
Revisiting the Roles of Filaggrin in Atopic Dermatitis

 , ,
, ,
Abstract
1. Filaggrin: A Look in the Rear-View Mirror
2. The Impact of the FLG Gene: From Ichthyosis Vulgaris to Atopic Dermatitis
3. FLG Null Mutations Are Strong Genetic Factors in AD
4. Decreased Filaggrin in Atopic Skin, Regardless of FLG Genotype
5. Filaggrin Deficiency Induces Subtle Epidermal Barrier Impairment in Non-Lesional AD
6. Filaggrin Deficiency Promotes Subclinical Inflammation in the Epidermis Nonspecific to AD
7. Does Filaggrin Deficiency in Atopic Skin Lead to Alkalization of the Skin?
8. Effects of Filaggrin Deficiency on the Skin Microbiota in AD
9. Filaggrin Deficiency per se Does Not Promote Colonization of Atopic Skin with Staphylococcus aureus
10. Filaggrin Deficiency Leads to Cellular Abnormalities in Keratinocytes: Potential Relevance in AD
11. FLG Null Mutations Regulate Skin Moisture in Non-Lesional AD
12. FLG Null Mutations Are Less Penetrant Genetic Factors in the Atopic March than in AD
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| AHR | aryl hydrocarbon receptor |
| AKT1 | RAC(Rho family)-alpha serine/threonine-protein kinase |
| ELOVL | elongation of very-long-chain fatty acids |
| FLG | Filaggrin |
| GATA3 | GATA binding protein 3 |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| Ig | immunoglobulin |
| IV | ichthyosis vulgaris |
| KC | keratinocyte |
| LEKTI | lympho-epithelial Kazal type inhibitor |
| MAPK | mitogen-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| NMF | natural moisturizing factor |
| OR | odds ratio |
| OVOL1 | ovo-like transcriptional repressor 1 |
| PCA | pyrrolidone carboxylic acid |
| PEP1 | proFLG endoproteinase 1 |
| PPAR | peroxisome proliferator-activated receptor |
| RAPTOR | regulatory-associated protein of mTOR |
| ROS | reactive oxygen species |
| SC | stratum corneum |
| SASPase | skin aspartic acid protease |
| SG | stratum granulosum |
| TEWL | transepidermal water loss |
| TSLP | thymic stromal lymphopoietin |
| UCA | trans-urocanic acid |
| UV | ultraviolet |
References
- Dale, B.A.; Lonsdale-Eccles, J.D.; Holbrook, K.A. Stratum corneum basic protein: An interfilamentous matrix protein of epidermal keratin. Curr. Probl. Dermatol. 1980, 10, 311–325. [Google Scholar] [PubMed]
- Dale, B.A.; Vadlamudi, B.; DeLap, L.W.; Bernstein, I.A. Similarities between stratum corneum basic protein and histidine-rich protein II from newborn rat epidermis. Biochim. Biophys. Acta 1981, 668, 98–106. [Google Scholar] [CrossRef]
- Mack, J.W.; Steven, A.C.; Steinert, P.M. The mechanism of interaction of filaggrin with intermediate filaments. The ionic zipper hypothesis. J. Mol. Biol. 1993, 232, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.A.; Presland, R.B.; Lewis, S.P.; Underwood, R.A.; Fleckman, P. Transient expression of epidermal filaggrin in cultured cells causes collapse of intermediate filament networks with alteration of cell shape and nuclear integrity. J. Investig. Dermatol. 1997, 108, 179–187. [Google Scholar] [CrossRef]
- Pendaries, V.; Malaisse, J.; Pellerin, L.; Le Lamer, M.; Nachat, R.; Kezic, S.; Schmitt, A.M.; Paul, C.; Poumay, Y.; Serre, G.; et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J. Investig. Dermatol. 2014, 134, 2938–2946. [Google Scholar] [CrossRef]
- Kawasaki, H.; Nagao, K.; Kubo, A.; Hata, T.; Shimizu, A.; Mizuno, H.; Yamada, T.; Amagai, M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J. Allergy Clin. Immunol. 2012, 129, 1538–1546. [Google Scholar] [CrossRef]
- Gruber, R.; Elias, P.M.; Crumrine, D.; Lin, T.K.; Brandner, J.M.; Hachem, J.P.; Presland, R.B.; Fleckman, P.; Janecke, A.R.; Sandilands, A.; et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am. J. Pathol. 2011, 178, 2252–2263. [Google Scholar] [CrossRef]
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291. [Google Scholar] [CrossRef]
- Steinert, P.M.; Cantieri, J.S.; Teller, D.C.; Lonsdale-Eccles, J.D.; Dale, B.A. Characterization of a class of cationic proteins that specifically interact with intermediate filaments. Proc. Natl. Acad. Sci. USA 1981, 78, 4097–4101. [Google Scholar] [CrossRef]
- Ramsden, M.; Loehren, D.; Balmain, A. Identification of a rapidly labelled 350K histidine-rich protein in neonatal mouse epidermis. Differentiation 1983, 23, 243–249. [Google Scholar] [CrossRef]
- Lonsdale-Eccles, J.D.; Teller, D.C.; Dale, B.A. Characterization of a phosphorylated form of the intermediate filament-aggregating protein filaggrin. Biochemistry 1982, 21, 5940–5948. [Google Scholar] [CrossRef]
- Meek, R.L.; Lonsdale-Eccles, J.D.; Dale, B.A. Epidermal filaggrin is synthesized on a large messenger ribonucleic acid as a high-molecular-weight precursor. Biochemistry 1983, 22, 4867–4871. [Google Scholar] [CrossRef]
- Lonsdale-Eccles, J.D.; Haugen, J.A.; Dale, B.A. A phosphorylated keratohyalin-derived precursor of epidermal stratum corneum basic protein. J. Biol. Chem. 1980, 255, 2235–2238. [Google Scholar] [CrossRef]
- Kuechle, M.K.; Thulin, C.D.; Presland, R.B.; Dale, B.A. Profilaggrin requires both linker and filaggrin peptide sequences to form granules: Implications for profilaggrin processing in vivo. J. Investig. Dermatol. 1999, 112, 843–852. [Google Scholar] [CrossRef]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef]
- Resing, K.A.; Walsh, K.A.; Dale, B.A. Identification of two intermediates during processing of profilaggrin to filaggrin in neonatal mouse epidermis. J. Cell Biol. 1984, 99, 1372–1378. [Google Scholar] [CrossRef]
- Gan, S.Q.; McBride, O.W.; Idler, W.W.; Markova, N.; Steinert, P.M. Organization, structure, and polymorphisms of the human profilaggrin gene. Biochemistry 1990, 29, 9432–9440. [Google Scholar] [CrossRef]
- Combarros, D.; Cadiergues, M.C.; Simon, M. Update on canine filaggrin: A review. Vet. Q. 2020, 40, 162–168. [Google Scholar] [CrossRef]
- Steinert, P.M.; Marekov, L.N. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J. Biol. Chem. 1995, 270, 17702–17711. [Google Scholar] [CrossRef]
- Manabe, M.; Sanchez, M.; Sun, T.T.; Dale, B.A. Interaction of filaggrin with keratin filaments during advanced stages of normal human epidermal differentiation and in ichthyosis vulgaris. Differentiation 1991, 48, 43–50. [Google Scholar] [CrossRef]
- Simon, M.; Sebbag, M.; Haftek, M.; Vincent, C.; Girbal-Neuhauser, E.; Rakotoarivony, J.; Sommé, G.; Schmitt, D.; Serre, G. Monoclonal antibodies to human epidermal filaggrin, some not recognizing profilaggrin. J. Investig. Dermatol. 1995, 105, 432–437. [Google Scholar] [CrossRef]
- Simon, M.; Haftek, M.; Sebbag, M.; Montézin, M.; Girbal-Neuhauser, E.; Schmitt, D.; Serre, G. Evidence that filaggrin is a component of cornified cell envelopes in human plantar epidermis. Biochem. J. 1996, 317, 173–177. [Google Scholar] [CrossRef]
- Albérola, G.; Schröder, J.M.; Froment, C.; Simon, M. The Amino-Terminal Part of Human FLG2 Is a Component of Cornified Envelopes. J. Investig. Dermatol. 2019, 139, 1395–1397. [Google Scholar] [CrossRef]
- Richards, S.; Scott, I.R.; Harding, C.R.; Liddell, J.E.; Powell, G.M.; Curtis, C.G. Evidence for filaggrin as a component of the cell envelope of the newborn rat. Biochem. J. 1988, 253, 153–160. [Google Scholar] [CrossRef]
- Kam, E.; Resing, K.A.; Lim, S.K.; Dale, B.A. Identification of rat epidermal profilaggrin phosphatase as a member of the protein phosphatase 2A family. J. Cell Sci. 1993, 106, 219–226. [Google Scholar] [CrossRef]
- Haugen-Scofield, J.; Resing, K.A.; Dale, B.A. Characterization of an epidermal phosphatase specific for filaggrin phosphorylated by casein kinase II. J. Investig. Dermatol. 1988, 91, 553–559. [Google Scholar] [CrossRef]
- Resing, K.A.; al-Alawi, N.; Blomquist, C.; Fleckman, P.; Dale, B.A. Independent regulation of two cytoplasmic processing stages of the intermediate filament-associated protein filaggrin and role of Ca2+ in the second stage. J. Biol. Chem. 1993, 268, 25139–25145. [Google Scholar] [CrossRef]
- Kamata, Y.; Taniguchi, A.; Yamamoto, M.; Nomura, J.; Ishihara, K.; Takahara, H.; Hibino, T.; Takeda, A. Neutral cysteine protease bleomycin hydrolase is essential for the breakdown of deiminated filaggrin into amino acids. J. Biol. Chem. 2009, 284, 12829–12836. [Google Scholar] [CrossRef] [PubMed]
- Kawada, A.; Hara, K.; Hiruma, M.; Noguchi, H.; Ishibashi, A. Rat epidermal cathepsin L-like proteinase: Purification and some hydrolytic properties toward filaggrin and synthetic substrates. J. Biochem. 1995, 118, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.S.; Tommasi, C.; Cole, C.; Brown, S.J.; Zhu, Y.; Way, B.; Willis Owen, S.A.; Moffatt, M.; Cookson, W.O.; Harper, J.I.; et al. A mechanistic target of rapamycin complex 1/2 (mTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 139, 1228–1241. [Google Scholar] [PubMed]
- Kashima, M.; Fukuyama, K.; Kikuchi, M.; Epstein, W.L. Limited proteolysis of high molecular weight histidine-rich protein of rat epidermis by epidermal proteinases. J. Investig. Dermatol. 1988, 90, 829–833. [Google Scholar] [CrossRef]
- Egberts, F.; Heinrich, M.; Jensen, J.M.; Winoto-Morbach, S.; Pfeiffer, S.; Wickel, M.; Schunck, M.; Steude, J.; Saftig, P.; Proksch, E.; et al. Cathepsin D is involved in the regulation of transglutaminase 1 and epidermal differentiation. J. Cell Sci. 2004, 117, 2295–2307. [Google Scholar] [CrossRef]
- Resing, K.A.; Thulin, C.; Whiting, K.; al-Alawi, N.; Mostad, S. Characterization of profilaggrin endoproteinase 1. A regulated cytoplasmic endoproteinase of epidermis. J. Biol. Chem. 1995, 270, 28193–28198. [Google Scholar] [CrossRef]
- Pearton, D.J.; Nirunsuksiri, W.; Rehemtulla, A.; Lewis, S.P.; Presland, R.B.; Dale, B.A. Proprotein convertase expression and localization in epidermis: Evidence for multiple roles and substrates. Exp. Dermatol. 2001, 10, 193–203. [Google Scholar] [CrossRef]
- List, K.; Szabo, R.; Wertz, P.W.; Segre, J.; Haudenschild, C.C.; Kim, S.Y.; Bugge, T.H. Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. J. Cell Biol. 2003, 163, 901–910. [Google Scholar] [CrossRef]
- Matsui, T.; Miyamoto, K.; Kubo, A.; Kawasaki, H.; Ebihara, T.; Hata, K.; Tanahashi, S.; Ichinose, S.; Imoto, I.; Inazawa, J.; et al. SASPase regulates stratum corneum hydration through profilaggrin-to-filaggrin processing. EMBO Mol. Med. 2011, 3, 320–333. [Google Scholar] [CrossRef]
- Donovan, M.; Salamito, M.; Thomas-Collignon, A.; Simonetti, L.; Desbouis, S.; Rain, J.C.; Formstecher, E.; Bernard, D. Filaggrin and filaggrin 2 processing are linked together through skin aspartic acid protease activation. PLoS ONE 2020, 15, e0232679. [Google Scholar] [CrossRef]
- Bernard, D.; Méhul, B.; Thomas-Collignon, A.; Delattre, C.; Donovan, M.; Schmidt, R. Identification and characterization of a novel retroviral-like aspartic protease specifically expressed in human epidermis. J. Investig. Dermatol. 2005, 125, 278–287. [Google Scholar] [CrossRef]
- Descargues, P.; Deraison, C.; Bonnart, C.; Kreft, M.; Kishibe, M.; Ishida-Yamamoto, A.; Elias, P.; Barrandon, Y.; Zambruno, G.; Sonnenberg, A.; et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 2005, 37, 56–65. [Google Scholar] [CrossRef]
- Hewett, D.R.; Simons, A.L.; Mangan, N.E.; Jolin, H.E.; Green, S.M.; Fallon, P.G.; McKenzie, A.N. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum. Mol. Genet. 2005, 14, 335–346. [Google Scholar] [CrossRef][Green Version]
- Yamamoto-Tanaka, M.; Makino, T.; Motoyama, A.; Miyai, M.; Tsuboi, R.; Hibino, T. Multiple pathways are involved in DNA degradation during keratinocyte terminal differentiation. Cell Death. Dis. 2014, 5, e1181. [Google Scholar] [CrossRef] [PubMed]
- Bonnart, C.; Deraison, C.; Lacroix, M.; Uchida, Y.; Besson, C.; Robin, A.; Briot, A.; Gonthier, M.; Lamant, L.; Dubus, P.; et al. Elastase 2 is expressed in human and mouse epidermis and impairs skin barrier function in Netherton syndrome through filaggrin and lipid misprocessing. J. Clin. Investig. 2010, 120, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Furio, L.; Hovnanian, A. Netherton syndrome: Defective kallikrein inhibition in the skin leads to skin inflammation and allergy. Biol. Chem. 2014, 395, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Barbieux, C.; Bonnet des Claustres, M.; Fahrner, M.; Petrova, E.; Tsoi, L.C.; Gouin, O.; Leturcq, F.; Nicaise-Roland, P.; Bole, C.; Béziat, V.; et al. Netherton syndrome subtypes share IL-17/IL-36 signature with distinct IFN-α and allergic responses. J. Allergy Clin. Immunol. 2022, 149, 1358–1372. [Google Scholar] [CrossRef] [PubMed]
- Méchin, M.C.; Cau, L.; Galliano, M.F.; Daunes-Marion, S.; Poigny, S.; Vidaluc, J.L.; Bessou-Touya, S.; Takahara, H.; Serre, G.; Duplan, H.; et al. Acefylline activates filaggrin deimination by peptidylarginine deiminases in the upper epidermis. J. Dermatol. Sci. 2016, 81, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Méchin, M.C.; Takahara, H.; Simon, M. Deimination and Peptidylarginine Deiminases in Skin Physiology and Diseases. Int. J. Mol. Sci. 2020, 21, 566. [Google Scholar] [CrossRef]
- Briot, J.; Simon, M.; Méchin, M.C. Deimination, Intermediate Filaments and Associated Proteins. Int J. Mol. Sci 2020, 21, 8746. [Google Scholar] [CrossRef]
- Rawlings, A.V.; Harding, C.R. Moisturization and skin barrier function. Dermatol. Ther. 2004, 17 (Suppl. 1), 43–48. [Google Scholar] [CrossRef]
- Barresi, C.; Stremnitzer, C.; Mlitz, V.; Kezic, S.; Kammeyer, A.; Ghannadan, M.; Posa-Markaryan, K.; Selden, C.; Tschachler, E.; Eckhart, L. Increased sensitivity of histidinemic mice to UVB radiation suggests a crucial role of endogenous urocanic acid in photoprotection. J. Investig. Dermatol. 2011, 131, 188–194. [Google Scholar] [CrossRef]
- Denecker, G.; Hoste, E.; Gilbert, B.; Hochepied, T.; Ovaere, P.; Lippens, S.; Van den Broecke, C.; Van Damme, P.; D’Herde, K.; Hachem, J.P.; et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat. Cell Biol. 2007, 9, 666–674. [Google Scholar] [CrossRef]
- Gibbs, N.K.; Tye, J.; Norval, M. Recent advances in urocanic acid photochemistry, photobiology and photoimmunology. Photochem. Photobiol. Sci. 2008, 7, 655–667. [Google Scholar] [CrossRef]
- Mildner, M.; Jin, J.; Eckhart, L.; Kezic, S.; Gruber, F.; Barresi, C.; Stremnitzer, C.; Buchberger, M.; Mlitz, V.; Ballaun, C.; et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J. Investig. Dermatol. 2010, 130, 2286–2294. [Google Scholar] [CrossRef]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; De Groote, P.; Roelandt, R.; et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J. Investig. Dermatol. 2011, 131, 2233–2241. [Google Scholar] [CrossRef]
- Devos, M.; Prawitt, J.; Staumont-Salle, D.; Hoste, E.; Fleury, S.; Bouchaert, E.; Gilbert, B.; Lippens, S.; Vandenabeele, P.; Dombrowicz, D.; et al. Filaggrin degradation by caspase-14 is required for UVB photoprotection but does not influence allergic sensitization in a mouse model of atopic dermatitis. J. Investig. Dermatol. 2012, 132, 2857–2860. [Google Scholar] [CrossRef]
- Scott, I.R.; Harding, C.R. Filaggrin breakdown to water binding compounds during development of the rat stratum corneum is controlled by the water activity of the environment. Dev. Biol. 1986, 115, 84–92. [Google Scholar] [CrossRef]
- Akiyama, K.; Senshu, T. Dynamic aspects of protein deimination in developing mouse epidermis. Exp. Dermatol. 1999, 8, 177–186. [Google Scholar] [CrossRef]
- Cau, L.; Pendaries, V.; Lhuillier, E.; Thompson, P.R.; Serre, G.; Takahara, H.; Méchin, M.C.; Simon, M. Lowering relative humidity level increases epidermal protein deimination and drives human filaggrin breakdown. J. Dermatol. Sci. 2017, 86, 106–113. [Google Scholar] [CrossRef]
- McAleer, M.A.; Jakasa, I.; Raj, N.; O’Donnell, C.P.F.; Lane, M.E.; Rawlings, A.V.; Voegeli, R.; McLean, W.H.I.; Kezic, S.; Irvine, A.D. Early-life regional and temporal variation in filaggrin-derived natural moisturizing factor, filaggrin-processing enzyme activity, corneocyte phenotypes and plasmin activity: Implications for atopic dermatitis. Br. J. Dermatol. 2018, 179, 431–441. [Google Scholar] [CrossRef]
- Harding, C.R.; Scott, I.R. Histidine-rich proteins (filaggrins): Structural and functional heterogeneity during epidermal differentiation. J. Mol. Biol. 1983, 170, 651–673. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Jakasa, I.; Riethmüller, C.; Schön, M.P.; Braun, A.; Haftek, M.; Fallon, P.G.; Wróblewski, J.; Jakubowski, H.; Eckhart, L.; et al. Filaggrin Expression and Processing Deficiencies Impair Corneocyte Surface Texture and Stiffness in Mice. J. Investig. Dermatol. 2020, 140, 615–623.e5. [Google Scholar] [CrossRef]
- Sybert, V.P.; Dale, B.A.; Holbrook, K.A. Ichthyosis vulgaris: Identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J. Investig. Dermatol. 1985, 84, 191–194. [Google Scholar] [CrossRef]
- Smith, F.J.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar] [CrossRef]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Seguchi, T.; Cui, C.Y.; Kusuda, S.; Takahashi, M.; Aisu, K.; Tezuka, T. Decreased expression of filaggrin in atopic skin. Arch. Dermatol. Res. 1996, 288, 442–446. [Google Scholar] [CrossRef]
- Pellerin, L.; Henry, J.; Hsu, C.Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.M.; Macchia, L.; et al. A Multicenter Study on the Prevalence of Clinical Patterns and Clinical Phenotypes in Adult Atopic Dermatitis. J. Investig. Allergol. Clin. Immunol. 2020, 30, 448–450. [Google Scholar] [CrossRef]
- Morelli, P.; Gaspari, M.; Gabriele, C.; Dastoli, S.; Bennardo, L.; Pavel, A.B.; Patruno, C.; Del Duca, E.; Nisticò, S.P. Proteomic analysis from skin swabs reveals a new set of proteins identifying skin impairment in atopic dermatitis. Exp. Dermatol. 2021, 30, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Deckers, I.A.; McLean, S.; Linssen, S.; Mommers, M.; van Schayck, C.P.; Sheikh, A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: A systematic review of epidemiological studies. PLoS ONE 2012, 7, e39803. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, S.M.; Thomsen, S.F. The prevalence of atopic dermatitis in adults: Systematic review on population studies. Dermatol. Online J. 2019, 25. [Google Scholar] [CrossRef]
- Nomura, T.; Kabashima, K. Advances in atopic dermatitis in 2019-2020: Endotypes from skin barrier, ethnicity, properties of antigen, cytokine profiles, microbiome, and engagement of immune cells. J. Allergy Clin. Immunol. 2021, 148, 1451–1462. [Google Scholar] [CrossRef]
- Esaki, H.; Brunner, P.M.; Renert-Yuval, Y.; Czarnowicki, T.; Huynh, T.; Tran, G.; Lyon, S.; Rodriguez, G.; Immaneni, S.; Johnson, D.B.; et al. Early-onset pediatric atopic dermatitis is T(H)2 but also T(H)17 polarized in skin. J. Allergy Clin. Immunol. 2016, 138, 1639–1651. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Del Duca, E.; Pavel, A.B.; Fang, M.; Lefferdink, R.; Wu, J.; Diaz, A.; Estrada, Y.D.; Canter, T.; Zhang, N.; et al. The molecular features of normal and atopic dermatitis skin in infants, children, adolescents, and adults. J. Allergy Clin. Immunol. 2021, 148, 148–163. [Google Scholar] [CrossRef]
- He, H.; Del Duca, E.; Diaz, A.; Kim, H.J.; Gay-Mimbrera, J.; Zhang, N.; Wu, J.; Beaziz, J.; Estrada, Y.; Krueger, J.G.; et al. Mild atopic dermatitis lacks systemic inflammation and shows reduced nonlesional skin abnormalities. J. Allergy Clin. Immunol. 2021, 147, 1369–1380. [Google Scholar] [CrossRef]
- Scott, I.R.; Harding, C.R.; Barrett, J.G. Histidine-rich protein of the keratohyalin granules. Source of the free amino acids, urocanic acid and pyrrolidone carboxylic acid in the stratum corneum. Biochim. Biophys. Acta 1982, 719, 110–117. [Google Scholar] [CrossRef]
- Sandilands, A.; O’Regan, G.M.; Liao, H.; Zhao, Y.; Terron-Kwiatkowski, A.; Watson, R.M.; Cassidy, A.J.; Goudie, D.R.; Smith, F.J.; McLean, W.H.; et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J. Investig. Dermatol. 2006, 126, 1770–1775. [Google Scholar] [CrossRef]
- Weidinger, S.; Illig, T.; Baurecht, H.; Irvine, A.D.; Rodriguez, E.; Diaz-Lacava, A.; Klopp, N.; Wagenpfeil, S.; Zhao, Y.; Liao, H.; et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J. Allergy Clin. Immunol. 2006, 118, 214–219. [Google Scholar] [CrossRef]
- Marenholz, I.; Nickel, R.; Rüschendorf, F.; Schulz, F.; Esparza-Gordillo, J.; Kerscher, T.; Grüber, C.; Lau, S.; Worm, M.; Keil, T.; et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J. Allergy Clin. Immunol. 2006, 118, 866–871. [Google Scholar] [CrossRef]
- Ruether, A.; Stoll, M.; Schwarz, T.; Schreiber, S.; Fölster-Holst, R. Filaggrin loss-of-function variant contributes to atopic dermatitis risk in the population of Northern Germany. Br. J. Dermatol. 2006, 155, 1093–1094. [Google Scholar] [CrossRef]
- Nomura, T.; Sandilands, A.; Akiyama, M.; Liao, H.; Evans, A.T.; Sakai, K.; Ota, M.; Sugiura, H.; Yamamoto, K.; Sato, H.; et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J. Allergy Clin. Immunol. 2007, 119, 434–440. [Google Scholar] [CrossRef]
- Sandilands, A.; Terron-Kwiatkowski, A.; Hull, P.R.; O’Regan, G.M.; Clayton, T.H.; Watson, R.M.; Carrick, T.; Evans, A.T.; Liao, H.; Zhao, Y.; et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat. Genet. 2007, 39, 650–654. [Google Scholar] [CrossRef]
- Baurecht, H.; Irvine, A.D.; Novak, N.; Illig, T.; Bühler, B.; Ring, J.; Wagenpfeil, S.; Weidinger, S. Toward a major risk factor for atopic eczema: Meta-analysis of filaggrin polymorphism data. J. Allergy Clin. Immunol. 2007, 120, 1406–1412. [Google Scholar] [CrossRef]
- Ekelund, E.; Liedén, A.; Link, J.; Lee, S.P.; D’Amato, M.; Palmer, C.N.; Kockum, I.; Bradley, M. Loss-of-function variants of the filaggrin gene are associated with atopic eczema and associated phenotypes in Swedish families. Acta Derm. Venereol. 2008, 88, 15–19. [Google Scholar] [CrossRef]
- Sasaki, T.; Kudoh, J.; Ebihara, T.; Shiohama, A.; Asakawa, S.; Shimizu, A.; Takayanagi, A.; Dekio, I.; Sadahira, C.; Amagai, M.; et al. Sequence analysis of filaggrin gene by novel shotgun method in Japanese atopic dermatitis. J. Dermatol. Sci. 2008, 51, 113–120. [Google Scholar] [CrossRef]
- Kang, T.W.; Lee, J.S.; Oh, S.W.; Kim, S.C. Filaggrin mutation c.3321delA in a Korean patient with ichthyosis vulgaris and atopic dermatitis. Dermatology 2009, 218, 186–187. [Google Scholar] [CrossRef]
- Müller, S.; Marenholz, I.; Lee, Y.A.; Sengler, C.; Zitnik, S.E.; Griffioen, R.W.; Meglio, P.; Wahn, U.; Nickel, R. Association of Filaggrin loss-of-function-mutations with atopic dermatitis and asthma in the Early Treatment of the Atopic Child (ETAC) population. Pediatr. Allergy Immunol. 2009, 20, 358–361. [Google Scholar] [CrossRef]
- Ching, G.K.; Hon, K.L.; Ng, P.C.; Leung, T.F. Filaggrin null mutations in childhood atopic dermatitis among the Chinese. Int J. Immunogenet. 2009, 36, 251–254. [Google Scholar] [CrossRef]
- Brown, S.J.; Relton, C.L.; Liao, H.; Zhao, Y.; Sandilands, A.; McLean, W.H.; Cordell, H.J.; Reynolds, N.J. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: Further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. Br. J. Dermatol. 2009, 161, 884–889. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, L.; Di, Z.H.; Zhao, L.P.; Lu, Y.N.; Xu, J.; Chen, H.D.; Gao, X.H. Association analysis of filaggrin gene mutations and atopic dermatitis in Northern China. Br. J. Dermatol. 2010, 162, 225–227. [Google Scholar] [CrossRef]
- Greisenegger, E.; Novak, N.; Maintz, L.; Bieber, T.; Zimprich, F.; Haubenberger, D.; Gleiss, A.; Stingl, G.; Kopp, T.; Zimprich, A. Analysis of four prevalent filaggrin mutations (R501X, 2282del4, R2447X and S3247X) in Austrian and German patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 607–610. [Google Scholar] [CrossRef]
- Ponińska, J.; Samoliński, B.; Tomaszewska, A.; Raciborski, F.; Samel-Kowalik, P.; Walkiewicz, A.; Lipiec, A.; Piekarska, B.; Komorowski, J.; Krzych-Fałta, E.; et al. Filaggrin gene defects are independent risk factors for atopic asthma in a Polish population: A study in ECAP cohort. PLoS ONE 2011, 6, e16933. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Common, J.E.; Haines, R.L.; Balakrishnan, A.; Brown, S.J.; Goh, C.S.; Cordell, H.J.; Sandilands, A.; Campbell, L.E.; Kroboth, K.; et al. Wide spectrum of filaggrin-null mutations in atopic dermatitis highlights differences between Singaporean Chinese and European populations. Br. J. Dermatol. 2011, 165, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Apter, A.J.; Gupta, J.; Hoffstad, O.; Papadopoulos, M.; Campbell, L.E.; Sandilands, A.; McLean, W.H.; Rebbeck, T.R.; Mitra, N. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J. Allergy Clin. Immunol. 2012, 130, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, M.S.; Ramamoorthy, P.; Reynolds, P.R.; Curran-Everett, D.; Leung, D.Y. Increased compound heterozygous filaggrin mutations in severe atopic dermatitis in the United States. J. Allergy Clin. Immunol. Pract. 2013, 1, 534–536. [Google Scholar] [CrossRef]
- Margolis, D.J.; Gupta, J.; Apter, A.J.; Hoffstad, O.; Papadopoulos, M.; Rebbeck, T.R.; Wubbenhorst, B.; Mitra, N. Exome sequencing of filaggrin and related genes in African-American children with atopic dermatitis. J. Investig. Dermatol. 2014, 134, 2272–2274. [Google Scholar] [CrossRef]
- Polcari, I.; Becker, L.; Stein, S.L.; Smith, M.S.; Paller, A.S. Filaggrin gene mutations in African Americans with both ichthyosis vulgaris and atopic dermatitis. Pediatr. Dermatol. 2014, 31, 489–492. [Google Scholar] [CrossRef]
- Sasaki, T.; Furusyo, N.; Shiohama, A.; Takeuchi, S.; Nakahara, T.; Uchi, H.; Hirota, T.; Tamari, M.; Shimizu, N.; Ebihara, T.; et al. Filaggrin loss-of-function mutations are not a predisposing factor for atopic dermatitis in an Ishigaki Island under subtropical climate. J. Dermatol. Sci. 2014, 76, 10–15. [Google Scholar] [CrossRef]
- Komova, E.G.; Shintyapina, A.B.; Makarova, S.I.; Ivanov, M.K.; Chekryga, E.A.; Kaznacheeva, L.F.; Vavilin, V.A. Filaggrin mutations in a Western siberian population and their association with atopic dermatitis in children. Genet. Test. Mol. Biomark. 2014, 18, 791–796. [Google Scholar] [CrossRef]
- Park, J.; Jekarl, D.W.; Kim, Y.; Kim, J.; Kim, M.; Park, Y.M. Novel FLG null mutations in Korean patients with atopic dermatitis and comparison of the mutational spectra in Asian populations. J. Dermatol. 2015, 42, 867–873. [Google Scholar] [CrossRef]
- Trisnowati, N.; Soebono, H.; Sadewa, A.H.; Kunisada, M.; Yogianti, F.; Nishigori, C. A novel filaggrin gene mutation 7487delC in an Indonesian (Javanese) patient with atopic dermatitis. Int. J. Dermatol. 2016, 55, 695–697. [Google Scholar] [CrossRef]
- Margolis, D.J.; Mitra, N.; Wubbenhorst, B.; D’Andrea, K.; Kraya, A.A.; Hoffstad, O.; Shah, S.; Nathanson, K.L. Association of Filaggrin Loss-of-Function Variants With Race in Children With Atopic Dermatitis. JAMA Dermatol. 2019, 155, 1269–1276. [Google Scholar] [CrossRef]
- González-Tarancón, R.; Sanmartín, R.; Lorente, F.; Salvador-Rupérez, E.; Hernández-Martín, A.; Rello, L.; Puzo, J.; Gilaberte, Y. Prevalence of FLG loss-of-function mutations R501X, 2282del4, and R2447X in Spanish children with atopic dermatitis. Pediatr. Dermatol. 2020, 37, 98–102. [Google Scholar] [CrossRef]
- Brown, S.J.; Sandilands, A.; Zhao, Y.; Liao, H.; Relton, C.L.; Meggitt, S.J.; Trembath, R.C.; Barker, J.N.; Reynolds, N.J.; Cordell, H.J.; et al. Prevalent and low-frequency null mutations in the filaggrin gene are associated with early-onset and persistent atopic eczema. J. Investig. Dermatol. 2008, 128, 1591–1594. [Google Scholar] [CrossRef]
- Brown, S.J.; Relton, C.L.; Liao, H.; Zhao, Y.; Sandilands, A.; Wilson, I.J.; Burn, J.; Reynolds, N.J.; McLean, W.H.; Cordell, H.J. Filaggrin null mutations and childhood atopic eczema: A population-based case-control study. J. Allergy Clin. Immunol. 2008, 121, 940–946. [Google Scholar] [CrossRef]
- Gruber, R.; Janecke, A.R.; Grabher, D.; Horak, E.; Schmuth, M.; Lercher, P. Lower prevalence of common filaggrin mutations in a community sample of atopic eczema: Is disease severity important? Wien. Klin. Wochenschr. 2010, 122, 551–557. [Google Scholar] [CrossRef]
- Morar, N.; Cookson, W.O.; Harper, J.I.; Moffatt, M.F. Filaggrin mutations in children with severe atopic dermatitis. J. Investig. Dermatol. 2007, 127, 1667–1672. [Google Scholar] [CrossRef]
- Henderson, J.; Northstone, K.; Lee, S.P.; Liao, H.; Zhao, Y.; Pembrey, M.; Mukhopadhyay, S.; Smith, G.D.; Palmer, C.N.; McLean, W.H.; et al. The burden of disease associated with filaggrin mutations: A population-based, longitudinal birth cohort study. J. Allergy Clin. Immunol. 2008, 121, 872–877. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Carlsen, B.C.; Bisgaard, H.; Giwercman, C.; Johansen, J.D.; Linneberg, A.; Meldgaard, M.; Szecsi, P.B.; Stender, S.; Menné, T. Individuals who are homozygous for the 2282del4 and R501X filaggrin null mutations do not always develop dermatitis and complete long-term remission is possible. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 386–389. [Google Scholar] [CrossRef]
- Giardina, E.; Paolillo, N.; Sinibaldi, C.; Novelli, G. R501X and 2282del4 filaggrin mutations do not confer susceptibility to psoriasis and atopic dermatitis in Italian patients. Dermatology 2008, 216, 83–84. [Google Scholar] [CrossRef]
- Cascella, R.; Foti Cuzzola, V.; Lepre, T.; Galli, E.; Moschese, V.; Chini, L.; Mazzanti, C.; Fortugno, P.; Novelli, G.; Giardina, E. Full sequencing of the FLG gene in Italian patients with atopic eczema: Evidence of new mutations, but lack of an association. J. Investig. Dermatol. 2011, 131, 982–984. [Google Scholar] [CrossRef]
- Nomura, T.; Akiyama, M.; Sandilands, A.; Nemoto-Hasebe, I.; Sakai, K.; Nagasaki, A.; Ota, M.; Hata, H.; Evans, A.T.; Palmer, C.N.; et al. Specific filaggrin mutations cause ichthyosis vulgaris and are significantly associated with atopic dermatitis in Japan. J. Investig. Dermatol. 2008, 128, 1436–1441. [Google Scholar] [CrossRef]
- Brown, S.J.; Kroboth, K.; Sandilands, A.; Campbell, L.E.; Pohler, E.; Kezic, S.; Cordell, H.J.; McLean, W.H.; Irvine, A.D. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J. Investig. Dermatol. 2012, 132, 98–104. [Google Scholar] [CrossRef]
- Barker, J.N.; Palmer, C.N.; Zhao, Y.; Liao, H.; Hull, P.R.; Lee, S.P.; Allen, M.H.; Meggitt, S.J.; Reynolds, N.J.; Trembath, R.C.; et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J. Investig. Dermatol. 2007, 127, 564–567. [Google Scholar] [CrossRef]
- Rice, N.E.; Patel, B.D.; Lang, I.A.; Kumari, M.; Frayling, T.M.; Murray, A.; Melzer, D. Filaggrin gene mutations are associated with asthma and eczema in later life. J. Allergy Clin. Immunol. 2008, 122, 834–836. [Google Scholar] [CrossRef]
- Tokura, Y.; Hayano, S. Subtypes of atopic dermatitis: From phenotype to endotype. Allergol. Int. 2021. [Google Scholar] [CrossRef]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; Debenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin barrier in atopic dermatitis: Beyond filaggrin. An. Bras. Dermatol. 2016, 91, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Dajnoki, Z.; Béke, G.; Mócsai, G.; Kapitány, A.; Gáspár, K.; Hajdu, K.; Emri, G.; Nagy, B.; Kovács, I.; Beke, L.; et al. Immune-mediated Skin Inflammation is Similar in Severe Atopic Dermatitis Patients With or Without Filaggrin Mutation. Acta Derm. Venereol. 2016, 96, 645–650. [Google Scholar] [CrossRef]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Hvid, M.; Johansen, C.; Deleuran, B.; Kemp, K.; Deleuran, M.; Vestergaard, C. Regulation of caspase 14 expression in keratinocytes by inflammatory cytokines--a possible link between reduced skin barrier function and inflammation? Exp. Dermatol. 2011, 20, 633–636. [Google Scholar] [CrossRef]
- Kim, J.H.; Bae, H.C.; Ko, N.Y.; Lee, S.H.; Jeong, S.H.; Lee, H.; Ryu, W.I.; Kye, Y.C.; Son, S.W. Thymic stromal lymphopoietin downregulates filaggrin expression by signal transducer and activator of transcription 3 (STAT3) and extracellular signal-regulated kinase (ERK) phosphorylation in keratinocytes. J. Allergy Clin. Immunol. 2015, 136, 205–208. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol. 2012, 21, 104–110. [Google Scholar] [CrossRef]
- Cornelissen, C.; Marquardt, Y.; Czaja, K.; Wenzel, J.; Frank, J.; Lüscher-Firzlaff, J.; Lüscher, B.; Baron, J.M. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol. 2012, 129, 426–433. [Google Scholar] [CrossRef]
- van Drongelen, V.; Haisma, E.M.; Out-Luiting, J.J.; Nibbering, P.H.; El Ghalbzouri, A. Reduced filaggrin expression is accompanied by increased Staphylococcus aureus colonization of epidermal skin models. Clin. Exp. Allergy 2014, 44, 1515–1524. [Google Scholar] [CrossRef]
- Seltmann, J.; Roesner, L.M.; von Hesler, F.W.; Wittmann, M.; Werfel, T. IL-33 impacts on the skin barrier by downregulating the expression of filaggrin. J. Allergy Clin. Immunol. 2015, 135, 1659–1661. [Google Scholar] [CrossRef]
- Vakirlis, E.; Lazaridou, E.; Tzellos, T.G.; Gerou, S.; Chatzidimitriou, D.; Ioannides, D. Investigation of cytokine levels and their association with SCORAD index in adults with acute atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 409–416. [Google Scholar] [CrossRef]
- Portugal-Cohen, M.; Horev, L.; Ruffer, C.; Schlippe, G.; Voss, W.; Ma’or, Z.; Oron, M.; Soroka, Y.; Frušić-Zlotkin, M.; Milner, Y.; et al. Non-invasive skin biomarkers quantification of psoriasis and atopic dermatitis: Cytokines, antioxidants and psoriatic skin auto-fluorescence. Biomed. Pharmacother. 2012, 66, 293–299. [Google Scholar] [CrossRef]
- Bitton, A.; Avlas, S.; Reichman, H.; Itan, M.; Karo-Atar, D.; Azouz, N.P.; Rozenberg, P.; Diesendruck, Y.; Nahary, L.; Rothenberg, M.E.; et al. A key role for IL-13 signaling via the type 2 IL-4 receptor in experimental atopic dermatitis. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Möbus, L.; Rodriguez, E.; Harder, I.; Stölzl, D.; Boraczynski, N.; Gerdes, S.; Kleinheinz, A.; Abraham, S.; Heratizadeh, A.; Handrick, C.; et al. Atopic dermatitis displays stable and dynamic skin transcriptome signatures. J. Allergy Clin. Immunol. 2021, 147, 213–223. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef]
- Okamoto, R.; Arikawa, J.; Ishibashi, M.; Kawashima, M.; Takagi, Y.; Imokawa, G. Sphingosylphosphorylcholine is upregulated in the stratum corneum of patients with atopic dermatitis. J. Lipid. Res. 2003, 44, 93–102. [Google Scholar] [CrossRef]
- Choi, H.; Kim, S.; Kim, H.J.; Kim, K.M.; Lee, C.H.; Shin, J.H.; Noh, M. Sphingosylphosphorylcholine down-regulates filaggrin gene transcription through NOX5-based NADPH oxidase and cyclooxygenase-2 in human keratinocytes. Biochem. Pharmacol. 2010, 80, 95–103. [Google Scholar] [CrossRef]
- Ming, M.; Zhao, B.; Shea, C.R.; Shah, P.; Qiang, L.; White, S.R.; Sims, D.M.; He, Y.Y. Loss of sirtuin 1 (SIRT1) disrupts skin barrier integrity and sensitizes mice to epicutaneous allergen challenge. J. Allergy Clin. Immunol. 2015, 135, 936–945. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; de La Serna, J.B.; Fritzsche, M.; Naeem, A.; Podobas, E.I.; Leeming, M.; Colin-York, H.; O’Shaughnessy, R.; Eggeling, C.; Ogg, G.S. Orchestrated control of filaggrin-actin scaffolds underpins cornification. Cell Death Dis. 2018, 9, 412. [Google Scholar] [CrossRef]
- Ziyab, A.H.; Karmaus, W.; Holloway, J.W.; Zhang, H.; Ewart, S.; Arshad, S.H. DNA methylation of the filaggrin gene adds to the risk of eczema associated with loss-of-function variants. J. Eur. Acad. Dermatol. Venereol. 2013, 27, e420–e423. [Google Scholar] [CrossRef]
- Lee, J.; Jang, A.; Seo, S.J.; Myung, S.C. Epigenetic Regulation of Filaggrin Gene Expression in Human Epidermal Keratinocytes. Ann. Dermatol. 2020, 32, 122–129. [Google Scholar] [CrossRef]
- Fortugno, P.; Furio, L.; Teson, M.; Berretti, M.; El Hachem, M.; Zambruno, G.; Hovnanian, A.; D’Alessio, M. The 420K LEKTI variant alters LEKTI proteolytic activation and results in protease deregulation: Implications for atopic dermatitis. Hum. Mol. Genet. 2012, 21, 4187–4200. [Google Scholar] [CrossRef]
- Tan, S.P.; Abdul-Ghaffar, S.; Weller, R.B.; Brown, S.B. Protease-antiprotease imbalance may be linked to potential defects in profilaggrin proteolysis in atopic dermatitis. Br. J. Dermatol. 2012, 166, 1137–1140. [Google Scholar] [CrossRef]
- Pellerin, L.; Paul, C.; Schmitt, A.M.; Serre, G.; Simon, M. Bleomycin hydrolase downregulation in lesional skin of adult atopic dermatitis patients is independent of FLG gene mutations. J. Allergy Clin. Immunol. 2014, 134, 1459–1461.e7. [Google Scholar] [CrossRef]
- Kamata, Y.; Yamamoto, M.; Kawakami, F.; Tsuboi, R.; Takeda, A.; Ishihara, K.; Hibino, T. Bleomycin hydrolase is regulated biphasically in a differentiation- and cytokine-dependent manner: Relevance to atopic dermatitis. J. Biol. Chem. 2011, 286, 8204–8212. [Google Scholar] [CrossRef] [PubMed]
- Winge, M.C.; Hoppe, T.; Berne, B.; Vahlquist, A.; Nordenskjöld, M.; Bradley, M.; Törmä, H. Filaggrin genotype determines functional and molecular alterations in skin of patients with atopic dermatitis and ichthyosis vulgaris. PLoS ONE 2011, 6, e28254. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Willenborg, S.; Bloch, W.; Wickström, S.A.; Wagle, P.; Brodesser, S.; Roers, A.; Jais, A.; Brüning, J.C.; Hall, M.N.; et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J. Allergy Clin. Immunol. 2020, 145, 283–300.e8. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Albanesi, C.; Madonna, S. Recent Updates on the Involvement of PI3K/AKT/mTOR Molecular Cascade in the Pathogenesis of Hyperproliferative Skin Disorders. Front. Med. 2021, 8, 665647. [Google Scholar] [CrossRef]
- Fanton, N.; Santoro, D.; Cornegliani, L.; Marsella, R. Increased filaggrin-metabolizing enzyme activity in atopic skin: A pilot study using a canine model of atopic dermatitis. Vet. Dermatol. 2017, 28, 479-e111. [Google Scholar] [CrossRef]
- Di, Z.H.; Ma, L.; Qi, R.Q.; Sun, X.D.; Huo, W.; Zhang, L.; Lyu, Y.N.; Hong, Y.X.; Chen, H.D.; Gao, X.H. T Helper 1 and T Helper 2 Cytokines Differentially Modulate Expression of Filaggrin and its Processing Proteases in Human Keratinocytes. Chin. Med. J. 2016, 129, 295–303. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kamata, Y.; Iida, T.; Fukushima, H.; Nomura, J.; Saito, M.; Tajima, M.; Okubo, Y.; Momoi, T.; Tsuboi, R.; et al. Quantification of activated and total caspase-14 with newly developed ELISA systems in normal and atopic skin. J. Dermatol. Sci. 2011, 61, 110–117. [Google Scholar] [CrossRef]
- Klapan, K.; Frangež, Ž.; Markov, N.; Yousefi, S.; Simon, D.; Simon, H.U. Evidence for Lysosomal Dysfunction within the Epidermis in Psoriasis and Atopic Dermatitis. J. Investig. Dermatol. 2021, 141, 2838–2848. [Google Scholar] [CrossRef]
- Scharschmidt, T.C.; Man, M.Q.; Hatano, Y.; Crumrine, D.; Gunathilake, R.; Sundberg, J.P.; Silva, K.A.; Mauro, T.M.; Hupe, M.; Cho, S.; et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J. Allergy Clin. Immunol. 2009, 124, 496–506.e1-6. [Google Scholar] [CrossRef]
- Fallon, P.G.; Sasaki, T.; Sandilands, A.; Campbell, L.E.; Saunders, S.P.; Mangan, N.E.; Callanan, J.J.; Kawasaki, H.; Shiohama, A.; Kubo, A.; et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet. 2009, 41, 602–608. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; Murphy, G.F.; Geha, R.S. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J. Allergy Clin. Immunol. 2009, 124, 485–493.e1. [Google Scholar] [CrossRef]
- Blunder, S.; Rühl, R.; Moosbrugger-Martinz, V.; Krimmel, C.; Geisler, A.; Zhu, H.; Crumrine, D.; Elias, P.M.; Gruber, R.; Schmuth, M.; et al. Alterations in Epidermal Eicosanoid Metabolism Contribute to Inflammation and Impaired Late Differentiation in FLG-Mutated Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 706–715. [Google Scholar] [CrossRef]
- Elias, M.S.; Long, H.A.; Newman, C.F.; Wilson, P.A.; West, A.; McGill, P.J.; Wu, K.C.; Donaldson, M.J.; Reynolds, N.J. Proteomic analysis of filaggrin deficiency identifies molecular signatures characteristic of atopic eczema. J. Allergy Clin. Immunol. 2017, 140, 1299–1309. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, J.J.; Gutowska-Owsiak, D.; Salimi, M.; Selvakumar, T.A.; Gwela, A.; Chen, L.Y.; Wang, Y.J.; Giannoulatou, E.; Ogg, G. Deficiency of filaggrin regulates endogenous cysteine protease activity, leading to impaired skin barrier function. Clin. Exp. Dermatol. 2017, 42, 622–631. [Google Scholar] [CrossRef]
- Igawa, S.; Kishibe, M.; Minami-Hori, M.; Honma, M.; Tsujimura, H.; Ishikawa, J.; Fujimura, T.; Murakami, M.; Ishida-Yamamoto, A. Incomplete KLK7 Secretion and Upregulated LEKTI Expression Underlie Hyperkeratotic Stratum Corneum in Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 449–456. [Google Scholar] [CrossRef]
- Jakasa, I.; Koster, E.S.; Calkoen, F.; McLean, W.H.; Campbell, L.E.; Bos, J.D.; Verberk, M.M.; Kezic, S. Skin barrier function in healthy subjects and patients with atopic dermatitis in relation to filaggrin loss-of-function mutations. J. Investig. Dermatol. 2011, 131, 540–542. [Google Scholar] [CrossRef]
- van Drongelen, V.; Alloul-Ramdhani, M.; Danso, M.O.; Mieremet, A.; Mulder, A.; van Smeden, J.; Bouwstra, J.A.; El Ghalbzouri, A. Knock-down of filaggrin does not affect lipid organization and composition in stratum corneum of reconstructed human skin equivalents. Exp. Dermatol. 2013, 22, 807–812. [Google Scholar] [CrossRef]
- van den Bogaard, E.; Ilic, D.; Dubrac, S.; Tomic-Canic, M.; Bouwstra, J.; Celli, A.; Mauro, T. Perspective and Consensus Opinion: Good Practices for Using Organotypic Skin and Epidermal Equivalents in Experimental Dermatology Research. J. Investig. Dermatol. 2021, 141, 203–205. [Google Scholar] [CrossRef]
- Pavel, P.; Leman, G.; Hermann, M.; Ploner, C.; Eichmann, T.O.; Minzaghi, D.; Radner, F.P.W.; Del Frari, B.; Gruber, R.; Dubrac, S. Peroxisomal Fatty Acid Oxidation and Glycolysis Are Triggered in Mouse Models of Lesional Atopic Dermatitis. JID Innov. 2021, 1, 100033. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kubo, A.; Kawasaki, H.; Yoshida, K.; Ishii, K.; Furuse, M.; Amagai, M. Epidermal tight junction barrier function is altered by skin inflammation, but not by filaggrin-deficient stratum corneum. J. Dermatol. Sci. 2015, 77, 28–36. [Google Scholar] [CrossRef]
- Leman, G.; Moosbrugger-Martinz, V.; Blunder, S.; Pavel, P.; Dubrac, S. 3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris. Cells 2019, 8, 489. [Google Scholar] [CrossRef]
- Angelova-Fischer, I.; Mannheimer, A.C.; Hinder, A.; Ruether, A.; Franke, A.; Neubert, R.H.; Fischer, T.W.; Zillikens, D. Distinct barrier integrity phenotypes in filaggrin-related atopic eczema following sequential tape stripping and lipid profiling. Exp. Dermatol. 2011, 20, 351–356. [Google Scholar] [CrossRef]
- Bandier, J.; Carlsen, B.C.; Rasmussen, M.A.; Petersen, L.J.; Johansen, J.D. Skin reaction and regeneration after single sodium lauryl sulfate exposure stratified by filaggrin genotype and atopic dermatitis phenotype. Br. J. Dermatol. 2015, 172, 1519–1529. [Google Scholar] [CrossRef]
- Jung, T.; Stingl, G. Atopic dermatitis: Therapeutic concepts evolving from new pathophysiologic insights. J. Allergy Clin. Immunol. 2008, 122, 1074–1081. [Google Scholar] [CrossRef]
- Smieszek, S.P.; Welsh, S.; Xiao, C.; Wang, J.; Polymeropoulos, C.; Birznieks, G.; Polymeropoulos, M.H. Correlation of age-of-onset of Atopic Dermatitis with Filaggrin loss-of-function variant status. Sci. Rep. 2020, 10, 2721. [Google Scholar] [CrossRef]
- Nemoto-Hasebe, I.; Akiyama, M.; Nomura, T.; Sandilands, A.; McLean, W.H.; Shimizu, H. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J. Investig. Dermatol. 2009, 129, 682–689. [Google Scholar] [CrossRef]
- Jungersted, J.M.; Scheer, H.; Mempel, M.; Baurecht, H.; Cifuentes, L.; Høgh, J.K.; Hellgren, L.I.; Jemec, G.B.; Agner, T.; Weidinger, S. Stratum corneum lipids, skin barrier function and filaggrin mutations in patients with atopic eczema. Allergy 2010, 65, 911–918. [Google Scholar] [CrossRef]
- O’Regan, G.M.; Kemperman, P.M.; Sandilands, A.; Chen, H.; Campbell, L.E.; Kroboth, K.; Watson, R.; Rowland, M.; Puppels, G.J.; McLean, W.H.; et al. Raman profiles of the stratum corneum define 3 filaggrin genotype-determined atopic dermatitis endophenotypes. J. Allergy Clin. Immunol. 2010, 126), 574–580. [Google Scholar] [CrossRef] [PubMed]
- Retraction notice. J. Allergy Clin. Immunol. 2021, 147, 1526. [CrossRef]
- Perusquía-Ortiz, A.M.; Oji, V.; Sauerland, M.C.; Tarinski, T.; Zaraeva, I.; Seller, N.; Metze, D.; Aufenvenne, K.; Hausser, I.; Traupe, H. Complete filaggrin deficiency in ichthyosis vulgaris is associated with only moderate changes in epidermal permeability barrier function profile. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Bouwstra, J.A.; Ponec, M. The skin barrier in healthy and diseased state. Biochim. Biophys. Acta 2006, 1758, 2080–2095. [Google Scholar] [CrossRef] [PubMed]
- Toncic, R.J.; Jakasa, I.; Hadzavdic, S.L.; Goorden, S.M.; Vlugt, K.J.G.; Stet, F.S.; Balic, A.; Petkovic, M.; Pavicic, B.; Zuzul, K.; et al. Altered Levels of Sphingosine, Sphinganine and Their Ceramides in Atopic Dermatitis Are Related to Skin Barrier Function, Disease Severity and Local Cytokine Milieu. Int. J. Mol. Sci. 2020, 21, 1958. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jia, Y.; Cheng, Z.W.; Gao, Y.; Zhang, G.L.; Li, J.Y.; He, C.F. Advancements in the maintenance of skin barrier/skin lipid composition and the involvement of metabolic enzymes. J. Cosmet. Dermatol. 2016, 15, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Hatano, Y.; Zhang, W.; Fujiwara, S.; Nishiyori, R. Knockdown of either filaggrin or loricrin increases the productions of interleukin (IL)-1α, IL-8, IL-18 and granulocyte macrophage colony-stimulating factor in stratified human keratinocytes. J. Dermatol. Sci. 2015, 80), 158–160. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Meng, X.; Dang, N. Knock-down of filaggrin influences the mitogen-activated protein kinases signaling pathway in normal human epidermal keratinocytes. Med. Sci. 2018, 34, 94–98. [Google Scholar] [CrossRef]
- Elias, M.S.; Wright, S.C.; Nicholson, W.V.; Morrison, K.D.; Prescott, A.R.; Ten Have, S.; Whitfield, P.D.; Lamond, A.I.; Brown, S.J. Functional and proteomic analysis of a full thickness filaggrin-deficient skin organoid model. Wellcome Open. Res. 2019, 4, 134. [Google Scholar] [CrossRef]
- Moosbrugger-Martinz, V.; Schmuth, M.; Dubrac, S. A Mouse Model for Atopic Dermatitis Using Topical Application of Vitamin D3 or of Its Analog MC903. Methods Mol. Biol. 2017, 1559, 91–106. [Google Scholar]
- Dubrac, S.; Schmuth, M.; Ebner, S. Atopic dermatitis: The role of Langerhans cells in disease pathogenesis. Immunol. Cell Biol. 2010, 88, 400–409. [Google Scholar] [CrossRef]
- Elentner, A.; Finke, D.; Schmuth, M.; Chappaz, S.; Ebner, S.; Malissen, B.; Kissenpfennig, A.; Romani, N.; Dubrac, S. Langerhans cells are critical in the development of atopic dermatitis-like inflammation and symptoms in mice. J. Cell Mol. Med. 2009, 13, 2658–2672. [Google Scholar] [CrossRef]
- Marschall, P.; Wei, R.; Segaud, J.; Yao, W.; Hener, P.; German, B.F.; Meyer, P.; Hugel, C.; Ada Da Silva, G.; Braun, R.; et al. Dual function of Langerhans cells in skin TSLP-promoted T(FH) differentiation in mouse atopic dermatitis. J. Allergy Clin. Immunol. 2021, 147, 1778–1794. [Google Scholar] [CrossRef]
- Li, M.; Hener, P.; Zhang, Z.; Kato, S.; Metzger, D.; Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 2006, 103, 11736–11741. [Google Scholar] [CrossRef]
- Wallmeyer, L.; Dietert, K.; Sochorová, M.; Gruber, A.D.; Kleuser, B.; Vávrová, K.; Hedtrich, S. TSLP is a direct trigger for T cell migration in filaggrin-deficient skin equivalents. Sci. Rep. 2017, 7, 774. [Google Scholar] [CrossRef]
- Saunders, S.P.; Moran, T.; Floudas, A.; Wurlod, F.; Kaszlikowska, A.; Salimi, M.; Quinn, E.M.; Oliphant, C.J.; Núñez, G.; McManus, R.; et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J. Allergy Clin. Immunol. 2016, 137, 482–491. [Google Scholar] [CrossRef]
- Lee, K.H.; Cho, K.A.; Kim, J.Y.; Kim, J.Y.; Baek, J.H.; Woo, S.Y.; Kim, J.W. Filaggrin knockdown and Toll-like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp. Dermatol. 2011, 20, 149–151. [Google Scholar] [CrossRef]
- Leitch, C.S.; Natafji, E.; Yu, C.; Abdul-Ghaffar, S.; Madarasingha, N.; Venables, Z.C.; Chu, R.; Fitch, P.M.; Muinonen-Martin, A.J.; Campbell, L.E.; et al. Filaggrin-null mutations are associated with increased maturation markers on Langerhans cells. J. Allergy Clin. Immunol. 2016, 138, 482–490. [Google Scholar] [CrossRef]
- Saunders, S.P.; Goh, C.S.; Brown, S.J.; Palmer, C.N.; Porter, R.M.; Cole, C.; Campbell, L.E.; Gierlinski, M.; Barton, G.J.; Schneider, G.; et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J. Allergy Clin. Immunol. 2013, 132, 1121–1129. [Google Scholar] [CrossRef]
- Malik, K.; He, H.; Huynh, T.N.; Tran, G.; Mueller, K.; Doytcheva, K.; Renert-Yuval, Y.; Czarnowicki, T.; Magidi, S.; Chou, M.; et al. Ichthyosis molecular fingerprinting shows profound T(H)17 skewing and a unique barrier genomic signature. J. Allergy Clin. Immunol. 2019, 143, 604–618. [Google Scholar] [CrossRef]
- Moosbrugger-Martinz, V.; Gruber, R.; Ladstätter, K.; Bellutti, M.; Blunder, S.; Schmuth, M.; Dubrac, S. Filaggrin null mutations are associated with altered circulating Tregs in atopic dermatitis. J. Cell Mol. Med. 2019, 23, 1288–1299. [Google Scholar] [CrossRef]
- Bonefeld, C.M.; Petersen, T.H.; Bandier, J.; Agerbeck, C.; Linneberg, A.; Ross-Hansen, K.; Stender, S.; Szecsi, P.B.; Johansen, J.D.; Geisler, C.; et al. Epidermal filaggrin deficiency mediates increased systemic T-helper 17 immune response. Br. J. Dermatol. 2016, 175, 706–712. [Google Scholar] [CrossRef]
- Jee, M.H.; Johansen, J.D.; Buus, T.B.; Petersen, T.H.; Gadsbøll, A.; Woetmann, A.; Ødum, N.; Thyssen, J.P.; White, A.J.; Anderson, G.; et al. Increased Production of IL-17A-Producing γδ T Cells in the Thymus of Filaggrin-Deficient Mice. Front. Immunol. 2018, 9, 988. [Google Scholar] [CrossRef]
- Laihia, J.K.; Taimen, P.; Kujari, H.; Leino, L. Topical cis-urocanic acid attenuates oedema and erythema in acute and subacute skin inflammation in the mouse. Br. J. Dermatol. 2012, 167, 506–513. [Google Scholar] [CrossRef]
- Miajlovic, H.; Fallon, P.G.; Irvine, A.D.; Foster, T.J. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J. Allergy Clin. Immunol. 2010, 126, 1184–1190. [Google Scholar] [CrossRef]
- Krien, P.M.; Kermici, M. Evidence for the existence of a self-regulated enzymatic process within the human stratum corneum -an unexpected role for urocanic acid. J. Investig. Dermatol. 2000, 115, 414–420. [Google Scholar] [CrossRef]
- Kanicky, J.R.; Shah, D.O. Effect of degree, type, and position of unsaturation on the pKa of long-chain fatty acids. J. Colloid. Interface Sci. 2002, 256, 201–207. [Google Scholar] [CrossRef]
- Zeeuwen, P.L.; Ederveen, T.H.; van der Krieken, D.A.; Niehues, H.; Boekhorst, J.; Kezic, S.; Hanssen, D.A.; Otero, M.E.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; et al. Gram-positive anaerobe cocci are underrepresented in the microbiome of filaggrin-deficient human skin. J. Allergy Clin. Immunol. 2017, 139, 1368–1371. [Google Scholar] [CrossRef]
- Pendaries, V.; Le Lamer, M.; Cau, L.; Hansmann, B.; Malaisse, J.; Kezic, S.; Serre, G.; Simon, M. In a three-dimensional reconstructed human epidermis filaggrin-2 is essential for proper cornification. Cell Death Dis. 2015, 6, e1656. [Google Scholar] [CrossRef]
- Fluhr, J.W.; Elias, P.M.; Man, M.Q.; Hupe, M.; Selden, C.; Sundberg, J.P.; Tschachler, E.; Eckhart, L.; Mauro, T.M.; Feingold, K.R. Is the filaggrin-histidine-urocanic acid pathway essential for stratum corneum acidification? J. Investig. Dermatol. 2010, 130, 2141–2144. [Google Scholar] [CrossRef]
- Voegeli, R.; Rawlings, A.V.; Breternitz, M.; Doppler, S.; Schreier, T.; Fluhr, J.W. Increased stratum corneum serine protease activity in acute eczematous atopic skin. Br. J. Dermatol. 2009, 161, 70–77. [Google Scholar] [CrossRef]
- Rawlings, A.V.; Voegeli, R. Stratum corneum proteases and dry skin conditions. Cell Tissue Res. 2013, 351, 217–235. [Google Scholar] [CrossRef]
- Bender, R.A. Regulation of the histidine utilization (hut) system in bacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 565–584. [Google Scholar] [CrossRef]
- Goto, T.; Yamashita, A.; Hirakawa, H.; Matsutani, M.; Todo, K.; Ohshima, K.; Toh, H.; Miyamoto, K.; Kuhara, S.; Hattori, M.; et al. Complete genome sequence of Finegoldia magna, an anaerobic opportunistic pathogen. DNA Res. 2008, 15, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.C.; Frick, I.M. Gram-positive anaerobic cocci--commensals and opportunistic pathogens. FEMS Microbiol. Rev. 2013, 37, 520–553. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, T.; Kawamura, Y.; Li, N.; Li, Z.Y.; Zhao, L.; Shu, S. Proposal of the genera Anaerococcus gen. nov., Peptoniphilus gen. nov. and Gallicola gen. nov. for members of the genus Peptostreptococcus. Int. J. Syst. Evol. Microbiol. 2001, 51, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Kubica, M.; Hildebrand, F.; Brinkman, B.M.; Goossens, D.; Del Favero, J.; Vercammen, K.; Cornelis, P.; Schröder, J.M.; Vandenabeele, P.; Raes, J.; et al. The skin microbiome of caspase-14-deficient mice shows mild dysbiosis. Exp. Dermatol. 2014, 23, 561–567. [Google Scholar] [CrossRef]
- Clausen, M.L.; Agner, T.; Lilje, B.; Edslev, S.M.; Johannesen, T.B.; Andersen, P.S. Association of Disease Severity With Skin Microbiome and Filaggrin Gene Mutations in Adult Atopic Dermatitis. JAMA. Dermatol. 2018, 154, 293–300. [Google Scholar] [CrossRef]
- Baurecht, H.; Rühlemann, M.C.; Rodríguez, E.; Thielking, F.; Harder, I.; Erkens, A.S.; Stölzl, D.; Ellinghaus, E.; Hotze, M.; Lieb, W.; et al. Epidermal lipid composition, barrier integrity, and eczematous inflammation are associated with skin microbiome configuration. J. Allergy Clin. Immunol. 2018, 141, 1668–1676.e16. [Google Scholar] [CrossRef]
- Francuzik, W.; Franke, K.; Schumann, R.R.; Heine, G.; Worm, M. Propionibacterium acnes Abundance Correlates Inversely with Staphylococcus aureus: Data from Atopic Dermatitis Skin Microbiome. Acta Derm. Venereol. 2018, 98, 490–495. [Google Scholar] [CrossRef]
- Moosbrugger-Martinz, V.; Hackl, H.; Gruber, R.; Pilecky, M.; Knabl, L.; Orth-Höller, D.; Dubrac, S. Initial Evidence of Distinguishable Bacterial and Fungal Dysbiosis in the Skin of Patients with Atopic Dermatitis or Netherton Syndrome. J. Investig. Dermatol. 2021, 141, 114–123. [Google Scholar] [CrossRef]
- Common, J.E.; Brown, S.J.; Haines, R.L.; Goh, C.S.; Chen, H.; Balakrishnan, A.; Munro, C.S.; Tan, A.W.; Tan, H.H.; Tang, M.B.; et al. Filaggrin null mutations are not a protective factor for acne vulgaris. J. Investig. Dermatol 2011, 131, 1378–1380. [Google Scholar] [CrossRef]
- Miao, H.; Dong, R.; Zhang, S.; Yang, L.; Liu, Y.; Wang, T. Inherited ichthyosis and fungal infection: An update on pathogenesis and treatment strategies. J. Dtsch. Dermatol. Ges. 2021, 19, 341–350. [Google Scholar] [CrossRef]
- Gao, P.S.; Rafaels, N.M.; Hand, T.; Murray, T.; Boguniewicz, M.; Hata, T.; Schneider, L.; Hanifin, J.M.; Gallo, R.L.; Gao, L.; et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J. Allergy Clin. Immunol. 2009, 124, 507–513. [Google Scholar]
- Clausen, M.L.; Edslev, S.M.; Andersen, P.S.; Clemmensen, K.; Krogfelt, K.A.; Agner, T. Staphylococcus aureus colonization in atopic eczema and its association with filaggrin gene mutations. Br. J. Dermatol. 2017, 177, 1394–1400. [Google Scholar] [CrossRef]
- Lopes, C.; Rocha, L.; Sokhatska, O.; Soares, J.; Tavaria, F.; Correia, O.; Pintado, M.; Fernandes, S.; Delgado, L.; Moreira, A. Filaggrin Polymorphism Pro478Ser Is Associated With the Severity of Atopic Dermatitis and Colonization by Staphylococcal aureus. J. Investig. Allergol. Clin. Immunol. 2016, 26, 70–72. [Google Scholar]
- Nath, S.; Kumari, N.; Bandyopadhyay, D.; Sinha, N.; Majumder, P.P.; Mitra, R.; Mukherjee, S. Dysbiotic Lesional Microbiome With Filaggrin Missense Variants Associate With Atopic Dermatitis in India. Front. Cell. Infect. Microbiol. 2020, 10, 570423. [Google Scholar] [CrossRef]
- Rippke, F.; Schreiner, V.; Doering, T.; Maibach, H.I. Stratum corneum pH in atopic dermatitis: Impact on skin barrier function and colonization with Staphylococcus Aureus. Am. J. Clin. Dermatol. 2004, 5, 217–223. [Google Scholar] [CrossRef]
- Tauber, M.; Balica, S.; Hsu, C.Y.; Jean-Decoster, C.; Lauze, C.; Redoules, D.; Viodé, C.; Schmitt, A.M.; Serre, G.; Simon, M.; et al. Staphylococcus aureus density on lesional and nonlesional skin is strongly associated with disease severity in atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 1272–1274. [Google Scholar] [CrossRef]
- Smits, J.P.H.; Ederveen, T.H.A.; Rikken, G.; van den Brink, N.J.M.; van Vlijmen-Willems, I.; Boekhorst, J.; Kamsteeg, M.; Schalkwijk, J.; van Hijum, S.; Zeeuwen, P.; et al. Targeting the Cutaneous Microbiota in Atopic Dermatitis by Coal Tar via AHR-Dependent Induction of Antimicrobial Peptides. J. Investig. Dermatol. 2020, 140, 415–424. [Google Scholar]
- van Mierlo, M.M.F.; Totté, J.E.E.; Fieten, K.B.; van den Broek, T.J.; Schuren, F.H.J.; Pardo, L.M.; Pasmans, S. The influence of treatment in alpine and moderate maritime climate on the composition of the skin microbiome in patients with difficult to treat atopic dermatitis. Clin. Exp. Allergy 2019, 49, 1437–1445. [Google Scholar] [CrossRef]
- Knor, T.; Meholjić-Fetahović, A.; Mehmedagić, A. Stratum corneum hydration and skin surface pH in patients with atopic dermatitis. Acta Dermatovenerol. Croat. 2011, 19, 242–247. [Google Scholar]
- Kezic, S.; O’Regan, G.M.; Lutter, R.; Jakasa, I.; Koster, E.S.; Saunders, S.; Caspers, P.; Kemperman, P.M.; Puppels, G.J.; Sandilands, A.; et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J. Allergy Clin. Immunol. 2012, 129, 1031–1039. [Google Scholar] [CrossRef]
- Korting, H.C.; Lukacs, A.; Vogt, N.; Urban, J.; Ehret, W.; Ruckdeschel, G. Influence of the pH-value on the growth of Staphylococcus epidermidis, Staphylococcus aureus and Propionibacterium acnes in continuous culture. Zentralblatt Hyg. Umweltmed. 1992, 193, 78–90. [Google Scholar]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus Lipoteichoic Acid Damages the Skin Barrier through an IL-1-Mediated Pathway. J. Investig. Dermatol. 2019, 139, 1753–1761. [Google Scholar] [CrossRef]
- Simpson, E.L.; Villarreal, M.; Jepson, B.; Rafaels, N.; David, G.; Hanifin, J.; Taylor, P.; Boguniewicz, M.; Yoshida, T.; De Benedetto, A.; et al. Patients with Atopic Dermatitis Colonized with Staphylococcus aureus Have a Distinct Phenotype and Endotype. J. Investig. Dermatol. 2018, 138, 2224–2233. [Google Scholar] [CrossRef]
- Smith, T.P.; Bailey, C.J. Epidermolytic toxin from Staphylococcus aureus binds to filaggrins. FEBS Lett. 1986, 194, 309–312. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Bin, L.; Kim, B.E.; Oyoshi, M.K.; Geha, R.S.; Goleva, E.; Leung, D.Y. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin-induced keratinocyte death. J. Allergy Clin. Immunol. 2013, 131, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Maintz, L.; Novak, N. Modifications of the innate immune system in atopic dermatitis. J. Innate Immun. 2011, 3, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Feuillie, C.; Vitry, P.; McAleer, M.A.; Kezic, S.; Irvine, A.D.; Geoghegan, J.A.; Dufrêne, Y.F. Adhesion of Staphylococcus aureus to Corneocytes from Atopic Dermatitis Patients Is Controlled by Natural Moisturizing Factor Levels. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Towell, A.M.; Feuillie, C.; Vitry, P.; Da Costa, T.M.; Mathelié-Guinlet, M.; Kezic, S.; Fleury, O.M.; McAleer, M.A.; Dufrêne, Y.F.; Irvine, A.D.; et al. Staphylococcus aureus binds to the N-terminal region of corneodesmosin to adhere to the stratum corneum in atopic dermatitis. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Bunick, C.G.; Presland, R.B.; Lawrence, O.T.; Pearton, D.J.; Milstone, L.M.; Steitz, T.A. Crystal Structure of Human Profilaggrin S100 Domain and Identification of Target Proteins Annexin II, Stratifin, and HSP27. J. Investig. Dermatol. 2015, 135, 1801–1809. [Google Scholar] [CrossRef]
- Kuechle, M.K.; Presland, R.B.; Lewis, S.P.; Fleckman, P.; Dale, B.A. Inducible expression of filaggrin increases keratinocyte susceptibility to apoptotic cell death. Cell Death Differ. 2000, 7, 566–573. [Google Scholar] [CrossRef]
- Dang, N.; Ma, X.; Meng, X.; An, L.; Pang, S. Dysregulated function of normal human epidermal keratinocytes in the absence of filaggrin. Mol. Med. Rep. 2016, 14, 2566–2572. [Google Scholar] [CrossRef][Green Version]
- Markova, N.G.; Marekov, L.N.; Chipev, C.C.; Gan, S.Q.; Idler, W.W.; Steinert, P.M. Profilaggrin is a major epidermal calcium-binding protein. Mol. Cell Biol. 1993, 13, 613–625. [Google Scholar]
- Matsui, M.S.; Chew, S.L.; DeLeo, V.A. Protein kinase C in normal human epidermal keratinocytes during proliferation and calcium-induced differentiation. J. Investig. Dermatol. 1992, 99, 565–571. [Google Scholar] [CrossRef]
- Aho, S.; Harding, C.R.; Lee, J.M.; Meldrum, H.; Bosko, C.A. Regulatory role for the profilaggrin N-terminal domain in epidermal homeostasis. J. Investig. Dermatol. 2012, 132, 2376–2385. [Google Scholar] [CrossRef]
- Asai, Y.; Greenwood, C.; Hull, P.R.; Alizadehfar, R.; Ben-Shoshan, M.; Brown, S.J.; Campbell, L.; Michel, D.L.; Bussières, J.; Rousseau, F.; et al. Filaggrin gene mutation associations with peanut allergy persist despite variations in peanut allergy diagnostic criteria or asthma status. J. Allergy Clin. Immunol. 2013, 132, 239–242. [Google Scholar] [CrossRef]
- Le Lamer, M.; Pellerin, L.; Reynier, M.; Cau, L.; Pendaries, V.; Leprince, C.; Méchin, M.C.; Serre, G.; Paul, C.; Simon, M. Defects of corneocyte structural proteins and epidermal barrier in atopic dermatitis. Biol. Chem. 2015, 396, 1163–1179. [Google Scholar] [CrossRef]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef]
- Sergeant, A.; Campbell, L.E.; Hull, P.R.; Porter, M.; Palmer, C.N.; Smith, F.J.; McLean, W.H.; Munro, C.S. Heterozygous null alleles in filaggrin contribute to clinical dry skin in young adults and the elderly. J. Investig. Dermatol. 2009, 129, 1042–1045. [Google Scholar] [CrossRef]
- Böhme, M.; Söderhäll, C.; Kull, I.; Bergström, A.; van Hage, M.; Wahlgren, C.F. Filaggrin mutations increase the risk for persistent dry skin and eczema independent of sensitization. J. Allergy Clin. Immunol. 2012, 129, 1153–1155. [Google Scholar] [CrossRef]
- Ginger, R.S.; Blachford, S.; Rowland, J.; Rowson, M.; Harding, C.R. Filaggrin repeat number polymorphism is associated with a dry skin phenotype. Arch. Dermatol. Res. 2005, 297, 235–241. [Google Scholar] [CrossRef]
- Mlitz, V.; Latreille, J.; Gardinier, S.; Jdid, R.; Drouault, Y.; Hufnagl, P.; Eckhart, L.; Guinot, C.; Tschachler, E. Impact of filaggrin mutations on Raman spectra and biophysical properties of the stratum corneum in mild to moderate atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 983–990. [Google Scholar] [CrossRef]
- Kezic, S.; Kemperman, P.M.; Koster, E.S.; de Jongh, C.M.; Thio, H.B.; Campbell, L.E.; Irvine, A.D.; McLean, W.H.; Puppels, G.J.; Caspers, P.J. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J. Investig. Dermatol. 2008, 128, 2117–2119. [Google Scholar] [CrossRef]
- Kezic, S.; Kammeyer, A.; Calkoen, F.; Fluhr, J.W.; Bos, J.D. Natural moisturizing factor components in the stratum corneum as biomarkers of filaggrin genotype: Evaluation of minimally invasive methods. Br. J. Dermatol. 2009, 161, 1098–1104. [Google Scholar] [CrossRef]
- Kezic, S.; O’Regan, G.M.; Yau, N.; Sandilands, A.; Chen, H.; Campbell, L.E.; Kroboth, K.; Watson, R.; Rowland, M.; McLean, W.H.; et al. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy 2011, 66, 934–940. [Google Scholar] [CrossRef]
- Ota, M.; Sasaki, T.; Ebihara, T.; Yokosawa, E.; Murakami, Y.; Matsunaka, H.; Chinuki, Y.; Amagai, M.; Morita, E. Filaggrin-gene mutation has minimal effect on the disease severity in the lesions of atopic dermatitis. J. Dermatol. 2021, 48, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, D.; Sjöberg, O.; Foucard, T. Development of allergies and asthma in infants and young children with atopic dermatitis--a prospective follow-up to 7 years of age. Allergy 2000, 55, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Akiyama, M.; Shimizu, H. Filaggrin: An emerging star in atopic march. J. Formos. Med. Assoc. 2008, 107, 429–431. [Google Scholar] [CrossRef][Green Version]
- Schuttelaar, M.L.; Kerkhof, M.; Jonkman, M.F.; Koppelman, G.H.; Brunekreef, B.; de Jongste, J.C.; Wijga, A.; McLean, W.H.; Postma, D.S. Filaggrin mutations in the onset of eczema, sensitization, asthma, hay fever and the interaction with cat exposure. Allergy 2009, 64, 1758–1765. [Google Scholar] [CrossRef]
- Spergel, J.M. From atopic dermatitis to asthma: The atopic march. Ann. Allergy Asthma Immunol. 2010, 105, 99–106, 107–109, 117. [Google Scholar] [CrossRef]
- Ziyab, A.H.; Karmaus, W.; Zhang, H.; Holloway, J.W.; Steck, S.E.; Ewart, S.; Arshad, S.H. Allergic sensitization and filaggrin variants predispose to the comorbidity of eczema, asthma, and rhinitis: Results from the Isle of Wight birth cohort. Clin. Exp. Allergy 2014, 44, 1170–1178. [Google Scholar] [CrossRef]
- Weidinger, S.; O’Sullivan, M.; Illig, T.; Baurecht, H.; Depner, M.; Rodriguez, E.; Ruether, A.; Klopp, N.; Vogelberg, C.; Weiland, S.K.; et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J. Allergy Clin. Immunol. 2008, 121, 1203–1209. [Google Scholar] [CrossRef]
- Rodríguez, E.; Baurecht, H.; Herberich, E.; Wagenpfeil, S.; Brown, S.J.; Cordell, H.J.; Irvine, A.D.; Weidinger, S. Meta-analysis of filaggrin polymorphisms in eczema and asthma: Robust risk factors in atopic disease. J. Allergy Clin. Immunol. 2009, 123, 1361–1370. [Google Scholar] [CrossRef]
- van den Oord, R.A.; Sheikh, A. Filaggrin gene defects and risk of developing allergic sensitisation and allergic disorders: Systematic review and meta-analysis. BMJ 2009, 339, b2433. [Google Scholar] [CrossRef]
- Rogers, A.J.; Celedón, J.C.; Lasky-Su, J.A.; Weiss, S.T.; Raby, B.A. Filaggrin mutations confer susceptibility to atopic dermatitis but not to asthma. J. Allergy Clin. Immunol. 2007, 120, 1332–1337. [Google Scholar] [CrossRef]
- Bocheva, G.S.; Slominski, R.M.; Slominski, A.T. Immunological Aspects of Skin Aging in Atopic Dermatitis. Int. J. Mol. Sci. 2021, 22, 5729. [Google Scholar] [CrossRef]
- Ziyab, A.H.; Karmaus, W.; Yousefi, M.; Ewart, S.; Schauberger, E.; Holloway, J.W.; Zhang, H.; Arshad, S.H. Interplay of filaggrin loss-of-function variants, allergic sensitization, and eczema in a longitudinal study covering infancy to 18 years of age. PLoS ONE 2012, 7, e32721. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Linneberg, A.; Ross-Hansen, K.; Carlsen, B.C.; Meldgaard, M.; Szecsi, P.B.; Stender, S.; Menné, T.; Johansen, J.D. Filaggrin mutations are strongly associated with contact sensitization in individuals with dermatitis. Contact Dermat. 2013, 68, 273–276. [Google Scholar] [CrossRef]
- Johansson, E.K.; Bergström, A.; Kull, I.; Lind, T.; Söderhäll, C.; van Hage, M.; Wickman, M.; Ballardini, N.; Wahlgren, C.F. IgE sensitization in relation to preschool eczema and filaggrin mutation. J. Allergy Clin. Immunol. 2017, 140, 1572–1579. [Google Scholar] [CrossRef]
- Elhaji, Y.; Sasseville, D.; Pratt, M.; Asai, Y.; Matheson, K.; McLean, W.H.I.; Hull, P.R. Filaggrin gene loss-of-function mutations constitute a factor in patients with multiple contact allergies. Contact Dermat. 2019, 80, 354–358. [Google Scholar] [CrossRef]
- Simpson, A.; Brough, H.A.; Haider, S.; Belgrave, D.; Murray, C.S.; Custovic, A. Early-life inhalant allergen exposure, filaggrin genotype, and the development of sensitization from infancy to adolescence. J. Allergy Clin. Immunol. 2020, 145, 993–1001. [Google Scholar] [CrossRef]
- Imoto, Y.; Enomoto, H.; Fujieda, S.; Okamoto, M.; Sakashita, M.; Susuki, D.; Okada, M.; Hirota, T.; Tamari, M.; Ebe, K.; et al. S2554X mutation in the filaggrin gene is associated with allergen sensitization in the Japanese population. J. Allergy Clin. Immunol. 2010, 12, 498–500. [Google Scholar] [CrossRef]
- Bisgaard, H.; Simpson, A.; Palmer, C.N.; Bønnelykke, K.; McLean, I.; Mukhopadhyay, S.; Pipper, C.B.; Halkjaer, L.B.; Lipworth, B.; Hankinson, J.; et al. Gene-environment interaction in the onset of eczema in infancy: Filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med. 2008, 5, e131. [Google Scholar] [CrossRef]
- McPherson, T.; Sherman, V.J.; Aslam, A.; Crack, L.; Chan, H.; Lloyd-Lavery, A.; Jones, L.; Ardern-Jones, M.; Ogg, G. Filaggrin null mutations associate with increased frequencies of allergen-specific CD4+ T-helper 2 cells in patients with atopic eczema. Br. J. Dermatol. 2010, 163, 544–549. [Google Scholar] [CrossRef]
- Brown, S.J.; Asai, Y.; Cordell, H.J.; Campbell, L.E.; Zhao, Y.; Liao, H.; Northstone, K.; Henderson, J.; Alizadehfar, R.; Ben-Shoshan, M.; et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J. Allergy Clin. Immunol. 2011, 127, 661–667. [Google Scholar] [CrossRef]
- Li, M.; Liu, J.B.; Liu, Q.; Yao, M.; Cheng, R.; Xue, H.; Zhou, H.; Yao, Z. Interactions between FLG mutations and allergens in atopic dermatitis. Arch. Dermatol. Res. 2012, 304, 787–793. [Google Scholar] [CrossRef]
- Brough, H.A.; Simpson, A.; Makinson, K.; Hankinson, J.; Brown, S.; Douiri, A.; Belgrave, D.C.; Penagos, M.; Stephens, A.C.; McLean, W.H.; et al. Peanut allergy: Effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J. Allergy Clin. Immunol. 2014, 134, 867–875. [Google Scholar] [CrossRef]
- Berdyshev, E.; Goleva, E.; Bronova, I.; Bronoff, A.S.; Hoffman, B.C.; Ramirez-Gama, M.A.; Garcia, S.L.; Crumrine, D.; Elias, P.M.; Cho, C.B.; et al. Unique skin abnormality in patients with peanut allergy but no atopic dermatitis. J. Allergy Clin. Immunol. 2021, 147, 361–367. [Google Scholar] [CrossRef]
- Schmitt, J.; Weidinger, S. Alternative models of comorbidity: A framework for the interpretation of epidemiological association studies. J. Investig. Dermatol. 2014, 134, 303–307. [Google Scholar] [CrossRef]
- Flohr, C.; Mann, J. New insights into the epidemiology of childhood atopic dermatitis. Allergy 2014, 69, 3–16. [Google Scholar] [CrossRef]
- Lerbaek, A.; Bisgaard, H.; Agner, T.; Ohm Kyvik, K.; Palmer, C.N.; Menné, T. Filaggrin null alleles are not associated with hand eczema or contact allergy. Br. J. Dermatol 2007, 157, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Cordell, H.J. Are filaggrin mutations associated with hand eczema or contact allergy?--we do not know. Br. J. Dermatol. 2008, 158, 1383–1384. [Google Scholar] [CrossRef]
- Carlsen, B.C.; Johansen, J.D.; Menné, T.; Meldgaard, M.; Szecsi, P.B.; Stender, S.; Thyssen, J.P. Filaggrin null mutations and association with contact allergy and allergic contact dermatitis: Results from a tertiary dermatology clinic. Contact Dermat. 2010, 63, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Hougaard, M.G.; Johansen, J.D.; Linneberg, A.; Bandier, J.; Stender, S.; Carlsen, B.C.; Szecsi, P.B.; Meldgaard, M.; Menné, T.; Thyssen, J.P. Skin prick test reactivity to aeroallergens by filaggrin mutation status. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Tang, L.; Husemoen, L.L.; Stender, S.; Szecsi, P.B.; Menné, T.; Johansen, J.D.; Linneberg, A. Filaggrin gene mutations are not associated with food and aeroallergen sensitization without concomitant atopic dermatitis in adults. J. Allergy Clin. Immunol. 2015, 135, 1375–1378. [Google Scholar] [CrossRef]
- Carlsen, B.C.; Meldgaard, M.; Hamann, D.; Hamann, Q.; Hamann, C.; Thyssen, J.P.; Meyer, D.M.; Gruninger, S.E.; Hamann, C. Latex allergy and filaggrin null mutations. J. Dent. 2011, 39, 128–132. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Johansen, J.D.; Zachariae, C.; Menné, T.; Linneberg, A. Xerosis is associated with atopic dermatitis, hand eczema and contact sensitization independent of filaggrin gene mutations. Acta Derm. Venereol. 2013, 93, 406–410. [Google Scholar] [CrossRef]
- Novak, N.; Baurecht, H.; Schäfer, T.; Rodriguez, E.; Wagenpfeil, S.; Klopp, N.; Heinrich, J.; Behrendt, H.; Ring, J.; Wichmann, E.; et al. Loss-of-function mutations in the filaggrin gene and allergic contact sensitization to nickel. J. Investig. Dermatol. 2008, 128, 1430–1435. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Johansen, J.D.; Linneberg, A.; Menné, T.; Nielsen, N.H.; Meldgaard, M.; Szecsi, P.B.; Stender, S.; Carlsen, B.C. The association between null mutations in the filaggrin gene and contact sensitization to nickel and other chemicals in the general population. Br. J. Dermatol 2010, 162, 1278–1285. [Google Scholar] [CrossRef]
- Ross-Hansen, K.; Menné, T.; Johansen, J.D.; Carlsen, B.C.; Linneberg, A.; Nielsen, N.H.; Stender, S.; Meldgaard, M.; Szecsi, P.B.; Thyssen, J.P. Nickel reactivity and filaggrin null mutations--evaluation of the filaggrin bypass theory in a general population. Contact Dermat. 2011, 64, 24–31. [Google Scholar] [CrossRef]
- Ross-Hansen, K.; Johansen, J.D.; Vølund, A.; Menné, T.; Thyssen, J.P. The nickel dose-response relationship by filaggrin genotype (FLG). Contact Dermat. 2014, 71, 49–53. [Google Scholar] [CrossRef]
- Petersen, T.H.; Jee, M.H.; Gadsbøll, A.; Schmidt, J.D.; Sloth, J.J.; Sonnenberg, G.F.; Geisler, C.; Thyssen, J.P.; Bonefeld, C.M. Mice with epidermal filaggrin deficiency show increased immune reactivity to nickel. Contact Dermat. 2019, 80, 139–148. [Google Scholar] [CrossRef]
- Ross-Hansen, K.; Østergaard, O.; Tanassi, J.T.; Thyssen, J.P.; Johansen, J.D.; Menné, T.; Heegaard, N.H.H. Filaggrin is a predominant member of the denaturation-resistant nickel-binding proteome of human epidermis. J. Investig. Dermatol. 2014, 134, 1164–1166. [Google Scholar] [CrossRef]
- Podobas, E.I.; Gutowska-Owsiak, D.; Moretti, S.; Poznański, J.; Kulińczak, M.; Grynberg, M.; Gruca, A.; Bonna, A.; Płonka, D.; Ogg, G.; et al. Ni(2+)-Assisted Hydrolysis May Affect the Human Proteome; Filaggrin Degradation Ex Vivo as an Example of Possible Consequences. Front. Mol. Biosci. 2022, 9, 828674. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef]
- Brown, S.J.; McLean, W.H. One remarkable molecule: Filaggrin. J. Investig. Dermatol. 2012, 132, 751–762. [Google Scholar] [CrossRef]




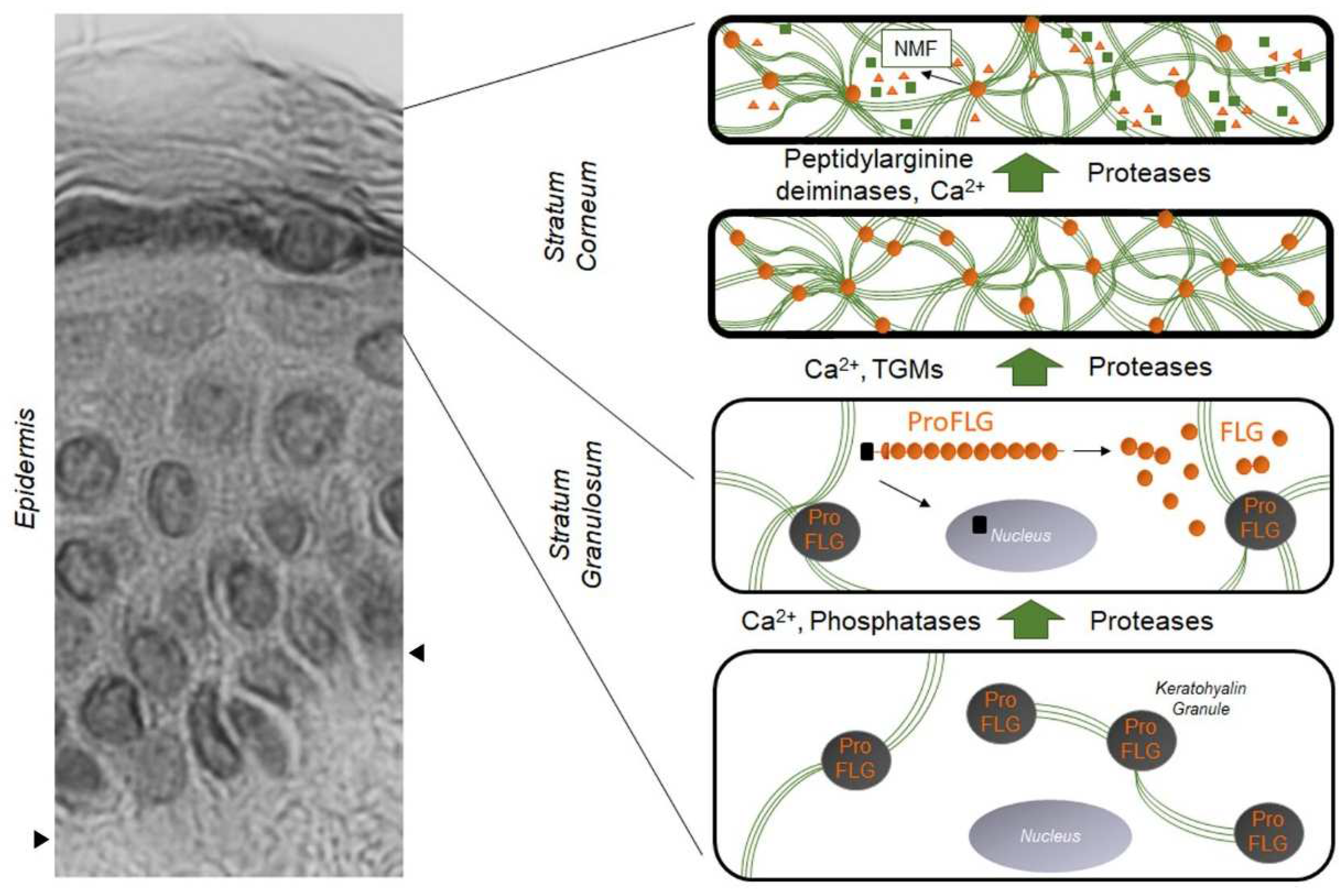{kind=link}
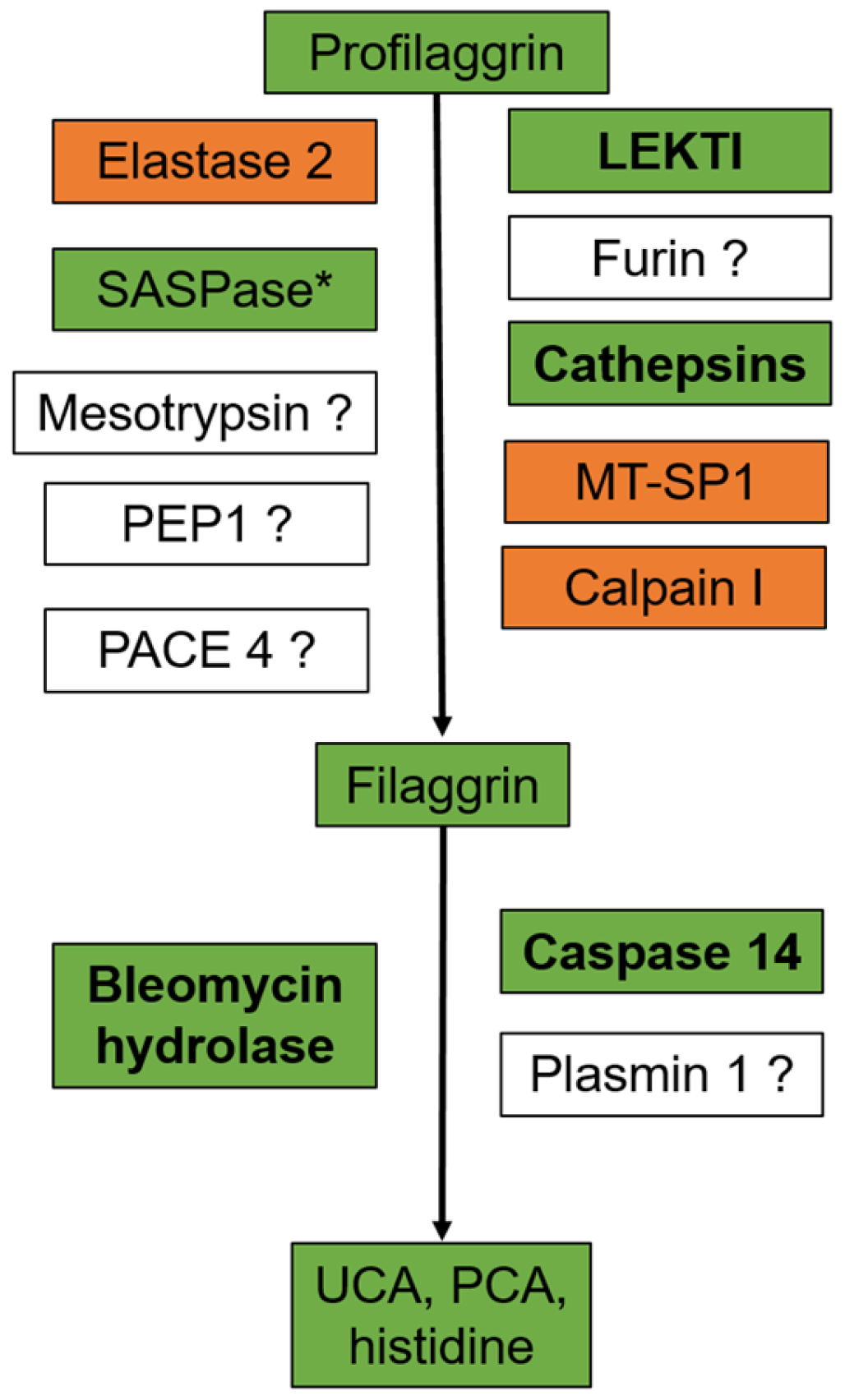{kind=link}
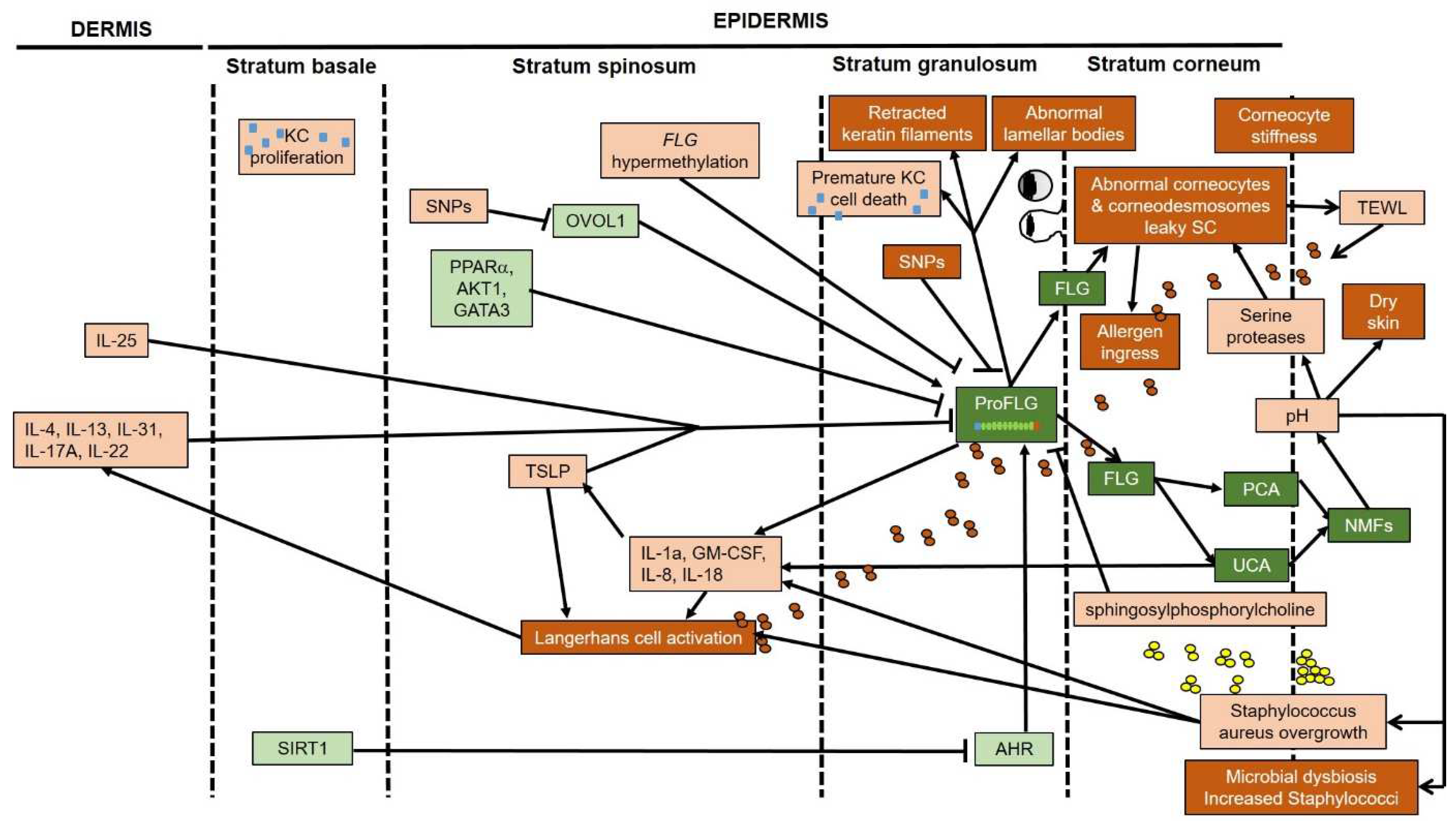{kind=link}
| In Vitro and Animal Models | in Humans | |
|---|---|---|
| Dephosphorylation of profilaggrin | Phosphatase of the protein phosphatase 2A family | |
| Profilaggrin processing | Calpain 1, cathepsins L-like and H, PEP1, furin, PACE4, MT-SP1, mesotrypsin, SASPase, LEKTI, elastase 2 | Calpain I, SASPase, mesotrypsin |
| Filaggrin citrullination in the stratum corneum | PAD1 and/or 3 | PAD1 and/or 3 |
| Proteolysis of filaggrin in the stratum corneum | Bleomycin hydrolase, calpain 1, caspase 14 | Bleomycin hydrolase, caspase 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moosbrugger-Martinz, V.; Leprince, C.; Méchin, M.-C.; Simon, M.; Blunder, S.; Gruber, R.; Dubrac, S. Revisiting the Roles of Filaggrin in Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 5318. https://doi.org/10.3390/ijms23105318
Moosbrugger-Martinz V, Leprince C, Méchin M-C, Simon M, Blunder S, Gruber R, Dubrac S. Revisiting the Roles of Filaggrin in Atopic Dermatitis. International Journal of Molecular Sciences. 2022; 23(10):5318. https://doi.org/10.3390/ijms23105318
Chicago/Turabian StyleMoosbrugger-Martinz, Verena, Corinne Leprince, Marie-Claire Méchin, Michel Simon, Stefan Blunder, Robert Gruber, and Sandrine Dubrac. 2022. "Revisiting the Roles of Filaggrin in Atopic Dermatitis" International Journal of Molecular Sciences 23, no. 10: 5318. https://doi.org/10.3390/ijms23105318
APA StyleMoosbrugger-Martinz, V., Leprince, C., Méchin, M.-C., Simon, M., Blunder, S., Gruber, R., & Dubrac, S. (2022). Revisiting the Roles of Filaggrin in Atopic Dermatitis. International Journal of Molecular Sciences, 23(10), 5318. https://doi.org/10.3390/ijms23105318