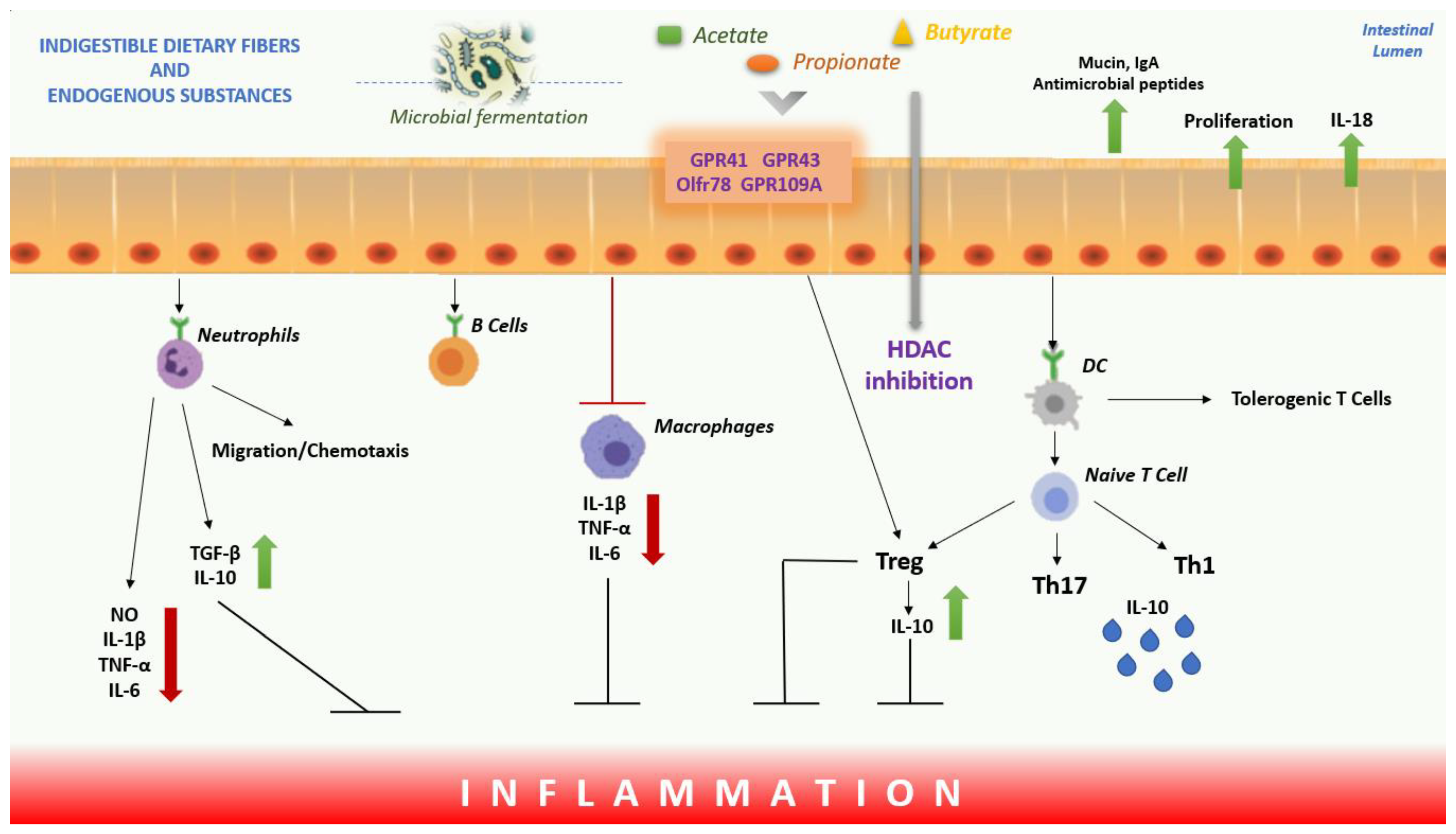
Mechanism, Role and Functions of SCFAs
3. Chronic Kidney Disease (CKD)
Gut Microbiota and SCFAs in CKD
4. Inflammation and Oxidative Stress in CKD
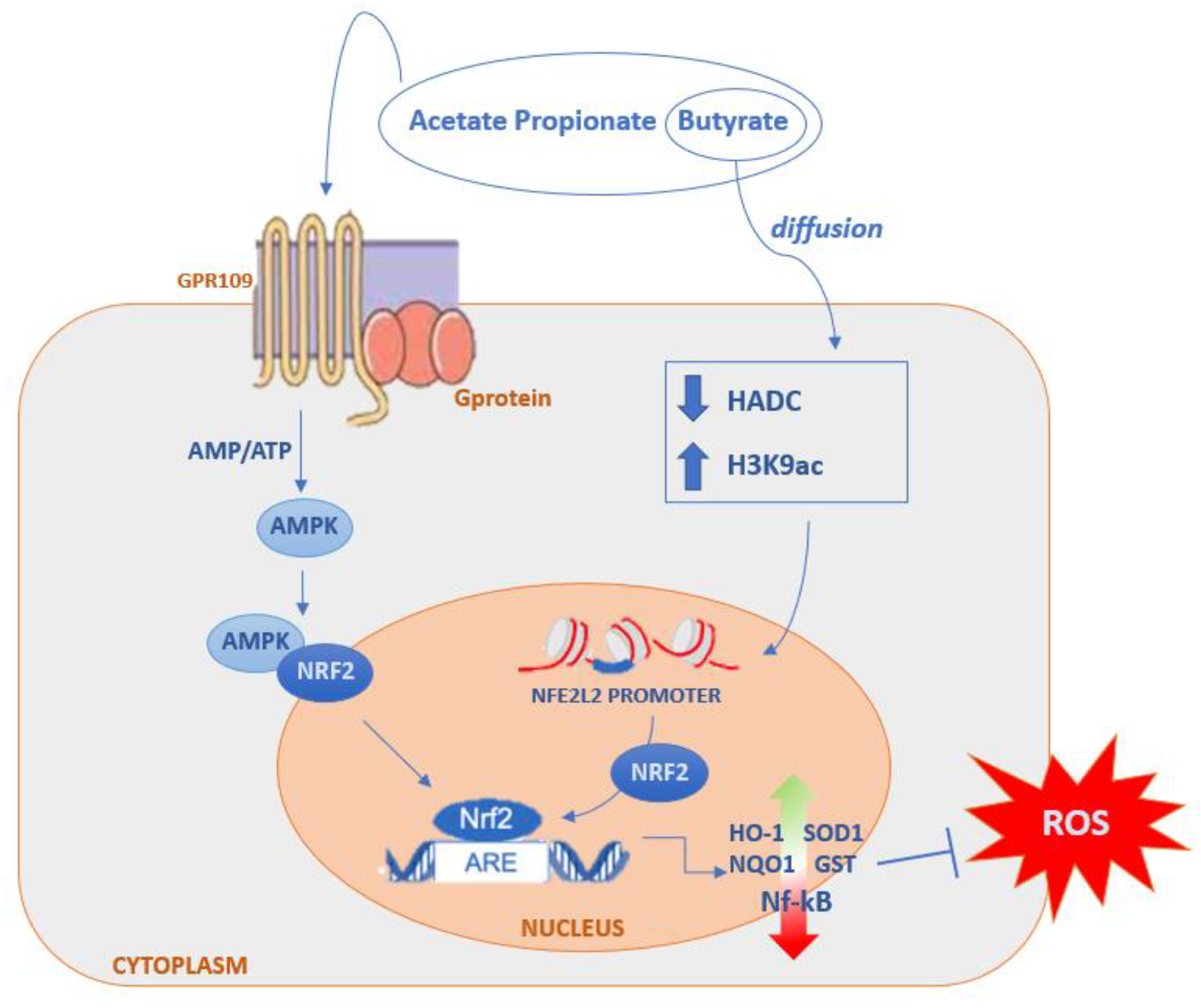
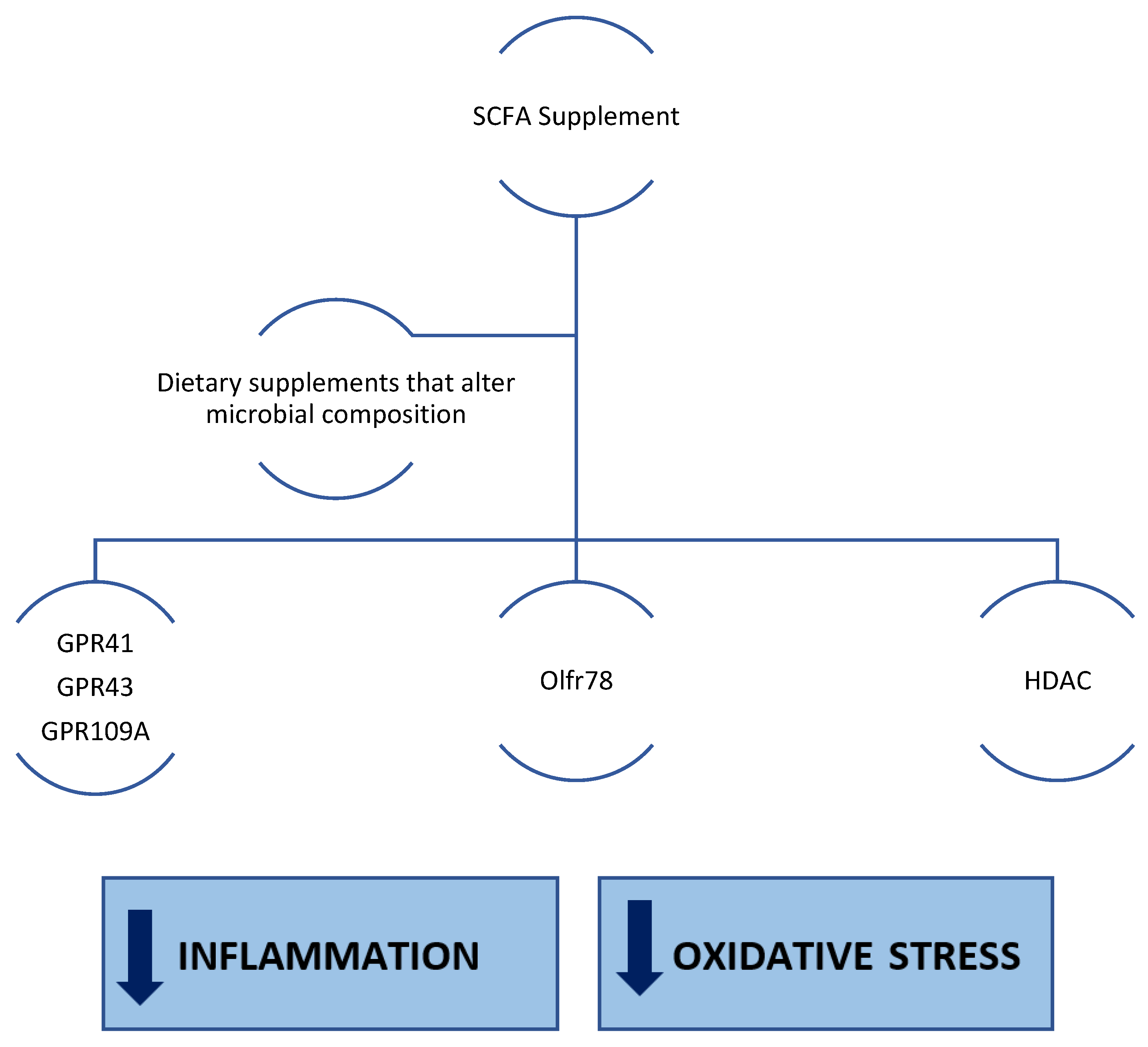
SCFAs, Inflammation and Oxidative Stress
5. Effects of SCFAs in CKD
5.1. Pre-Clinical Observations
5.2. Clinical Observations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miele, L.; Giorgio, V.; Alberelli, M.A.; De Candia, E.; Gasbarrini, A.; Grieco, A. Impact of Gut Microbiota on Obesity, Diabetes, and Cardiovascular Disease Risk. Curr. Cardiol. Rep. 2015, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen 2019, 4, e00678. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; De Santis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef]
- Moraes, C.; Fouque, D.; Amaral, A.C.; Mafra, D. Trimethylamine N-oxide from gut microbiota in chronic kidney disease patients: Focus on diet. J. Ren. Nutr. 2015, 25, 459–465. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Marzocco, S.; Nardone, L.; Sirico, M.; De Simone, E.; Di Natale, G.; Di Micco, L. Urea and impairment of the Gut-Kidney axis in Chronic Kidney Disease. G Ital. Nefrol. 2017, 34, 1–12. [Google Scholar]
- Cosola, C.; Rocchetti, M.T.; Sabatino, A.; Fiaccadori, E.; Di Iorio, B.R.; Gesualdo, L. Microbiota issue in CKD: How promising are gut-targeted approaches? J. Nephrol. 2019, 32, 27–37. [Google Scholar] [CrossRef]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.P.; Rosado, M.; Laganà, B.; D’Amelio, R. Microbiota and chronic inflammatory arthritis: An interwoven link. J. Transl. Med. 2016, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediat. Inflamm. 2014, 2014, 162021. [Google Scholar] [CrossRef]
- Pisano, A.; D’Arrigo, G.; Coppolino, G.; Bolignano, D. Biotic Supplements for Renal Patients: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 1224. [Google Scholar] [CrossRef]
- Pei, M.; Wei, L.; Hu, S.; Yang, B.; Si, J.; Yang, H.; Zhai, J. Probiotics, prebiotics and synbiotics for chronic kidney disease: Protocol for a systematic review and meta-analysis. BMJ Open 2018, 8, e020863. [Google Scholar] [CrossRef]
- Harig, J.M.; Soergel, K.H.; Komorowski, R.A.; Wood, C.M. Treatment of diversion colitis with short-chain-fatty acid irrigation. N. Engl. J. Med. 1989, 320, 23–28. [Google Scholar] [CrossRef]
- Khanna, S. Microbiota Replacement Therapies: Innovation in Gastrointestinal Care. Clin. Pharm. Ther. 2018, 103, 102–111. [Google Scholar] [CrossRef]
- Ochoa-Repáraz, J.; Kirby, T.O.; Kasper, L.H. The Gut Microbiome and Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029017. [Google Scholar] [CrossRef]
- Iraporda, C.; Errea, A.; Romanin, D.E.; Cayet, D.; Pereyra, E.; Pignataro, O.; Sirard, J.C.; Garrote, G.L.; Abraham, A.G.; Rumbo, M. Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 2015, 220, 1161–1169. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Richards, J.L.; Yap, Y.A.; McLeod, K.H.; Mackay, C.R.; Mariño, E. Dietary metabolites and the gut microbiota: An alternative approach to control inflammatory and autoimmune diseases. Clin. Transl. Immunol. 2016, 5, e82. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef]
- Bourriaud, C.; Robins, R.J.; Martin, L.; Kozlowski, F.; Tenailleau, E.; Cherbut, C.; Michel, C. Lactate is mainly fermented to butyrate by human intestinal microfloras but inter-individual variation is evident. J. Appl. Microbiol. 2005, 99, 201–212. [Google Scholar] [CrossRef]
- Scott, K.P.; Martin, J.C.; Campbell, G.; Mayer, C.D.; Flint, H.J. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J. Bacteriol. 2006, 188, 4340–4349. [Google Scholar] [CrossRef]
- Duncan, S.H.; Barcenilla, A.; Stewart, C.S.; Pryde, S.E.; Flint, H.J. Acetate utilization and butyryl coenzyme A (CoA): Acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 2002, 68, 5186–5190. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.; Ross, R.P.; Shanahan, F.; O’Mahony, L.; O’Mahony, C.; Coakley, M.; Hart, O.; Lawlor, P.; Quigley, E.M.; Kiely, B.; et al. Metabolic activity of the enteric microbiota influences the fatty acid composition of murine and porcine liver and adipose tissues. Am. J. Clin. Nutr. 2009, 89, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Kappo, K.A.; Petzke, K.J.; Kipp, A.P.; Blaut, M.; Klaus, S. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol. Nutr. Food Res. 2016, 60, 2611–2621. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Miyamoto, J.; Hasegawa, S.; Kasubuchi, M.; Ichimura, A.; Nakajima, A.; Kimura, I. Nutritional Signaling via Free Fatty Acid Receptors. Int. J. Mol. Sci. 2016, 17, 450. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Haase, S.; Haghikia, A.; Wilck, N.; Müller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short-and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, L.; Fu, P. Gut microbiota-derived short-chain fatty acids and kidney diseases. Drug Des. Dev. Ther. 2017, 11, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Hong, Y.H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.C.; Feng, D.D.; Chen, C.; Lee, H.G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; de Roos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Adom, D.; Nie, D. Regulation of autophagy by short chain fatty acids in colon cancer cells. In Autophagy—A Double-Edged Sword—Cell Survival or Death? Bailly, Y., Ed.; InTech: London, UK, 2013. [Google Scholar]
- Nøhr, M.K.; Egerod, K.L.; Christiansen, S.H.; Gille, A.; Offermanns, S.; Schwartz, T.W.; Møller, M. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 2015, 290, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; Macia, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 7320. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 10–1002. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalizations. N. Engl. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66, s03–s09. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther. 2016, 12, S41–S48. [Google Scholar] [CrossRef]
- Schrimpf, C.; Duffield, J.S. Mechanisms of fibrosis: The role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011, 20, 297–305. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Zafrani, L.; Ince, C. Microcirculation in Acute and Chronic Kidney Diseases. Am. J. Kidney Dis. 2015, 66, 1083–1094. [Google Scholar] [CrossRef]
- Körner, A.; Eklöf, A.C.; Celsi, G.; Aperia, A. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes 1994, 43, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Schofield, Z.V.; Woodruff, T.M.; Halai, R.; Wu, M.C.; Cooper, M.A. Neutrophils--a key component of ischemia-reperfusion injury. Shock 2013, 40, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, A.; Braga, T.T.; Correa-Costa, M.; Aguiar, C.F.; Bassi, Ê.J.; Correa-Silva, R.; Elias, R.M.; Salvador, F.; Moraes-Vieira, P.M.; Cenedeze, M.A.; et al. TLR2, TLR4 and the MYD88 signaling pathway are crucial for neutrophil migration in acute kidney injury induced by sepsis. PLoS ONE 2012, 7, e37584. [Google Scholar] [CrossRef] [PubMed]
- Dendooven, A.; Ishola DAJr Nguyen, T.Q.; Van der Giezen, D.M.; Kok, R.J.; Goldschmeding, R.; Joles, J.A. Oxidative stress in obstructive nephropathy. Int. J. Exp. Pathol. 2011, 92, 202–210. [Google Scholar] [CrossRef]
- Drożdż, D.; Kwinta, P.; Sztefko, K.; Kordon, Z.; Drożdż, T.; Łątka, M.; Miklaszewska, M.; Zachwieja, K.; Rudziński, A.; Pietrzyk, J.A. Oxidative Stress Biomarkers and Left Ventricular Hypertrophy in Children with Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2016, 2016, 7520231. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Reis, T.; Husain-Syed, F.; Vanholder, R.; Hutchison, C.; Stenvinkel, P.; Blankestijn, P.J.; Cozzolino, M.; Juillard, L.; Kashani, K.; et al. Classification of Uremic Toxins and Their Role in Kidney Failure. Clin. J. Am. Soc. Nephrol. 2021, 16, 1918–1928. [Google Scholar] [CrossRef]
- Basile, C.; Libutti, P.; Teutonico, A.; Lomonte, C. Tossine uremiche: Il caso dei “protein-bound compounds” [Uremic toxins: The case of protein-bound compounds]. G Ital. Nefrol. 2010, 27, 498–507. [Google Scholar]
- El Chamieh, C.; Liabeuf, S.; Massy, Z. Uremic Toxins and Cardiovascular Risk in Chronic Kidney Disease: What Have We Learned Recently beyond the Past Findings? Toxins 2022, 14, 280. [Google Scholar] [CrossRef]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Piaz, F.D.; Raffaele Di Iorio, B.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Ribecco, M.T.; Sorrentino, R.; Domenico, B.; Genesio, M.; Pinto, A.; Autore, G.; Cuzzocrea, S. Effect of methylguanidine in a model of septic shock induced by LPS. Free Radic. Res. 2004, 38, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Lisowska-Myjak, B. Uremic toxins and their effects on multiple organ systems. Nephron Clin. Pract. 2014, 128, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Borges, N.A.; Barros, A.F.; Nakao, L.S.; Dolenga, C.J.; Fouque, D.; Mafra, D. Protein-Bound Uremic Toxins from Gut Microbiota and Inflammatory Markers in Chronic Kidney Disease. J. Ren. Nutr. 2016, 26, 396–400. [Google Scholar] [CrossRef]
- Rapa, S.F.; Prisco, F.; Popolo, A.; Iovane, V.; Autore, G.; Di Iorio, B.R.; Dal Piaz, F.; Paciello, O.; Nishijima, F.; Marzocco, S. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. Int. J. Mol. Sci. 2021, 22, 1135. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Genovese, T.; Sorrentino, R.; Britti, D.; Scollo, G.; Pinto, A.; Cuzzocrea, S.; Autore, G. Methylguanidine reduces the development of non septic shock induced by zymosan in mice. Life Sci. 2004, 75, 1417–1433. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Serraino, I.; Sorrentino, R.; Meli, R.; Mattaceraso, G.; Cuzzocrea, S.; Pinto, A.; Autore, G. Effect of methylguanidine in carrageenan-induced acute inflammation in the rats. Eur. J. Pharmacol. 2004, 484, 341–350. [Google Scholar] [CrossRef]
- Ahlawat, S.; Asha Sharma, K.K. Gut-organ axis: A microbial outreach and networking. Lett. Appl. Microbiol. 2021, 72, 636–668. [Google Scholar] [CrossRef]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2017, 32, 2005–2014. [Google Scholar] [CrossRef]
- Meštrović, T.; Matijašić, M.; Perić, M.; Čipčić Paljetak, H.; Barešić, A.; Verbanac, D. The Role of Gut, Vaginal, and Urinary Microbiome in Urinary Tract Infections: From Bench to Bedside. Diagnostics 2020, 11, 7. [Google Scholar] [CrossRef]
- Bien, J.; Sokolova, O.; Bozko, P. Role of Uropathogenic Escherichia coli Virulence Factors in Development of Urinary Tract Infection and Kidney Damage. Int. J. Nephrol. 2012, 2012, 681473. [Google Scholar] [CrossRef] [PubMed]
- Magruder, M.; Sholi, A.N.; Gong, C.; Zhang, L.; Edusei, E.; Huang, J.; Albakry, S.; Satlin, M.J.; Westblade, L.F.; Crawford, C.; et al. Gut uropathogen abundance is a risk factor for development of bacteriuria and urinary tract infection. Nat. Commun. 2019, 10, 5521. [Google Scholar] [CrossRef] [PubMed]
- Belyayeva, M.; Jeong, J.M. Acute pyelonephritis. In StatPearls; Stat Pearls Publishing: Treasure Island, FL, USA, 2020; pp. 2–3. [Google Scholar]
- Nallu, A.; Sharma, S.; Ramezani, A.; Muralidharan, J.; Raj, D. Gut microbiome in chronic kidney disease: Challenges and opportunities. Transl. Res. 2017, 179, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Barrows, I.R.; Ramezani, A.; Raj, D.S. Gut Feeling in AKI: The Long Arm of Short–Chain Fatty Acids. J. Am. Soc. Nephrol. 2015, 26, 1755–1757. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Al Khodor, S.; Shatat, I.F. Gut microbiome and kidney disease: A bidirectional relationship. Pediatr. Nephrol. 2017, 32, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Steenbeke, M.; Valkenburg, S.; Gryp, T.; Van Biesen, W.; Delanghe, J.R.; Speeckaert, M.M.; Glorieux, G. Gut Microbiota and Their Derived Metabolites, a Search for Potential Targets to Limit Accumulation of Protein-Bound Uremic Toxins in Chronic Kidney Disease. Toxins 2021, 13, 809. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic kidney disease and the gut microbiome. Am. J. Physiol.-Ren. Physiol. 2019, 316, F1211–F1217. [Google Scholar] [CrossRef]
- Vaziri, N.D. Effect of synbiotic therapy on gut–derived uremic toxins and the intestinal microbiome in patients with CKD. J. Am. Soc. Nephrol. 2016, 11, 199–201. [Google Scholar] [CrossRef]
- Pluznick, J.L. Gut microbiota in renal physiology: Focus on short-chain fatty acids and their receptors. Kidney Int. 2016, 90, 1191–1198. [Google Scholar] [CrossRef]
- Mahmoodpoor, F.; Rahbar Saadat, Y.; Barzegari, A.; Ardalan, M.; Zununi Vahed, S. The impact of gut microbiota on kidney function and pathogenesis. Biomed. Pharmacother. 2017, 93, 412–419. [Google Scholar] [CrossRef]
- Vijay, A.; Valdes, A.M. Role of the gut microbiome in chronic diseases: A narrative review. Eur. J. Clin. Nutr. 2021, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Irazabal, M.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Zipeto, D.; Popescu, I.D. Inflammation in Cancer: Part of the Problem or Part of the Solution? J. Immunol. Res. 2019, 2019, 5403910. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Roubicek, T.; Bartlova, M.; Krajickova, J.; Haluzikova, D.; Mraz, M.; Lacinova, Z.; Kudla, M.; Teplan, V.; Haluzik, M. Increased production of proinflammatory cytokines in adipose tissue of patients with end-stage renal disease. Nutrition 2009, 25, 762–768. [Google Scholar] [CrossRef]
- Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. J. Clin. Med. 2018, 7, 365. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Saldanha, J.F.; Yi, D.; Mafra, D.; Fouque, D.; Soulage, C.O. The uremic toxin indoxyl sulfate exacerbates reactive oxygen species production and inflammation in 3T3-L1 adipose cells. Free Radic. Res. 2016, 50, 337–344. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Kao, M.P.; Ang, D.S.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Descamps-Latscha, B.; Drüeke, T.; Witko-Sarsat, V. Dialysis-induced oxidative stress: Biological aspects, clinical consequences, and therapy. Semin. Dial. 2001, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004, 24, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Tanea, T.; Venditti, P.; Manuel, V. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Birben, E.; Murat, U.; Md, S.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. WAO J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Krata, N.; Zagożdżon, R.; Foroncewicz, B.; Mucha, K. Oxidative Stress in Kidney Diseases: The Cause or the Consequence? Arch. Immunol. Et. Ther. Exp. 2018, 66, 211–220. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Guijarro, C.; Egido, J. Transcription factor-κB (NF-κB) and renal disease. Kidney Int. 2001, 59, 415–424. [Google Scholar] [CrossRef]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in Renal Inflammation. JASN 2010, 21, 1254–1262. [Google Scholar] [CrossRef]
- Yiu, W.H.; Wong, D.W.L.; Chan, L.Y.Y.; Leung, J.C.K.; Chan, K.W.; Lan, H.Y.; Lai, K.N.; Tang, S.C.W. Tissue Kallikrein Mediates Pro-Inflammatory Pathways and Activation of Protease-Activated Receptor-4 in Proximal Tubular Epithelial Cells. PLoS ONE 2014, 9, e88894. [Google Scholar] [CrossRef]
- Kinugasa, E. Markers and possible uremic toxins: Japanese experiences. Contrib. Nephrol. 2011, 168, 134–138. [Google Scholar] [PubMed]
- Boulanger, E.; Wautier, M.P.; Wautier, J.L.; Boval, B.; Panis, Y.; Wernert, N.; Danze, P.M.; Dequiedt, P. AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Int. 2002, 61, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.E.; Forman, H.J. Introduction to Special Issue on Nrf2 Regulated Redox Signaling and Metabolism in Physiology and Medicine. Free Radic. Biol. Med. 2015, 88, 91–92. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Wang, X.; He, G.; Peng, Y.; Zhong, W.; Wang, Y.; Zhang, B. Sodium butyrate alleviates adipocyte inflammation by inhibiting NLRP3 pathway. Sci. Rep. 2015, 5, 12676. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, L.; Bhat, O.M.; Lohner, H.; Li, P.L. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: Antioxidant action of butyrate. Redox Biol. 2018, 16, 21–31. [Google Scholar] [CrossRef]
- Stempelj, M.; Kedinger, M.; Augenlicht, L.; Klampfer, L. Essential role of the JAK/STAT1 signaling pathway in the expression of inducible nitric-oxide synthase in intestinal epithelial cells and its regulation by butyrate. J. Biol. Chem. 2007, 282, 9797–9804. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, X.; Li, Y.; Li, N.; Shi, X.; Ding, H.; Zhang, Y.; Li, X.; Liu, G.; Wang, Z. Non-esterified fatty acids activate the ROS-p38-p53/Nrf2 signaling pathway to induce bovine hepatocyte apoptosis in vitro. Apoptosis 2014, 19, 984–997. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Li, J.; Wang, G.; Li, L. Non-Esterified Fatty Acid-Induced Reactive Oxygen Species Mediated Granulosa Cells Apoptosis Is Regulated by Nrf2/p53 Signaling Pathway. Antioxidants 2020, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, S.; Mao, L.; Leak, R.K.; Shi, Y.; Zhang, W.; Hu, X.; Sun, B.; Cao, G.; Gao, Y.; et al. Omega-3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J. Neurosci. 2014, 34, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiang, Z.; Zhang, H.; Liang, W.; Huang, W.; Zhang, H.; Li, Y.; Wang, Z.; Wang, J.; Jia, Y.; et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 2018, 124, 454–465. [Google Scholar] [CrossRef]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with Nrf2 and p53. Mol. Cell. Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020, 152, 728–742. [Google Scholar] [CrossRef]
- Srivastava, S.; Alfieri, A.; Siow, R.C.; Mann, G.E.; Fraser, P.A. Temporal and spatial distribution of Nrf2 in rat brain following stroke: Quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J. Physiol. 2013, 591, 3525–3538. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood-brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, E.; Sun, G.; Yan, H.Q.; Foley, L.M.; Andrzejczuk, L.A.; Attarwala, I.Y.; Hitchens, T.K.; Kiselyov, K.; Dixon, C.E.; et al. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Chen, J.H.; Chiang, C.K. Uremic Toxins and Protein-Bound Therapeutics in AKI and CKD: Up-to-Date Evidence. Toxins 2021, 14, 8. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef]
- Cheng, T.H.; Ma, M.C.; Liao, M.T.; Zheng, C.M.; Lu, K.C.; Liao, C.H.; Hou, Y.C.; Liu, W.C.; Lu, C.L. Indoxyl Sulfate, a Tubular Toxin, Contributes to the Development of Chronic Kidney Disease. Toxins 2020, 12, 684. [Google Scholar] [CrossRef]
- Walther, C.P.; Nambi, V.; Hanania, N.A.; Navaneethan, S.D. Diagnosis and Management of Pulmonary Hypertension in Patients With CKD. Am. J. Kidney Dis. 2020, 75, 935–945. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Fouque, D.; Soulage, C.O.; Croze, M.; Mafra, D. Indoxyl sulfate and p-cresyl sulfate in chronic kidney disease. Could these toxins modulate the antioxidant Nrf2-Keap1 pathway? J. Ren. Nutr. 2014, 24, 286–291. [Google Scholar] [CrossRef]
- Huang, W.; Guo, H.L.; Deng, X.; Zhu, T.T.; Xiong, J.F.; Xu, Y.H.; Xu, Y. Short-Chain Fatty Acids Inhibit Oxidative Stress and Inflammation in Mesangial Cells Induced by High Glucose and Lipopolysaccharide. Exp. Clin. Endocrinol. Diabetes 2017, 125, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; de Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Chen, Y.A.; Chen, S.A.; Chen, Y.J.; Lin, Y.K. Uremic Toxins—Novel Arrhythmogenic Factor in Chronic Kidney Disease—Related Atrial Fibrillation. Acta Cardiol. Sin. 2016, 32, 259–264. [Google Scholar] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, F.; Tang, Y.; Xiao, J.; Li, C.; Ouyang, Y.; Hou, Y. Valproic acid regulates Ang II-induced pericyte-myofibroblast trans-differentiation via MAPK/ERK pathway. Am. J. Transl. Res. 2018, 10, 1976–1989. [Google Scholar]
- Matsumoto, N.; Riley, S.; Fraser, D.; Al-Assaf, S.; Ishimura, E.; Wolever, T.; Phillips, G.O.; Phillips, A.O. Butyrate modulates TGF-beta1 generation and function: Potential renal benefit for Acacia(sen) SUPERGUM (gum arabic)? Kidney Int. 2006, 69, 257–265. [Google Scholar] [CrossRef][Green Version]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; Alotaibi, M.R.; Al-Asmari, A.F.; Alanazi, W.A.; Al-Harbi, M.M.; El-Sherbeeny, A.M.; Ibrahim, K.E. Short chain fatty acid, acetate ameliorates sepsis-induced acute kidney injury by inhibition of NADPH oxidase signaling in T cells. Int. Immunopharmacol. 2018, 58, 24–31. [Google Scholar] [CrossRef]
- Machado, R.A.; Constantino Lde, S.; Tomasi, C.D.; Rojas, H.A.; Vuolo, F.S.; Vitto, M.F.; Cesconetto, P.A.; de Souza, C.T.; Ritter, C.; Dal-Pizzol, F. Sodium butyrate decreases the activation of NF-κB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol. Dial. Transplant. 2012, 27, 3136–3140. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.J.; Loh, Y.W.; Singer, J.; Zhu, W.; Macia, L.; Mackay, C.R.; Wang, W.; Chadban, S.J.; Wu, H. Fiber Derived Microbial Metabolites Prevent Acute Kidney Injury Through G-Protein Coupled Receptors and HDAC Inhibition. Front. Cell Dev. Biol. 2021, 9, 648639. [Google Scholar] [CrossRef]
- Huang, W.; Man, Y.; Gao, C.; Zhou, L.; Gu, J.; Xu, H.; Wan, Q.; Long, Y.; Chai, L.; Xu, Y.; et al. Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF-κB Signaling. Oxidative Med. Cell. Longev. 2020, 2020, 4074832. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Felizardo, R.J.F.; de Almeida, D.C.; Pereira, R.L.; Watanabe, I.K.M.; Doimo, N.T.S.; Ribeiro, W.R.; Cenedeze, M.A.; Hiyane, M.I.; Amano, M.T.; Braga, T.T.; et al. Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic- and GPR109a-mediated mechanisms. FASEB J. 2019, 33, 11894–11908. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Z.; Peng, R.; Zhang, L.; Liu, H.; Wang, X.; Tian, Y.; Sun, Y. RNA-Seq analysis reveals critical transcriptome changes caused by sodium butyrate in DN mouse models. Biosci. Rep. 2021, 41, BSR20203005. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.V.; Suzuki, T. Dietary Fermentable Fibers Attenuate Chronic Kidney Disease in Mice by Protecting the Intestinal Barrier. J. Nutr. 2018, 148, 552–561. [Google Scholar] [CrossRef]
- Yang, J.; Li, Q.; Henning, S.M.; Zhong, J.; Hsu, M.; Lee, R.; Long, J.; Chan, B.; Nagami, G.T.; Heber, D.; et al. Effects of Prebiotic Fiber Xylooligosaccharide in Adenine-Induced Nephropathy in Mice. Mol. Nutr. Food Res. 2018, 62, e1800014. [Google Scholar] [CrossRef]
- Marzocco, S.; Fazeli, G.; Di Micco, L.; Autore, G.; Adesso, S.; Dal Piaz, F.; Heidland, A.; Di Iorio, B. Supplementation of Short-Chain Fatty Acid, Sodium Propionate, in Patients on Maintenance Hemodialysis: Beneficial Effects on Inflammatory Parameters and Gut-Derived Uremic Toxins, A Pilot Study (PLAN Study). J. Clin. Med. 2018, 7, 315. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Esgalhado, M.; Kemp, J.A.; Damasceno, N.R.; Fouque, D.; Mafra, D. Short-chain fatty acids: A link between prebiotics and microbiota in chronic kidney disease. Future Microbiol. 2017, 12, 1413–1425. [Google Scholar] [CrossRef]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef]
- Weber, G.J.; Foster, J.; Pushpakumar, S.B.; Sen, U. Altered microRNA regulation of short chain fatty acid receptors in the hypertensive kidney is normalized with hydrogen sulfide supplementation. Pharmacol. Res. 2018, 134, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Pevsner-Fischer, M.; Blacher, E.; Tatirovsky, E.; Ben-Dov, I.Z.; Elinav, E. The gut microbiome and hypertension. Curr. Opin. Nephrol. Hypertens. 2017, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jadoon, A.; Mathew, A.V.; Byun, J.; Gadegbeku, C.A.; Gipson, D.S.; Afshinnia, F.; Pennathur, S. For the Michigan Kidney Translational Core CPROBE Investigator Group. Gut Microbial Product Predicts Cardiovascular Risk in Chronic Kidney Disease Patients. Am. J. Nephrol. 2018, 48, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.H.; Schönfelder, T.; Brandão, I.; Wilms, E.; Hörmann, N.; Jäckel, S.; Schüler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5, e003698. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Marzocco, S.; Bellasi, A.; De Simone, E.; Dal Piaz, F.; Rocchetti, M.T.; Cosola, C.; Di Micco, L.; Gesualdo, L. Nutritional therapy reduces protein carbamylation through urea lowering in chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Di Micco, L.; Marzocco, S.; De Simone, E.; De Blasio, A.; Sirico, M.L.; Nardone, L. UBI Study Group. Very Low-Protein Diet (VLPD) Reduces Metabolic Acidosis in Subjects with Chronic Kidney Disease: The “Nutritional Light Signal” of the Renal Acid Load. Nutrients 2017, 9, 69. [Google Scholar] [CrossRef]
- Bellasi, A.; Di Micco, L.; Santoro, D.; Marzocco, S.; De Simone, E.; Cozzolino, M.; Di Lullo, L.; Guastaferro, P.; Di Iorio, B. UBI study investigators. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016, 17, 158. [Google Scholar] [CrossRef]
- Rocchetti, M.T.; Di Iorio, B.R.; Vacca, M.; Cosola, C.; Marzocco, S.; di Bari, I.; Calabrese, F.M.; Ciarcia, R.; De Angelis, M.; Gesualdo, L. Ketoanalogs’ Effects on Intestinal Microbiota Modulation and Uremic Toxins Serum Levels in Chronic Kidney Disease (Medika2 Study). J. Clin. Med. 2021, 10, 840. [Google Scholar] [CrossRef]
- Marzocco, S.; Dal Piaz, F.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef]
- Kemp, J.A.; Regis de Paiva, B.; Fragoso Dos Santos, H.; Emiliano de Jesus, H.; Craven, H.; ZIjaz, U.; Alvarenga Borges, N.; GShiels, P.; Mafra, D. The Impact of Enriched Resistant Starch Type-2 Cookies on the Gut Microbiome in Hemodialysis Patients: A Randomized Controlled Trial. Mol. Nutr. Food Res. 2021, 65, e2100374. [Google Scholar] [CrossRef]
- Di Iorio, B.R.; Rocchetti, M.T.; De Angelis, M.; Cosola, C.; Marzocco, S.; Di Micco, L.; di Bari, I.; Accetturo, M.; Vacca, M.; Gobbetti, M.; et al. Nutritional Therapy Modulates Intestinal Microbiota and Reduces Serum Levels of Total and Free Indoxyl Sulfate and P-Cresyl Sulfate in Chronic Kidney Disease (Medika Study). J. Clin. Med. 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Lee, C.C.; Hsu, H.J.; Sun, C.Y.; Chen, Y.C.; Yang, K.J.; Yang, C.W.; Chung, W.H.; Lai, H.C.; Chang, L.C.; et al. Compositional and Functional Adaptations of Intestinal Microbiota and Related Metabolites in CKD Patients Receiving Dietary Protein Restriction. Nutrients 2020, 12, 2799. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SCFAs | Producers | Binding | Intestinal Expression | Non Intestinal Expression |
|---|---|---|---|---|
| Acetate | Akkermansia muciniphila, Bacterioides spp., Bifidobacterium spp., Prevotella spp., Ruminococcus spp., Blautia hydrogenotrophica, Clostridium spp., Streptococcus spp. | GPR43 | Colonic, Small intestinal epithelium, Colonic lamina propria cells, Leukocytes in small intestinal | Polymorphonuclear cells, Adipocytes, Skeletal muscle, Heart, Spleen |
| Olfr78 | ||||
| Propionate | Bacterioides spp., Phascolarctobacterium succinatutens, Dialister spp., Veilonella spp., Megasphera elsdenii, Coprococcus catus, Ruminococcus obeum, Salmonella spp., Roseburia inulinivorans | GPR43 | Colonic, Small intestinal epithelium, Colonic lamina propria cells (mast cells), Pancreas, Gut enteroendocrine cells located in the crypts and lower part of the villi | Spleen, Bone marrow, Lymph nodes, Adipose tissue, Periportal afferent system, Peripheral nervous system, Peripheral blood mononuclear cells |
| GPR41 | ||||
| Olfr78 | ||||
| Butyrate | Coprococcus viene, Coprococcus eutacus, Anaerostipes spp., Coprococcus catus, Eubacterium rectale, Eubacterium hallii, Faecalibacterium prausnitzii, Roseburia spp. | GPR109A | Apical membrane of colonic/small intestinal epithelium, Macrophages, Monocytes, Neutrophils, Dendritic cells | Adipocytes (white and brown), Epidermal Langerhans cells, Retinal pigment epithelium |
| GPR41 | ||||
| GPR43 |
| Pre-Clinical Studies | Treatment | Parameters Evaluated | Effect | References |
|---|---|---|---|---|
| Mouse glomerular mesangial cells (SV-40 MES 13) | Acetate, Butyrate, GPR43 agonist | Cell viability assays, detection of ROS, MDA and SOD, MCP-1, IL-1β and ICAM-1 | Butyrate, Acetate, and GPR43 agonist reduced inflammatory markers | Huang et al., 2017 [132] |
| Renal tubular epithelial cells | Butyrate, Propionate, Acetate | NF-κB activation, NO production, ROS production | SCFAs reduced inflammation and hypoxia and also modulated immune response | Andrade-Olivera et al., 2015 [133] |
| Bone marrow dendritic cells | Molecules CD80 and CD40 | |||
| Antigen-presenting cells from RAGKO mice | Proliferation of CD8+ and CD4+ cells | |||
| Male C57BL/6 mice with AKI | Apoptosis assessment, immunohistochemical analysis, mitochondrial DNA, DNA methylation, NF-κB levels, TLR-4, IL-6, IFN-γ, TNF-α, TGF-β1, MCP-1, IL-1β, GSS/GSSH ratio | Acetate reducing ROS, cytokines and chemokines; Low mRNA levels of TLR-4, lower activation of the NF-κB pathway, low levels of activated neutrophils and macrophages, a low frequency of infiltrating macrophages and a low frequency of activated DCs were observed. Acetate also increased the expression of GPR43 and inhibited the activity of HDACs | ||
| Immortalised human renal proximal tubular epithelial cells (HK-2 cells) | Butyrate | TGF-β1 levels and expression | Butyrate reduces the basal generation of TGF-β1 through inhibition of the ERK/MAP kinase | Matsumoto et al., 2006 [137] |
| Wild-type and knockout C57BL/6 mice for GPR41, GPR43, or GPR109A receptors with diabetic nephropathy | Propionate, Butyrate | Assessment of serum creatinine, histological examination of renal tissues, evaluation of microbial composition, and SCFA levels, gene expression analysis of TLR-2, TLR-4, NLRP3, TNF-α, IL-6, IL-18, IL-1β, IL-4, IL-10, IFNγ, CXCL2, CCL2, CXCL10, iNOS, KIM1, MMP2, MMP9, TGFβ1, HDAC1-11, and GAPDH | Reduced inflammation and kidney injury, increased Bifidobacterium and Prevotella that increased faecal and serum SCFA concentrations | Liu et al., 2021 [140] |
| Mouse glomerular mesangial cells (SV-40 MES 13) | Acetate, Propionate, Butyrate | Cell viability assays, detection of ROS, MDA and SOD, Western blot analysis or ELISA for GPR43, β-arrestin-2, NF-κB, p65, MCP-1, IL-1β, I-κBα, GAPDH | Butyrate restored high glucose concentrations, oxidative stress, NF-κB signalling, and interaction between β -arrestin-2 and I-κB α-induced GPR43 | Huang et al., 2020 [141] |
| Eight-week-old male C57BL/6 mice with type 2 diabetes induced by streptozotocin, diabetic nephropathy | Acetate, Propionate, Butyrate | FBG levels, ACR, FINS, BUN, SCr, serum cystatin C, TC, TG, LDL and LDL-C, renal glomerular histology, immunohistochemical staining for GPR43, β-arrestin-2, NF-κB, p65, MCP-1 | Butyrate improved hyperglycemia, improved insulin resistance of T2D, prevented renal dysfunction in T2D, inhibited DT2-induced renal NF-κB activation and regulated GPR43- β -Arrestin2-signalling | |
| Renal tubular epithelial cells and podocytes were isolated from C57BL/6 mice | Acetate, Propionate, Butyrate | mRNA expression of IL-6, IFN-γ, TNF-α, CCL2, CXCL10 | Butyrate and propionate significantly inhibited inflammation | Li et al., 2020 [142] |
| Wild-type and knockout C57BL/6 mice lacking genes for GPR43 or GPR109A with diabetic nephropathy | Fibre-rich diets, Sodium Acetate, Sodium Propionate, and Sodium Butyrate | Immunohistochemical analysis, histological analysis, real-time PCR for TLR-2, TLR-4, IL-6, IFN-γ, TNF-α, CCL2, CXCL10, TGF-β1, fibronectin, and GAPDH, bacterial DNA sequencing analysis, analysis of SCFA levels | Increased Prevotella and Bifidobacterium, increased SCFA, modulation of inflammation in renal tubular cells and podocytes under hyperglycemic conditions | |
| 33 7-week-old male ICR with CKD adenine-induced | DF, GG, PHGG | Evaluation of TNF- α, MCP-1, IL-1β, IL-6, Tgfb1, Col1A1, Acta2, TLR-4 and Myd88 mRNA levels, expression levels of ZO-1, ZO-2, occludin, JAMA, claudin 3, claudin 4, and claudin 7, serum creatinine and urea analysis, immunohistochemistry analysis, microbiota composition analysis, SCFA levels, IgA of the mucosa and the mucus itself | Colonic barrier protection and reduced endotoxemia, restoration of tight junction protein expression and localisation, increased Bifidobacterium, increased propionic and butyric acid production correlated with a reduction in pro-inflammatory parameters | Hung et Suzuki, 2018 [145] |
| Seven-week-old male C57BL/6J mice with CKD adenine-induced | XOS | Col1A1, Cgtf, IL-6, TNF-α, Arg, Ym1, Defa, Pla2g2a, Reg3γ, caecal SCFAs, histological analysis of renal tissue | Reduced gene expression of markers observed in CKD, including lColA1 and Cgtf, IL-6 and the M2 macrophage marker, Defa5, reduced IS and pCS and increased SCFA-producing bacteria, and improved renal function | Yang et al., 2018 [144] |
| Male isogenic Balb/c mice and C57BL/6 with nephropathy | Diet of high amylose butyrate-releasing corn starch | Immunohistochemical analysis, DNA expression, analysis TGF-β1, Fsp1, ActaII, Col4α1, Mmp9, Timp1, urinary albumin analysis, monoclonal antibody generation for GPR109A and GPR43, analysis of SCFAs concentrations, measurement of pro-inflammatory cytokines, isolation and polarisation of bone marrow-derived macrophages, HDAC activity, DNA methylation | Butyrate attenuates inflammation and renal fibrosis through its receptors GPR109A, GPR43, and GPR41 | Felizardo et al., 2019 [143] |
| Clinical Studies | Treatment | Parameters Evaluated | Effect | References |
|---|---|---|---|---|
| 20 patients with CKD and related complications | Sodium Propionate | Biochemical analyse on sera for hs-CRP, IL-2, IL-6, IL-10, IL-17a, TNF-α, INFγ, TGF-β, IL-10, MDA, and endotoxins/lipopolysaccharides | Sodium Propionate reduced inflammatory markers and improved anti-inflammatory parameters | Marzocco et al., 2018 [147] |
| 43 patients with CKD | LPD | Analysis of bacterial populations and IS and pCS levels | LPD modulates gut dysbiosis and positively impacts the outcome of patients with CKD | Wu et al., 2020 [158] |
| Patients in HD with CKD | RS2 | Evaluation of biochemical and clinical parameters, analysis of serum and plasma samples, genomic sequencing analysis | Mitigate inflammation and oxidative stress in hemodialysis patients by positively altering SCFA-producing bacteria | Kemp et al., 2021 [156] |
| 214 patients with CKD and CVD | Addition of valerate | Evaluation of anthropometric, biochemical and clinical parameters, measurement of plasma levels of SCFAs | The addition of valerate to a model of hypertension, diabetes mellitus, and other complications significantly improved there conditions of patients | Jadoon et al., 2019 [154] |
| 60 patients with grade 3B-4 CKD | FD, MD, VLPD | Anthropometri, clinical, and biochemical parameters obtained from stool and serum samples | Increased Lachnospiraceae, Ruminococcaceae, Prevotellaceae, Bifidobacteriaceae, Coprococcus, and Roseburia forming butyrate, increased anti-inflammatory potential, improved intestinal permeability and systolic blood pressure, reduced Enterobacteriaceae pathogens and circulating levels of IS, pCS, and D-Lactate | Di Iorio et al., 2018 [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magliocca, G.; Mone, P.; Di Iorio, B.R.; Heidland, A.; Marzocco, S. Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation. Int. J. Mol. Sci. 2022, 23, 5354. https://doi.org/10.3390/ijms23105354
Magliocca G, Mone P, Di Iorio BR, Heidland A, Marzocco S. Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation. International Journal of Molecular Sciences. 2022; 23(10):5354. https://doi.org/10.3390/ijms23105354
Chicago/Turabian StyleMagliocca, Giorgia, Pasquale Mone, Biagio Raffaele Di Iorio, August Heidland, and Stefania Marzocco. 2022. "Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation" International Journal of Molecular Sciences 23, no. 10: 5354. https://doi.org/10.3390/ijms23105354
APA StyleMagliocca, G., Mone, P., Di Iorio, B. R., Heidland, A., & Marzocco, S. (2022). Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation. International Journal of Molecular Sciences, 23(10), 5354. https://doi.org/10.3390/ijms23105354