
The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
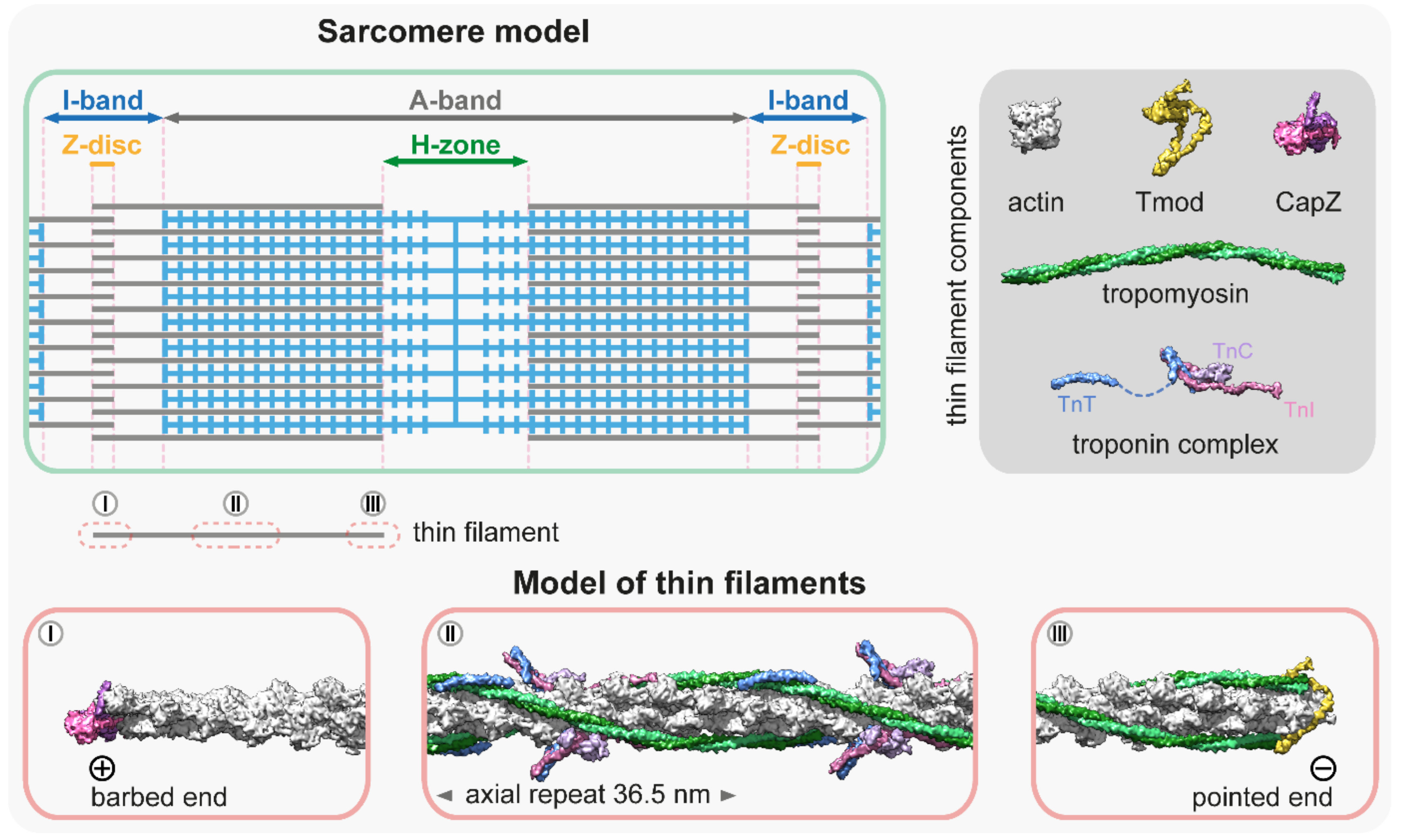
2. Thin Filament Structure
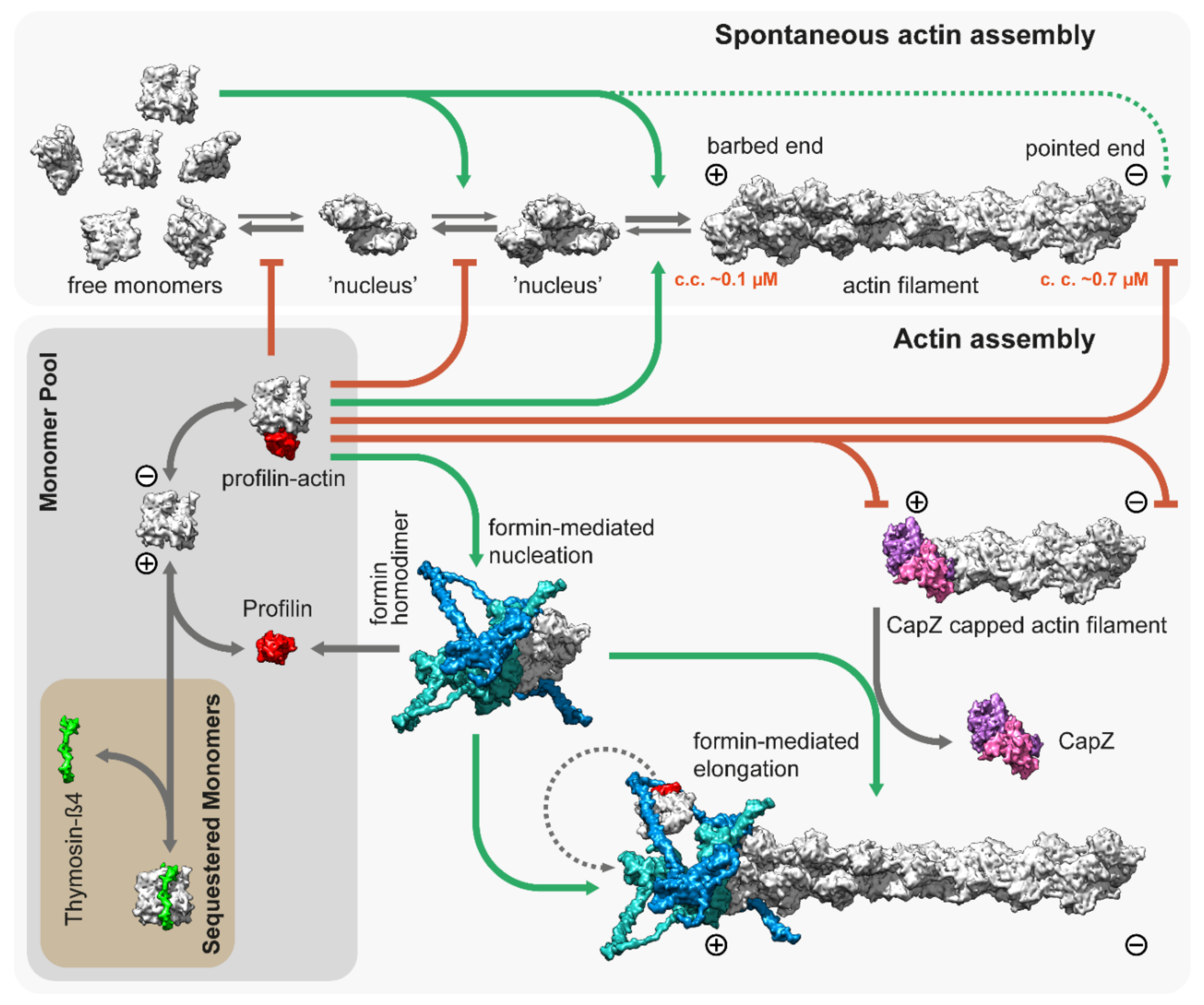
3. Thin Filament Assembly
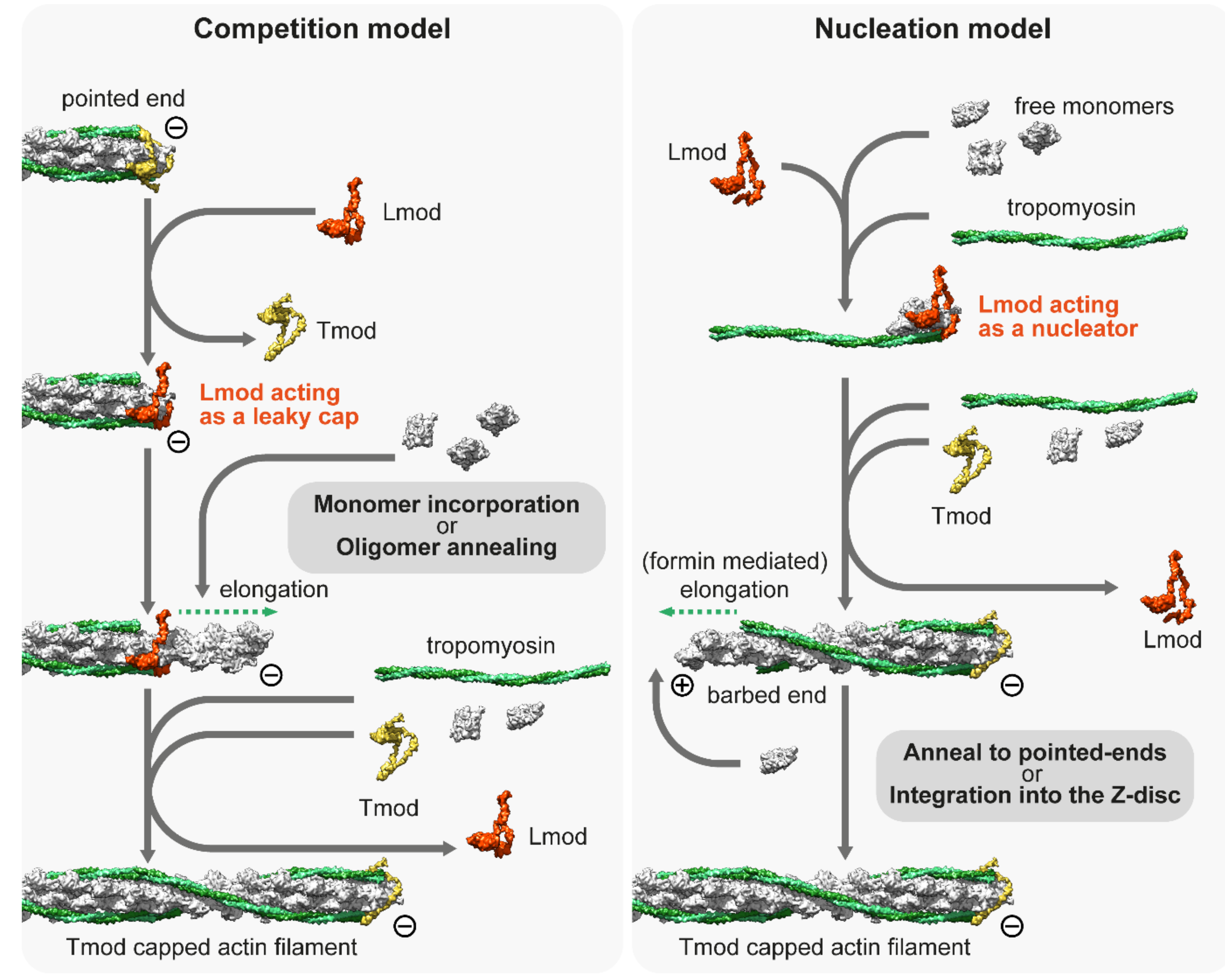
4. Thin Filament Elongation
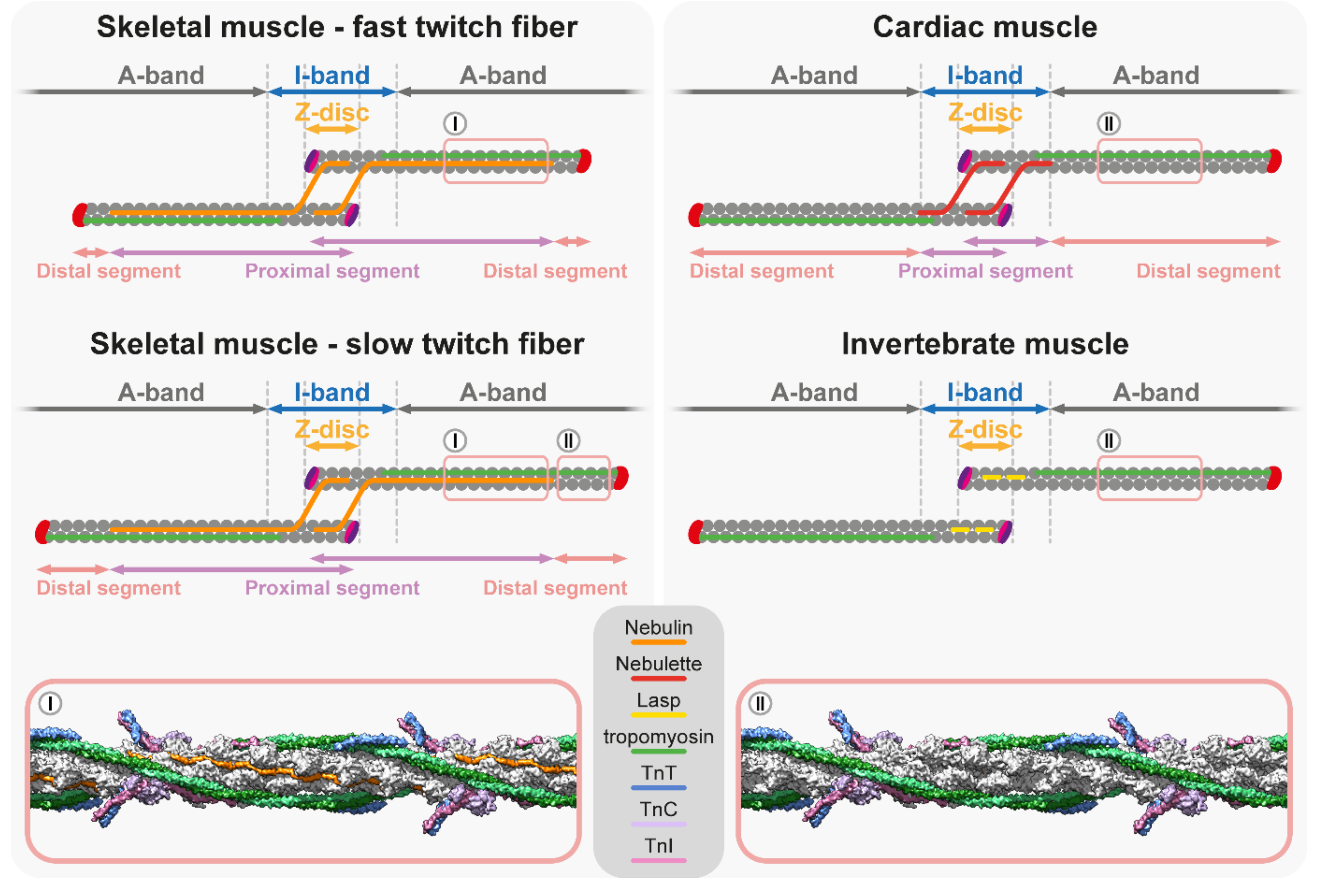
5. Thin Filament Length Determination
5.1. Nebulin Ruler Hypothesis
5.2. Titin/Myosin Scaffold Model
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Huxley, A.F.; Niedergerke, R. Structural changes in muscle during contraction; interference microscopy of living muscle fibres. Nature 1954, 173, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Edman, K.A. The relation between sarcomere length and active tension in isolated semitendinosus fibres of the frog. J. Physiol. 1966, 183, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.M.; Huxley, A.F.; Julian, F.J. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J. Physiol. 1966, 184, 170–192. [Google Scholar] [CrossRef]
- Castillo, A.; Nowak, R.; Littlefield, K.P.; Fowler, V.M.; Littlefield, R.S. A nebulin ruler does not dictate thin filament lengths. Biophys. J. 2009, 96, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Kim, N.E.; Lewis, S.A.; Hoenecke, H.R.; D’Lima, D.D.; Fowler, V.M. Thin-filament length correlates with fiber type in human skeletal muscle. Am. J. Physiol. Cell Physiol. 2011, 302, C555–C565. [Google Scholar] [CrossRef]
- Gokhin, D.S.; Lewis, R.A.; McKeown, C.R.; Nowak, R.B.; Kim, N.E.; Littlefield, R.S.; Lieber, R.L.; Fowler, V.M. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J. Cell Biol. 2010, 189, 95–109. [Google Scholar] [CrossRef]
- Granzier, H.L.; Akster, H.A.; Ter Keurs, H.E. Effect of thin filament length on the force-sarcomere length relation of skeletal muscle. Am. J. Physiol. Cell Physiol. 1991, 260, C1060–C1070. [Google Scholar] [CrossRef]
- Ono, S. Regulation of structure and function of sarcomeric actin filaments in striated muscle of the nematode Caenorhabditis elegans. Anat. Rec. 2014, 297, 1548–1559. [Google Scholar] [CrossRef]
- Gieseler, K.; Qadota, H.; Benian, G.M. Development, Structure, and Maintenance of C. Elegans Body Wall Muscle. WormBook Online Rev. C. Elegans Biol. 2017, 2017, 1–59. [Google Scholar] [CrossRef]
- Prill, K.; Dawson, J.F. Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. Int. J. Mol. Sci. 2020, 21, 542. [Google Scholar] [CrossRef]
- De Winter, J.M.; Ottenheijm, C.A.C. Sarcomere Dysfunction in Nemaline Myopathy. J. Neuromuscul. Dis. 2017, 4, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Laitila, J.; Wallgren-Pettersson, C. Recent advances in nemaline myopathy. Neuromuscul. Disord. NMD 2021, 31, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Whitby, F.G.; Phillips, G.N., Jr. Crystal structure of tropomyosin at 7 Angstroms resolution. Proteins 2000, 38, 49–59. [Google Scholar] [CrossRef]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maéda, Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, M.V.; Stone, D.B.; Malanina, G.G.; Karatzaferi, C.; Cooke, R.; Mendelson, R.A.; Fletterick, R.J. Ca(2+)-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA 2005, 102, 5038–5043. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Iwane, A.H.; Yanagida, T.; Namba, K. Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 2010, 467, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Lehman, W.; Craig, R.; Vibert, P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature 1994, 368, 65–67. [Google Scholar] [CrossRef]
- Xu, C.; Craig, R.; Tobacman, L.; Horowitz, R.; Lehman, W. Tropomyosin Positions in Regulated Thin Filaments Revealed by Cryoelectron Microscopy. Biophys. J. 1999, 77, 985–992. [Google Scholar] [CrossRef]
- Narita, A.; Yasunaga, T.; Ishikawa, T.; Mayanagi, K.; Wakabayashi, T. Ca(2+)-induced switching of troponin and tropomyosin on actin filaments as revealed by electron cryo-microscopy. J. Mol. Biol. 2001, 308, 241–261. [Google Scholar] [CrossRef]
- Oda, T.; Iwasa, M.; Aihara, T.; Maéda, Y.; Narita, A. The nature of the globular- to fibrous-actin transition. Nature 2009, 457, 441–445. [Google Scholar] [CrossRef]
- Paul, D.M.; Squire, J.M.; Morris, E.P. A novel approach to the structural analysis of partially decorated actin based filaments. J. Struct. Biol. 2010, 170, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Barbu-Tudoran, L.; Orzechowski, M.; Craig, R.; Trinick, J.; White, H.; Lehman, W. Three-dimensional organization of troponin on cardiac muscle thin filaments in the relaxed state. Biophys. J. 2014, 106, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.M.; Squire, J.M.; Morris, E.P. Relaxed and active thin filament structures; a new structural basis for the regulatory mechanism. J. Struct. Biol. 2017, 197, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca(2+) levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef]
- Miller, A.; Tregear, E.T. Structure of insect fibrillar flight muscle in the presence and absence of ATP. J. Mol. Biol. 1972, 70, 85–104. [Google Scholar] [CrossRef]
- Reedy, M.K.; Reedy, M.C. Rigor crossbridge structure in tilted single filament layers and flared-X formations from insect flight muscle. J. Mol. Biol. 1985, 185, 145–176. [Google Scholar] [CrossRef]
- Schmitz, H.; Lucaveche, C.; Reedy, M.K.; Taylor, K.A. Oblique section 3-D reconstruction of relaxed insect flight muscle reveals the cross-bridge lattice in helical registration. Biophys. J. 1994, 67, 1620–1633. [Google Scholar] [CrossRef]
- Cooper, J.A. Actin Dynamics: Tropomyosin Provides Stability. Curr. Biol. 2002, 12, R523–R525. [Google Scholar] [CrossRef]
- Orzechowski, M.; Li, X.E.; Fischer, S.; Lehman, W. An atomic model of the tropomyosin cable on F-actin. Biophys. J. 2014, 107, 694–699. [Google Scholar] [CrossRef]
- Marston, S.; Zamora, J.E. Troponin structure and function: A view of recent progress. J. Muscle Res. Cell Motil. 2020, 41, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Von der Ecken, J.; Heissler, S.M.; Pathan-Chhatbar, S.; Manstein, D.J.; Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 2016, 534, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Madasu, Y.; Dominguez, R. Mechanism of actin filament pointed-end capping by tropomodulin. Science 2014, 345, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.; Merino, F.; Schaks, M.; Rottner, K.; Raunser, S.; Bieling, P. A barbed end interference mechanism reveals how capping protein promotes nucleation in branched actin networks. Nat. Commun. 2021, 12, 5329. [Google Scholar] [CrossRef]
- Murakami, K.; Stewart, M.; Nozawa, K.; Tomii, K.; Kudou, N.; Igarashi, N.; Shirakihara, Y.; Wakatsuki, S.; Yasunaga, T.; Wakabayashi, T. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl. Acad. Sci. USA 2008, 105, 7200–7205. [Google Scholar] [CrossRef]
- Hanson, J. Axial Period of Actin Filaments. Nature 1967, 213, 353–356. [Google Scholar] [CrossRef]
- Egelman, E.H.; Francis, N.; DeRosier, D.J. F-actin is a helix with a random variable twist. Nature 1982, 298, 131–135. [Google Scholar] [CrossRef]
- Narita, A.; Oda, T.; Maéda, Y. Structural basis for the slow dynamics of the actin filament pointed end. EMBO J. 2011, 30, 1230–1237. [Google Scholar] [CrossRef]
- Zsolnay, V.; Katkar, H.H.; Chou, S.Z.; Pollard, T.D.; Voth, G.A. Structural basis for polarized elongation of actin filaments. Proc. Natl. Acad. Sci. USA 2020, 117, 30458–30464. [Google Scholar] [CrossRef]
- Bugyi, B.; Papp, G.; Hild, G.; Lõrinczy, D.; Nevalainen, E.M.; Lappalainen, P.; Somogyi, B.; Nyitrai, M. Formins regulate actin filament flexibility through long range allosteric interactions. J. Biol. Chem. 2006, 281, 10727–10736. [Google Scholar] [CrossRef]
- Papp, G.; Bugyi, B.; Ujfalusi, Z.; Barkó, S.; Hild, G.; Somogyi, B.; Nyitrai, M. Conformational changes in actin filaments induced by formin binding to the barbed end. Biophys. J. 2006, 91, 2564–2572. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of F-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Galkin, V.E.; Orlova, A.; Lukoyanova, N.; Wriggers, W.; Egelman, E.H. Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 2001, 153, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Grintsevich, E.E.; Phillips, M.L.; Reisler, E.; Gimzewski, J.K. Atomic force microscopy reveals drebrin induced remodeling of f-actin with subnanometer resolution. Nano Lett. 2011, 11, 825–827. [Google Scholar] [CrossRef]
- Burbaum, L.; Schneider, J.; Scholze, S.; Böttcher, R.T.; Baumeister, W.; Schwille, P.; Plitzko, J.M.; Jasnin, M. Molecular-scale visualization of sarcomere contraction within native cardiomyocytes. Nat. Commun. 2021, 12, 4086. [Google Scholar] [CrossRef]
- Wang, Z.; Grange, M.; Wagner, T.; Kho, A.L.; Gautel, M.; Raunser, S. The molecular basis for sarcomere organization in vertebrate skeletal muscle. Cell 2021, 184, 2135–2150.e2113. [Google Scholar] [CrossRef]
- Szikora, S.; Gajdos, T.; Novák, T.; Farkas, D.; Földi, I.; Lenart, P.; Erdélyi, M.; Mihály, J. Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. J. Cell Biol. 2020, 219, e201907026. [Google Scholar] [CrossRef]
- Schueder, F.; Mangeol, P.; Chan, E.H.; Rees, R.; Schünemann, J.; Jungmann, R.; Görlich, D.; Schnorrer, F. Nanobodies combined with DNA-PAINT super-resolution reveal a staggered titin nano-architecture in flight muscles. bioRxiv 2022. [Google Scholar] [CrossRef]
- Sigal, Y.M.; Zhou, R.; Zhuang, X. Visualizing and discovering cellular structures with super-resolution microscopy. Science 2018, 361, 880–887. [Google Scholar] [CrossRef]
- Szikora, S.; Görög, P.; Kozma, C.; Mihály, J. Drosophila Models Rediscovered with Super-Resolution Microscopy. Cells 2021, 10, 1924. [Google Scholar] [CrossRef]
- Carlier, M.F.; Pantaloni, D. Control of actin assembly dynamics in cell motility. J. Biol. Chem. 2007, 282, 23005–23009. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Schutt, C.E.; Myslik, J.C.; Rozycki, M.D.; Goonesekere, N.C.W.; Lindberg, U. The structure of crystalline profilin–β-actin. Nature 1993, 365, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Yamashita, M.; Higashi, T.; Suetsugu, S.; Sato, Y.; Ikeda, T.; Shirakawa, R.; Kita, T.; Takenawa, T.; Horiuchi, H.; Fukai, S.; et al. Crystal structure of human DAAM1 formin homology 2 domain. Genes Cells 2007, 12, 1255–1265. [Google Scholar] [CrossRef]
- Kursula, P.; Kursula, I.; Massimi, M.; Song, Y.H.; Downer, J.; Stanley, W.A.; Witke, W.; Wilmanns, M. High-resolution structural analysis of mammalian profilin 2a complex formation with two physiological ligands: The formin homology 1 domain of mDia1 and the proline-rich domain of VASP. J. Mol. Biol. 2008, 375, 270–290. [Google Scholar] [CrossRef]
- Otomo, T.; Tomchick, D.R.; Otomo, C.; Panchal, S.C.; Machius, M.; Rosen, M.K. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 2005, 433, 488–494. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef]
- Funk, J.; Merino, F.; Venkova, L.; Heydenreich, L.; Kierfeld, J.; Vargas, P.; Raunser, S.; Piel, M.; Bieling, P. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife 2019, 8, e50963. [Google Scholar] [CrossRef]
- Shimizu, N.; Obinata, T. Presence of Three Actin Types in Skeletal Muscle of Chick Embryos. Dev. Growth Differ. 1980, 22, 789–796. [Google Scholar] [CrossRef]
- Shimizu, N.; Obinata, T. Actin Concentration and Monomer-Polymer Ratio in Developing Chicken Skeletal Muscle 1. J. Biochem. 1986, 99, 751–759. [Google Scholar] [CrossRef]
- Safer, D.; Nachmias, V.T. Beta thymosins as actin binding peptides. BioEssays News Rev. Mol. Cell. Dev. Biol. 1994, 16, 473–479. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, E.M.; Ostap, E.M.; Brundage, R.A.; Reddy, K.S.; Sweeney, H.L.; Safer, D. Thymosin-β4 Changes the Conformation and Dynamics of Actin Monomers. Biophys. J. 2000, 78, 2516–2527. [Google Scholar] [CrossRef][Green Version]
- Babcock, G.; Rubenstein, P.A. Control of profilin and actin expression in muscle and nonmuscle cells. Cell Motil. Cytoskelet. 1993, 24, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, R.; Minami, N.; Hayakawa, K.; Abe, H.; Obinata, T. Quantitative analysis of low molecular weight G-actin-binding proteins, cofilin, ADF and profilin, expressed in developing and degenerating chicken skeletal muscles. J. Muscle Res. Cell Motil. 1996, 17, 463–473. [Google Scholar] [CrossRef]
- Kooij, V.; Viswanathan, M.C.; Lee, D.I.; Rainer, P.P.; Schmidt, W.; Kronert, W.A.; Harding, S.E.; Kass, D.A.; Bernstein, S.I.; Van Eyk, J.E.; et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 110, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R. The WH2 Domain and Actin Nucleation: Necessary but Insufficient. Trends Biochem. Sci. 2016, 41, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Higgs, H.N.; Peterson, K.J. Phylogenetic analysis of the formin homology 2 domain. Mol. Biol. Cell 2005, 16, 1–13. [Google Scholar] [CrossRef]
- Pring, M.; Evangelista, M.; Boone, C.; Yang, C.; Zigmond, S.H. Mechanism of formin-induced nucleation of actin filaments. Biochemistry 2003, 42, 486–496. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.S.; Pollard, T.D. Review of the mechanism of processive actin filament elongation by formins. Cell Motil. Cytoskelet. 2009, 66, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Kovar, D.R.; Kuhn, J.R.; Tichy, A.L.; Pollard, T.D. The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J. Cell Biol. 2003, 161, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, S.H.; Evangelista, M.; Boone, C.; Yang, C.; Dar, A.C.; Sicheri, F.; Forkey, J.; Pring, M. Formin leaky cap allows elongation in the presence of tight capping proteins. Curr. Biol. CB 2003, 13, 1820–1823. [Google Scholar] [CrossRef]
- Moseley, J.B.; Sagot, I.; Manning, A.L.; Xu, Y.; Eck, M.J.; Pellman, D.; Goode, B.L. A Conserved Mechanism for Bni1- and mDia1-induced Actin Assembly and Dual Regulation of Bni1 by Bud6 and Profilin. Mol. Biol. Cell 2003, 15, 896–907. [Google Scholar] [CrossRef]
- Gutsche-Perelroizen, I.; Lepault, J.; Ott, A.; Carlier, M.F. Filament assembly from profilin-actin. J. Biol. Chem. 1999, 274, 6234–6243. [Google Scholar] [CrossRef]
- Kinosian, H.; Selden, L.; Gershman, L.; Estes, J. Actin Filament Barbed End Elongation with Nonmuscle MgATP−Actin and MgADP−Actin in the Presence of Profilin. Biochemistry 2002, 41, 6734–6743. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry 1984, 23, 6631–6641. [Google Scholar] [CrossRef]
- Pring, M.; Weber, A.; Bubb, M.R. Profilin-actin complexes directly elongate actin filaments at the barbed end. Biochemistry 1992, 31, 1827–1836. [Google Scholar] [CrossRef]
- Bartolini, F.; Gundersen, G.G. Formins and microtubules. Biochim. Biophys. Acta 2010, 1803, 164–173. [Google Scholar] [CrossRef]
- Bartolini, F.; Moseley, J.B.; Schmoranzer, J.; Cassimeris, L.; Goode, B.L.; Gundersen, G.G. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 2008, 181, 523–536. [Google Scholar] [CrossRef]
- Gaillard, J.; Ramabhadran, V.; Neumanne, E.; Gurel, P.; Blanchoin, L.; Vantard, M.; Higgs, H.N. Differential interactions of the formins INF2, mDia1, and mDia2 with microtubules. Mol. Biol. Cell 2011, 22, 4575–4587. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.S.; Higgs, H.N. Biochemical Analysis of Mammalian Formin Effects on Actin Dynamics. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2006; Volume 406, pp. 190–214. [Google Scholar]
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 2004, 6, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Vig, A.T.; Földi, I.; Szikora, S.; Migh, E.; Gombos, R.; Tóth, M.; Huber, T.; Pintér, R.; Talián, G.C.; Mihály, J.; et al. The activities of the C-terminal regions of the formin protein disheveled-associated activator of morphogenesis (DAAM) in actin dynamics. J. Biol. Chem. 2017, 292, 13566–13583. [Google Scholar] [CrossRef] [PubMed]
- Szikora, S.; Földi, I.; Tóth, K.; Migh, E.; Vig, A.; Bugyi, B.; Maléth, J.; Hegyi, P.; Kaltenecker, P.; Sanchez-Soriano, N.; et al. The formin DAAM is required for coordination of the actin and microtubule cytoskeleton in axonal growth cones. J. Cell Sci. 2017, 130, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Foldi, I.; Szikora, S.; Mihály, J. Formin’ bridges between microtubules and actin filaments in axonal growth cones. Neural Regen. Res. 2017, 12, 1971–1973. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical roles for multiple formins during cardiac myofibril development and repair. Mol. Biol. Cell 2014, 25, 811–827. [Google Scholar] [CrossRef]
- Spletter, M.L.; Barz, C.; Yeroslaviz, A.; Zhang, X.; Lemke, S.B.; Bonnard, A.; Brunner, E.; Cardone, G.; Basler, K.; Habermann, B.H.; et al. A transcriptomics resource reveals a transcriptional transition during ordered sarcomere morphogenesis in flight muscle. eLife 2018, 7, e34058. [Google Scholar] [CrossRef]
- Al Haj, A.; Mazur, A.J.; Radaszkiewicz, K.; Radaszkiewicz, T.; Makowiecka, A.; Stopschinski, B.E.; Schönichen, A.; Geyer, M.; Mannherz, H.G. Distribution of formins in cardiac muscle: FHOD1 is a component of intercalated discs and costameres. Eur. J. Cell Biol. 2015, 94, 101–113. [Google Scholar] [CrossRef]
- Dwyer, J.; Pluess, M.; Iskratsch, T.; Dos Remedios, C.G.; Ehler, E. The formin FHOD1 in cardiomyocytes. Anat. Rec. 2014, 297, 1560–1570. [Google Scholar] [CrossRef]
- Kanaya, H.; Takeya, R.; Takeuchi, K.; Watanabe, N.; Jing, N.; Sumimoto, H. Fhos2, a novel formin-related actin-organizing protein, probably associates with the nestin intermediate filament. Genes Cells 2005, 10, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan, O.M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Lange, S.; Dwyer, J.; Kho, A.L.; dos Remedios, C.; Ehler, E. Formin follows function: A muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J. Cell Biol. 2010, 191, 1159–1172. [Google Scholar] [CrossRef]
- Kan-o, M.; Takeya, R.; Taniguchi, K.; Tanoue, Y.; Tominaga, R.; Sumimoto, H. Expression and Subcellular Localization of Mammalian Formin Fhod3 in the Embryonic and Adult Heart. PLoS ONE 2012, 7, e34765. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Wooten, E.C.; Hebl, V.B.; Wolf, M.J.; Greytak, S.R.; Orr, N.M.; Draper, I.; Calvino, J.E.; Kapur, N.K.; Maron, M.S.; Kullo, I.J.; et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circulation. Cardiovasc. Genet. 2013, 6, 10–18. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Dos Remedios, C.G.; Li, A.; Lal, S. Non-sarcomeric causes of heart failure: A Sydney Heart Bank perspective. Biophys. Rev. 2018, 10, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan, O.M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef]
- Shwartz, A.; Dhanyasi, N.; Schejter, E.D.; Shilo, B.-Z. The Drosophila formin Fhos is a primary mediator of sarcomeric thin-filament array assembly. eLife 2016, 5, e16540. [Google Scholar] [CrossRef]
- Schönichen, A.; Mannherz, H.G.; Behrmann, E.; Mazur, A.J.; Kühn, S.; Silván, U.; Schoenenberger, C.-A.; Fackler, O.T.; Raunser, S.; Dehmelt, L.; et al. FHOD1 is a combined actin filament capping and bundling factor that selectively associates with actin arcs and stress fibers. J. Cell Sci. 2013, 126, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Oztug Durer, Z.A.; van Loon, A.P.; Bremer, K.V.; Quinlan, M.E. Drosophila and human FHOD family formin proteins nucleate actin filaments. J. Biol. Chem. 2018, 293, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Kan, O.M.; Ushijima, T.; Kage, Y.; Tominaga, R.; Sumimoto, H.; Takeya, R. Transgenic Expression of the Formin Protein Fhod3 Selectively in the Embryonic Heart: Role of Actin-Binding Activity of Fhod3 and Its Sarcomeric Localization during Myofibrillogenesis. PLoS ONE 2016, 11, e0148472. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Kage, Y.; Fujimoto, N.; Ushijima, T.; Tsuruda, T.; Kitamura, K.; Shiose, A.; Asada, Y.; Sumimoto, H.; Takeya, R. Interaction between cardiac myosin-binding protein C and formin Fhod3. Proc. Natl. Acad. Sci. USA 2018, 115, E4386–E4395. [Google Scholar] [CrossRef]
- Li, D.; Hallett, M.A.; Zhu, W.; Rubart, M.; Liu, Y.; Yang, Z.; Chen, H.; Haneline, L.S.; Chan, R.J.; Schwartz, R.J.; et al. Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development 2011, 138, 303–315. [Google Scholar] [CrossRef]
- Bao, B.; Zhang, L.; Hu, H.; Yin, S.; Liang, Z. Deletion of a single-copy DAAM1 gene in congenital heart defect: A case report. BMC Med. Genet. 2012, 13, 63. [Google Scholar] [CrossRef]
- Ajima, R.; Bisson, J.A.; Helt, J.-C.; Nakaya, M.-A.; Habas, R.; Tessarollo, L.; He, X.; Morrisey, E.E.; Yamaguchi, T.P.; Cohen, E.D. DAAM1 and DAAM2 are co-required for myocardial maturation and sarcomere assembly. Dev. Biol. 2015, 408, 126–139. [Google Scholar] [CrossRef]
- Molnár, I.; Migh, E.; Szikora, S.; Kalmár, T.; Végh, A.G.; Deák, F.; Barkó, S.; Bugyi, B.; Orfanos, Z.; Kovács, J.; et al. DAAM Is Required for Thin Filament Formation and Sarcomerogenesis during Muscle Development in Drosophila. PLoS Genet. 2014, 10, e1004166. [Google Scholar] [CrossRef]
- Higashi, T.; Ikeda, T.; Shirakawa, R.; Kondo, H.; Kawato, M.; Horiguchi, M.; Okuda, T.; Okawa, K.; Fukai, S.; Nureki, O.; et al. Biochemical characterization of the Rho GTPase-regulated actin assembly by diaphanous-related formins, mDia1 and Daam1, in platelets. J. Biol. Chem. 2008, 283, 8746–8755. [Google Scholar] [CrossRef]
- Barkó, S.; Bugyi, B.; Carlier, M.-F.; Gombos, R.; Matusek, T.; Mihály, J.; Nyitrai, M. Characterization of the Biochemical Properties and Biological Function of the Formin Homology Domains of Drosophila DAAM*. J. Biol. Chem. 2010, 285, 13154–13169. [Google Scholar] [CrossRef]
- Deng, S.; Silimon, R.L.; Balakrishnan, M.; Bothe, I.; Juros, D.; Soffar, D.B.; Baylies, M.K. The actin polymerization factor Diaphanous and the actin severing protein Flightless I collaborate to regulate sarcomere size. Dev. Biol. 2021, 469, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Bothe, I.; Baylies, M.K. The Formin Diaphanous Regulates Myoblast Fusion through Actin Polymerization and Arp2/3 Regulation. PLoS Genet. 2015, 11, e1005381. [Google Scholar] [CrossRef] [PubMed]
- Mi-Mi, L.; Votra, S.; Kemphues, K.; Bretscher, A.; Pruyne, D. Z-line formins promote contractile lattice growth and maintenance in striated muscles of C. elegans. J. Cell Biol. 2012, 198, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Pappas, C.T.; Mayfield, R.M.; Henderson, C.; Jamilpour, N.; Cover, C.; Hernandez, Z.; Hutchinson, K.R.; Chu, M.; Nam, K.H.; Valdez, J.M.; et al. Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc. Natl. Acad. Sci. USA 2015, 112, 13573–13578. [Google Scholar] [CrossRef]
- Kolb, J.; Li, F.; Methawasin, M.; Adler, M.; Escobar, Y.-N.; Nedrud, J.; Pappas, C.T.; Harris, S.P.; Granzier, H. Thin filament length in the cardiac sarcomere varies with sarcomere length but is independent of titin and nebulin. J. Mol. Cell. Cardiol. 2016, 97, 286–294. [Google Scholar] [CrossRef]
- Littlefield, R.; Almenar-Queralt, A.; Fowler, V.M. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 2001, 3, 544–551. [Google Scholar] [CrossRef]
- Mardahl-Dumesnil, M.; Fowler, V.M. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J. Cell Biol. 2001, 155, 1043–1053. [Google Scholar] [CrossRef]
- Michele, D.E.; Albayya, F.P.; Metzger, J.M. Thin Filament Protein Dynamics in Fully Differentiated Adult Cardiac Myocytes: Toward A Model of Sarcomere Maintenance. J. Cell Biol. 1999, 145, 1483–1495. [Google Scholar] [CrossRef]
- Schafer, D.A.; Hug, C.; Cooper, J.A. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 1995, 128, 61–70. [Google Scholar] [CrossRef]
- Weber, A.; Pennise, C.R.; Babcock, G.G.; Fowler, V.M. Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol. 1994, 127, 1627–1635. [Google Scholar] [CrossRef]
- Yamashiro, S.; Gokhin, D.S.; Kimura, S.; Nowak, R.B.; Fowler, V.M. Tropomodulins: Pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton 2012, 69, 337–370. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, C.C.; Weber, A.; Bondad, M.; Pennise, C.R.; Fowler, V.M. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature 1995, 377, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Kostyukova, A.S.; Choy, A.; Rapp, B.A. Tropomodulin binds two tropomyosins: A novel model for actin filament capping. Biochemistry 2006, 45, 12068–12075. [Google Scholar] [CrossRef]
- Sussman, M.A.; Welch, S.; Cambon, N.; Klevitsky, R.; Hewett, T.E.; Price, R.; Witt, S.A.; Kimball, T.R. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J. Clin. Investig. 1998, 101, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Tierney, M.T.; Sui, Z.; Sacco, A.; Fowler, V.M. Calpain-mediated proteolysis of tropomodulin isoforms leads to thin filament elongation in dystrophic skeletal muscle. Mol. Biol. Cell 2014, 25, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Ochala, J.; Domenighetti, A.A.; Fowler, V.M. Tropomodulin 1 directly controls thin filament length in both wild-type and tropomodulin 4-deficient skeletal muscle. Development 2015, 142, 4351–4362. [Google Scholar] [CrossRef]
- Fowler, V.M.; Dominguez, R. Tropomodulins and Leiomodins: Actin Pointed End Caps and Nucleators in Muscles. Biophys. J. 2017, 112, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243. [Google Scholar] [CrossRef]
- Tsukada, T.; Pappas, C.T.; Moroz, N.; Antin, P.B.; Kostyukova, A.S.; Gregorio, C.C. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J. Cell Sci. 2010, 123, 3136–3145. [Google Scholar] [CrossRef]
- Yuen, M.; Sandaradura, S.A.; Dowling, J.J.; Kostyukova, A.S.; Moroz, N.; Quinlan, K.G.; Lehtokari, V.L.; Ravenscroft, G.; Todd, E.J.; Ceyhan-Birsoy, O.; et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Investig. 2014, 124, 4693–4708. [Google Scholar] [CrossRef]
- Boczkowska, M.; Rebowski, G.; Kremneva, E.; Lappalainen, P.; Dominguez, R. How Leiomodin and Tropomodulin use a common fold for different actin assembly functions. Nat. Commun. 2015, 6, 8314. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mo, K.; Tian, H.; Chu, C.; Sun, S.; Tian, L.; Ding, S.; Li, T.-R.; Wu, X.; Liu, F.; et al. Lmod2 piggyBac mutant mice exhibit dilated cardiomyopathy. Cell Biosci. 2016, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Nicklas, R.C.; Pappas, C.T.; Farman, G.P.; Mayfield, R.M.; Larrinaga, T.M.; Medne, L.; Ritter, A.; Krantz, I.D.; Murali, C.; Lin, K.Y.; et al. Disruption of cardiac thin filament assembly arising from a mutation in LMOD2: A novel mechanism of neonatal dilated cardiomyopathy. Sci. Adv. 2019, 5, eaax2066. [Google Scholar] [CrossRef]
- Cenik, B.K.; Garg, A.; McAnally, J.R.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N.; Liu, N. Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Investig. 2015, 125, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Halim, D.; Wilson, M.P.; Oliver, D.; Brosens, E.; Verheij, J.B.; Han, Y.; Nanda, V.; Lyu, Q.; Doukas, M.; Stoop, H.; et al. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. USA 2017, 114, E2739–E2747. [Google Scholar] [CrossRef] [PubMed]
- Skwarek-Maruszewska, A.; Boczkowska, M.; Zajac, A.L.; Kremneva, E.; Svitkina, T.; Dominguez, R.; Lappalainen, P. Different localizations and cellular behaviors of leiomodin and tropomodulin in mature cardiomyocyte sarcomeres. Mol. Biol. Cell 2010, 21, 3352–3361. [Google Scholar] [CrossRef]
- Tolkatchev, D.; Smith, G.E., Jr.; Schultz, L.E.; Colpan, M.; Helms, G.L.; Cort, J.R.; Gregorio, C.C.; Kostyukova, A.S. Leiomodin creates a leaky cap at the pointed end of actin-thin filaments. PLoS Biol. 2020, 18, e3000848. [Google Scholar] [CrossRef]
- Pappas, C.T.; Farman, G.P.; Mayfield, R.M.; Konhilas, J.P.; Gregorio, C.C. Cardiac-specific knockout of Lmod2 results in a severe reduction in myofilament force production and rapid cardiac failure. J. Mol. Cell. Cardiol. 2018, 122, 88–97. [Google Scholar] [CrossRef]
- Bai, J.; Hartwig, J.H.; Perrimon, N. SALS, a WH2-Domain-Containing Protein, Promotes Sarcomeric Actin Filament Elongation from Pointed Ends during Drosophila Muscle Growth. Dev. Cell 2007, 13, 828–842. [Google Scholar] [CrossRef]
- Tolkatchev, D.; Gregorio, C.C.; Kostyukova, A.S. The role of leiomodin in actin dynamics: A new road or a secret gate. FEBS J. 2021. [Google Scholar] [CrossRef]
- Ly, T.; Moroz, N.; Pappas, C.T.; Novak, S.M.; Tolkatchev, D.; Wooldridge, D.; Mayfield, R.M.; Helms, G.; Gregorio, C.C.; Kostyukova, A.S. The N-terminal tropomyosin- and actin-binding sites are important for leiomodin 2’s function. Mol. Biol. Cell 2016, 27, 2565–2575. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tóth, M.Á.; Majoros, A.K.; Vig, A.T.; Migh, E.; Nyitrai, M.; Mihály, J.; Bugyi, B. Biochemical Activities of the Wiskott-Aldrich Syndrome Homology Region 2 Domains of Sarcomere Length Short (SALS) Protein*. J. Biol. Chem. 2016, 291, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Watanabe-Takano, H.; Suetsugu, S.; Kurita, S.; Tsujita, K.; Kimura, S.; Karatsu, T.; Takenawa, T.; Endo, T. Nebulin and N-WASP cooperate to cause IGF-1-induced sarcomeric actin filament formation. Science 2010, 330, 1536–1540. [Google Scholar] [CrossRef]
- Boczkowska, M.; Yurtsever, Z.; Rebowski, G.; Eck, M.J.; Dominguez, R. Crystal Structure of Leiomodin 2 in Complex with Actin: A Structural and Functional Reexamination. Biophys. J. 2017, 113, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Paunola, E.; Mattila, P.K.; Lappalainen, P. WH2 domain: A small, versatile adapter for actin monomers. FEBS Lett. 2002, 513, 92–97. [Google Scholar] [CrossRef]
- Conley, C.A.; Fritz-Six, K.L.; Almenar-Queralt, A.; Fowler, V.M. Leiomodins: Larger members of the tropomodulin (Tmod) gene family. Genomics 2001, 73, 127–139. [Google Scholar] [CrossRef]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef]
- Szatmári, D.; Bugyi, B.; Ujfalusi, Z.; Grama, L.; Dudás, R.; Nyitrai, M. Cardiac leiomodin2 binds to the sides of actin filaments and regulates the ATPase activity of myosin. PLoS ONE 2017, 12, e0186288. [Google Scholar] [CrossRef]
- Li, F.; Barton, E.R.; Granzier, H. Deleting nebulin’s C-terminus reveals its importance to sarcomeric structure and function and is sufficient to invoke nemaline myopathy. Hum. Mol. Genet. 2019, 28, 1709–1725. [Google Scholar] [CrossRef]
- Yamamoto, D.L.; Vitiello, C.; Zhang, J.; Gokhin, D.S.; Castaldi, A.; Coulis, G.; Piaser, F.; Filomena, M.C.; Eggenhuizen, P.J.; Kunderfranco, P.; et al. The nebulin SH3 domain is dispensable for normal skeletal muscle structure but is required for effective active load bearing in mouse. J. Cell Sci. 2013, 126, 5477–5489. [Google Scholar] [CrossRef]
- Kremneva, E.; Makkonen, M.H.; Skwarek-Maruszewska, A.; Gateva, G.; Michelot, A.; Dominguez, R.; Lappalainen, P. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev. Cell 2014, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, M.; Yu, S.F.; Chin, S.M.; Soffar, D.B.; Windner, S.E.; Goode, B.L.; Baylies, M.K. Cofilin Loss in Drosophila Muscles Contributes to Muscle Weakness through Defective Sarcomerogenesis during Muscle Growth. Cell Rep. 2020, 32, 107893. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Parast, M.; Alberico, C.; Benian, G.M.; Ono, S. Specific requirement for two ADF/cofilin isoforms in distinct actin-dependent processes in Caenorhabditis elegans. J. Cell Sci. 2003, 116, 2073–2085. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Sato, N.; Nakagaki, T.; Abe, H.; Ono, S.; Obinata, T. Two mouse cofilin isoforms, muscle-type (MCF) and non-muscle type (NMCF), interact with F-actin with different efficiencies. J. Biochem. 2005, 138, 519–526. [Google Scholar] [CrossRef]
- Gurniak, C.B.; Chevessier, F.; Jokwitz, M.; Jönsson, F.; Perlas, E.; Richter, H.; Matern, G.; Boyl, P.P.; Chaponnier, C.; Fürst, D.; et al. Severe protein aggregate myopathy in a knockout mouse model points to an essential role of cofilin2 in sarcomeric actin exchange and muscle maintenance. Eur. J. Cell Biol. 2014, 93, 252–266. [Google Scholar] [CrossRef]
- Agrawal, P.B.; Greenleaf, R.S.; Tomczak, K.K.; Lehtokari, V.L.; Wallgren-Pettersson, C.; Wallefeld, W.; Laing, N.G.; Darras, B.T.; Maciver, S.K.; Dormitzer, P.R.; et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 2007, 80, 162–167. [Google Scholar] [CrossRef]
- Ockeloen, C.W.; Gilhuis, H.J.; Pfundt, R.; Kamsteeg, E.J.; Agrawal, P.B.; Beggs, A.H.; Hama-Amin, A.D.; Diekstra, A.; Knoers, N.V.; Lammens, M.; et al. Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul. Disord. NMD 2012, 22, 632–639. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Mustonen, T.; Mattila, P.K.; Ojala, P.J.; Thesleff, I.; Partanen, J.; Lappalainen, P. The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics. Mol. Biol. Cell 2002, 13, 183–194. [Google Scholar] [CrossRef]
- Nomura, K.; Ono, K.; Ono, S. CAS-1, a C. elegans cyclase-associated protein, is required for sarcomeric actin assembly in striated muscle. J. Cell Sci. 2012, 125, 4077–4089. [Google Scholar] [CrossRef]
- Effendi, K.; Yamazaki, K.; Mori, T.; Masugi, Y.; Makino, S.; Sakamoto, M. Involvement of hepatocellular carcinoma biomarker, cyclase-associated protein 2 in zebrafish body development and cancer progression. Exp. Cell Res. 2013, 319, 35–44. [Google Scholar] [CrossRef]
- Field, J.; Vojtek, A.; Ballester, R.; Bolger, G.; Colicelli, J.; Ferguson, K.; Gerst, J.; Kataoka, T.; Michaeli, T.; Powers, S.; et al. Cloning and characterization of CAP, the S. cerevisiae gene encoding the 70 kd adenylyl cyclase-associated protein. Cell 1990, 61, 319–327. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell Sci. 2002, 115, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Little, K.; Talarico, L.; Quintero-Monzon, O.; Goode, B.L. A central role for the WH2 domain of Srv2/CAP in recharging actin monomers to drive actin turnover in vitro and in vivo. Cytoskeleton 2010, 67, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Kotila, T.; Wioland, H.; Enkavi, G.; Kogan, K.; Vattulainen, I.; Jégou, A.; Romet-Lemonne, G.; Lappalainen, P. Mechanism of synergistic actin filament pointed end depolymerization by cyclase-associated protein and cofilin. Nat. Commun. 2019, 10, 5320. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Chung, J.; Kondev, J.; Gelles, J.; Goode, B.L. Synergy between Cyclase-associated protein and Cofilin accelerates actin filament depolymerization by two orders of magnitude. Nat. Commun. 2019, 10, 5319. [Google Scholar] [CrossRef]
- Purde, V.; Busch, F.; Kudryashova, E.; Wysocki, V.H.; Kudryashov, D.S. Oligomerization Affects the Ability of Human Cyclase-Associated Proteins 1 and 2 to Promote Actin Severing by Cofilins. Int. J. Mol. Sci. 2019, 20, 5647. [Google Scholar] [CrossRef]
- Kotila, T.; Kogan, K.; Enkavi, G.; Guo, S.; Vattulainen, I.; Goode, B.L.; Lappalainen, P. Structural basis of actin monomer re-charging by cyclase-associated protein. Nat. Commun. 2018, 9, 1892. [Google Scholar] [CrossRef]
- Bertling, E.; Hotulainen, P.; Mattila, P.K.; Matilainen, T.; Salminen, M.; Lappalainen, P. Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 2004, 15, 2324–2334. [Google Scholar] [CrossRef]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schröder, R.; Schleicher, M.; Holak, T.A.; Clemen, C.S.; Ramanath, Y.B.; Pfitzer, G.; et al. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell. Mol. Life Sci. CMLS 2007, 64, 2702–2715. [Google Scholar] [CrossRef]
- Colpan, M.; Iwanski, J.; Gregorio, C.C. CAP2 is a regulator of actin pointed end dynamics and myofibrillogenesis in cardiac muscle. Commun. Biol. 2021, 4, 365. [Google Scholar] [CrossRef]
- Kepser, L.J.; Damar, F.; De Cicco, T.; Chaponnier, C.; Prószyński, T.J.; Pagenstecher, A.; Rust, M.B. CAP2 deficiency delays myofibril actin cytoskeleton differentiation and disturbs skeletal muscle architecture and function. Proc. Natl. Acad. Sci. USA 2019, 116, 8397–8402. [Google Scholar] [CrossRef] [PubMed]
- Peche, V.S.; Holak, T.A.; Burgute, B.D.; Kosmas, K.; Kale, S.P.; Wunderlich, F.T.; Elhamine, F.; Stehle, R.; Pfitzer, G.; Nohroudi, K.; et al. Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell. Mol. LifeSci. CMLS 2013, 70, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Field, J.; Ye, D.Z.; Shinde, M.; Liu, F.; Schillinger, K.J.; Lu, M.; Wang, T.; Skettini, M.; Xiong, Y.; Brice, A.K.; et al. CAP2 in cardiac conduction, sudden cardiac death and eye development. Sci. Rep. 2015, 5, 17256. [Google Scholar] [CrossRef] [PubMed]
- Aspit, L.; Levitas, A.; Etzion, S.; Krymko, H.; Slanovic, L.; Zarivach, R.; Etzion, Y.; Parvari, R. CAP2 mutation leads to impaired actin dynamics and associates with supraventricular tachycardia and dilated cardiomyopathy. J. Med. Genet. 2019, 56, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Fowler, V.M. Regulation of actin filament length in erythrocytes and striated muscle. Curr. Opin. Cell Biol. 1996, 8, 86–96. [Google Scholar] [CrossRef]
- Wang, K.; Wright, J. Architecture of the sarcomere matrix of skeletal muscle: Immunoelectron microscopic evidence that suggests a set of parallel inextensible nebulin filaments anchored at the Z line. J. Cell Biol. 1988, 107, 2199–2212. [Google Scholar] [CrossRef]
- Maruyama, K.; Matsuno, A.; Higuchi, H.; Shimaoka, S.; Kimura, S.; Shimizu, T. Behaviour of connectin (titin) and nebulin in skinned muscle fibres released after extreme stretch as revealed by immunoelectron microscopy. J. Muscle Res. Cell Motil. 1989, 10, 350–359. [Google Scholar] [CrossRef]
- Pierobon-Bormioli, S.; Betto, R.; Salviati, G. The organization of titin (connectin) and nebulin in the sarcomeres: An immunocytolocalization study. J. Muscle Res. Cell Motil. 1989, 10, 446–456. [Google Scholar] [CrossRef]
- Kruger, M.; Wright, J.; Wang, K. Nebulin as a length regulator of thin filaments of vertebrate skeletal muscles: Correlation of thin filament length, nebulin size, and epitope profile. J. Cell Biol. 1991, 115, 97–107. [Google Scholar] [CrossRef]
- Jin, J.P.; Wang, K. Nebulin as a giant actin-binding template protein in skeletal muscle sarcomere. Interaction of actin and cloned human nebulin fragments. FEBS Lett. 1991, 281, 93–96. [Google Scholar] [CrossRef]
- Wright, J.; Huang, Q.Q.; Wang, K. Nebulin is a full-length template of actin filaments in the skeletal muscle sarcomere: An immunoelectron microscopic study of its orientation and span with site-specific monoclonal antibodies. J. Muscle Res. Cell Motil. 1993, 14, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Kolmerer, B. The complete primary structure of human nebulin and its correlation to muscle structure. J. Mol. Biol. 1995, 248, 308–315. [Google Scholar] [CrossRef]
- Lehtokari, V.L.; Pelin, K.; Sandbacka, M.; Ranta, S.; Donner, K.; Muntoni, F.; Sewry, C.; Angelini, C.; Bushby, K.; Van den Bergh, P.; et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum. Mutat. 2006, 27, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.B.; Lehtokari, V.L.; Quijano-Roy, S.; Monnier, N.; Claeys, K.G.; Carlier, R.Y.; Pellegrini, N.; Orlikowski, D.; Barois, A.; Laing, N.G.; et al. Core-rod myopathy caused by mutations in the nebulin gene. Neurology 2009, 73, 1159–1161. [Google Scholar] [CrossRef]
- Scoto, M.; Cullup, T.; Cirak, S.; Yau, S.; Manzur, A.Y.; Feng, L.; Jacques, T.S.; Anderson, G.; Abbs, S.; Sewry, C.; et al. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur. J. Hum. Genet. EJHG 2013, 21, 1249–1252. [Google Scholar] [CrossRef]
- Pappas, C.T.; Bliss, K.T.; Zieseniss, A.; Gregorio, C.C. The Nebulin family: An actin support group. Trends Cell Biol. 2011, 21, 29–37. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Granzier, H.; Labeit, S. The sarcomeric protein nebulin: Another multifunctional giant in charge of muscle strength optimization. Front. Physiol 2012, 3, 37. [Google Scholar] [CrossRef][Green Version]
- Labeit, S.; Gibson, T.; Lakey, A.; Leonard, K.; Zeviani, M.; Knight, P.; Wardale, J.; Trinick, J. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett. 1991, 282, 313–316. [Google Scholar] [CrossRef]
- Donner, K.; Sandbacka, M.; Lehtokari, V.L.; Wallgren-Pettersson, C.; Pelin, K. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur. J. Hum. Genet. EJHG 2004, 12, 744–751. [Google Scholar] [CrossRef]
- Laitila, J.; Hanif, M.; Paetau, A.; Hujanen, S.; Keto, J.; Somervuo, P.; Huovinen, S.; Udd, B.; Wallgren-Pettersson, C.; Auvinen, P.; et al. Expression of multiple nebulin isoforms in human skeletal muscle and brain. Muscle Nerve 2012, 46, 730–737. [Google Scholar] [CrossRef]
- Marttila, M.; Hanif, M.; Lemola, E.; Nowak, K.J.; Laitila, J.; Grönholm, M.; Wallgren-Pettersson, C.; Pelin, K. Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet. Muscle 2014, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Pappas, C.T.; Bhattacharya, N.; Cooper, J.A.; Gregorio, C.C. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol. Biol. Cell 2008, 19, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, A.S.; Kolmerer, B.; Fowler, V.M.; Labeit, S.; Gregorio, C.C. The N-terminal end of nebulin interacts with tropomodulin at the pointed ends of the thin filaments. J. Biol. Chem. 2001, 276, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.; Hudson, B.D.; Ottenheijm, C.A.; Labeit, S.; Granzier, H. Differential splicing of the large sarcomeric protein nebulin during skeletal muscle development. J. Struct. Biol. 2010, 170, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Grange, M.; Pospich, S.; Wagner, T.; Kho, A.L.; Gautel, M.; Raunser, S. Structures from intact myofibrils reveal mechanism of thin filament regulation through nebulin. Science 2022, 375, eabn1934. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.-L.; Li, X.; Littlefield, R.; Bremner, S.; Thor, A.; Knowlton, K.U.; Lieber, R.L.; Chen, J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 2006, 173, 905–916. [Google Scholar] [CrossRef]
- Witt, C.C.; Burkart, C.; Labeit, D.; McNabb, M.; Wu, Y.; Granzier, H.; Labeit, S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. 2006, 25, 3843–3855. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Witt, C.C.; Stienen, G.J.; Labeit, S.; Beggs, A.H.; Granzier, H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum. Mol. Genet. 2009, 18, 2359–2369. [Google Scholar] [CrossRef]
- Pappas, C.T.; Krieg, P.A.; Gregorio, C.C. Nebulin regulates actin filament lengths by a stabilization mechanism. J. Cell Biol. 2010, 189, 859–870. [Google Scholar] [CrossRef]
- Gokhin, D.S.; Fowler, V.M. A two-segment model for thin filament architecture in skeletal muscle. Nat. Rev. Mol. Cell Biol. 2013, 14, 113–119. [Google Scholar] [CrossRef]
- Kiss, B.; Lee, E.J.; Ma, W.; Li, F.W.; Tonino, P.; Mijailovich, S.M.; Irving, T.C.; Granzier, H.L. Nebulin stiffens the thin filament and augments cross-bridge interaction in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, 10369–10374. [Google Scholar] [CrossRef] [PubMed]
- Kiss, B.; Gohlke, J.; Tonino, P.; Hourani, Z.; Kolb, J.; Strom, J.; Alekhina, O.; Smith, J.E., 3rd; Ottenheijm, C.; Gregorio, C.; et al. Nebulin and Lmod2 are critical for specifying thin-filament length in skeletal muscle. Sci. Adv. 2020, 6, eabc1992. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Zhang, J.Q.; Nguyen, T.P.; Herrera, A.H.; Paterson, B.; Horowits, R. Complete cDNA sequence and tissue localization of N-RAP, a novel nebulin-related protein of striated muscle. Cell Motil. Cytoskelet. 1997, 38, 75–90. [Google Scholar] [CrossRef]
- Schreiber, V.; Moog-Lutz, C.; Régnier, C.H.; Chenard, M.-P.; Boeuf, H.; Vonesch, J.-L.; Tomasetto, C.; Rio, M.-C. Lasp-1, a Novel Type of Actin-Binding Protein Accumulating in Cell Membrane Extensions. Mol. Med. 1998, 4, 675–687. [Google Scholar] [CrossRef]
- Li, B.; Zhuang, L.; Trueb, B. Zyxin interacts with the SH3 domains of the cytoskeletal proteins LIM-nebulette and Lasp-1. J. Biol. Chem. 2004, 279, 20401–20410. [Google Scholar] [CrossRef]
- Wu, H.; Reynolds, A.B.; Kanner, S.B.; Vines, R.R.; Parsons, J.T. Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol. Cell. Biol. 1991, 11, 5113–5124. [Google Scholar] [CrossRef]
- Moncman, C.L.; Wang, K. Nebulette: A 107 kD nebulin-like protein in cardiac muscle. Cell Motil. Cytoskelet. 1995, 32, 205–225. [Google Scholar] [CrossRef]
- Bang, M.L.; Chen, J. Roles of Nebulin Family Members in the Heart. Circ. J. 2015, 79, 2081–2087. [Google Scholar] [CrossRef]
- Kazmierski, S.T.; Antin, P.B.; Witt, C.C.; Huebner, N.; McElhinny, A.S.; Labeit, S.; Gregorio, C.C. The complete mouse nebulin gene sequence and the identification of cardiac nebulin. J. Mol. Biol. 2003, 328, 835–846. [Google Scholar] [CrossRef]
- Moncman, C.L.; Wang, K. Functional dissection of nebulette demonstrates actin binding of nebulin-like repeats and Z-line targeting of SH3 and linker domains. Cell Motil Cytoskelet. 1999, 44, 1–22. [Google Scholar] [CrossRef]
- Arimura, T.; Nakamura, T.; Hiroi, S.; Satoh, M.; Takahashi, M.; Ohbuchi, N.; Ueda, K.; Nouchi, T.; Yamaguchi, N.; Akai, J.; et al. Characterization of the human nebulette gene: A polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum. Genet. 2000, 107, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Purevjav, E.; Varela, J.; Morgado, M.; Kearney, D.L.; Li, H.; Taylor, M.D.; Arimura, T.; Moncman, C.L.; McKenna, W.; Murphy, R.T.; et al. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J. Am. Coll. Cardiol. 2010, 56, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Maiellaro-Rafferty, K.; Wansapura, J.P.; Mendsaikhan, U.; Osinska, H.; James, J.F.; Taylor, M.D.; Robbins, J.; Kranias, E.G.; Towbin, J.A.; Purevjav, E. Altered regional cardiac wall mechanics are associated with differential cardiomyocyte calcium handling due to nebulette mutations in preclinical inherited dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 60, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Tomasov, P.; Villard, E.; Faludi, R.; Melacini, P.; Lossie, J.; Lohmann, N.; Richard, P.; De Bortoli, M.; Angelini, A.; et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch. Med. Sci. AMS 2016, 12, 263–278. [Google Scholar] [CrossRef]
- Ogut, O.; Hossain, M.M.; Jin, J.P. Interactions between nebulin-like motifs and thin filament regulatory proteins. J. Biol. Chem. 2003, 278, 3089–3097. [Google Scholar] [CrossRef]
- Esham, M.; Bryan, K.; Milnes, J.; Holmes, W.B.; Moncman, C.L. Expression of nebulette during early cardiac development. Cell Motil. Cytoskelet. 2007, 64, 258–273. [Google Scholar] [CrossRef]
- Mastrototaro, G.; Liang, X.; Li, X.; Carullo, P.; Piroddi, N.; Tesi, C.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Poggesi, C.; et al. Nebulette knockout mice have normal cardiac function, but show Z-line widening and up-regulation of cardiac stress markers. Cardiovasc. Res. 2015, 107, 216–225. [Google Scholar] [CrossRef]
- Fernandes, I.; Schöck, F. The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. J. Cell Biol. 2014, 206, 559–572. [Google Scholar] [CrossRef]
- Wang, K.; McClure, J.; Tu, A. Titin: Major myofibrillar components of striated muscle. Proc. Natl. Acad. Sci. USA 1979, 76, 3698–3702. [Google Scholar] [CrossRef]
- Fürst, D.O.; Osborn, M.; Nave, R.; Weber, K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: A map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 1988, 106, 1563–1572. [Google Scholar] [CrossRef]
- Trombitás, K.; Jin, J.P.; Granzier, H. The mechanically active domain of titin in cardiac muscle. Circ. Res. 1995, 77, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Kellermayer, M.S.; Smith, S.B.; Granzier, H.L.; Bustamante, C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science 1997, 276, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Yoshioka, T.; Higuchi, H.; Ohashi, K.; Kimura, S.; Natori, R. Connectin filaments link thick filaments and Z lines in frog skeletal muscle as revealed by immunoelectron microscopy. J. Cell Biol. 1985, 101, 2167–2172. [Google Scholar] [CrossRef] [PubMed]
- Whiting, A.; Wardale, J.; Trinick, J. Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 1989, 205, 263–268. [Google Scholar] [CrossRef]
- Bennett, P.M.; Gautel, M. Titin Domain Patterns Correlate with the Axial Disposition of Myosin at the End of the Thick Filament. J. Mol. Biol. 1996, 259, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Bennett, P.; Rees, M.; Gautel, M. The Axial Alignment of Titin on the Muscle Thick Filament Supports Its Role as a Molecular Ruler. J. Mol. Biol. 2020, 432, 4815–4829. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J. Titin and Nebulin in Thick and Thin Filament Length Regulation. Subcell. Biochem. 2017, 82, 285–318. [Google Scholar] [CrossRef]
- Gregorio, C.C.; Trombitás, K.; Centner, T.; Kolmerer, B.; Stier, G.; Kunke, K.; Suzuki, K.; Obermayr, F.; Herrmann, B.; Granzier, H.; et al. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 1998, 143, 1013–1027. [Google Scholar] [CrossRef]
- Peckham, M.; Young, P.; Gautel, M. Constitutive and variable regions of Z-disk titin/connectin in myofibril formation: A dominant-negative screen. Cell Struct. Funct. 1997, 22, 95–101. [Google Scholar] [CrossRef][Green Version]
- Linke, W.A.; Ivemeyer, M.; Mundel, P.; Stockmeier, M.R.; Kolmerer, B. Nature of PEVK-titin elasticity in skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 8052–8057. [Google Scholar] [CrossRef]
- Opitz, C.A.; Kulke, M.; Leake, M.C.; Neagoe, C.; Hinssen, H.; Hajjar, R.J.; Linke, W.A. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc. Natl. Acad. Sci. USA 2003, 100, 12688–12693. [Google Scholar] [CrossRef]
- Granzier, H.L.; Labeit, S. Titin and its associated proteins: The third myofilament system of the sarcomere. Adv. Protein Chem. 2005, 71, 89–119. [Google Scholar] [CrossRef]
- Muhle-Goll, C.; Habeck, M.; Cazorla, O.; Nilges, M.; Labeit, S.; Granzier, H. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1—A possible role in the Frank-Starling mechanism of the heart. J. Mol. Biol. 2001, 313, 431–447. [Google Scholar] [CrossRef]
- Freiburg, A.; Gautel, M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 1996, 235, 317–323. [Google Scholar] [CrossRef]
- Obermann, W.M.; Gautel, M.; Weber, K.; Fürst, D.O. Molecular structure of the sarcomeric M band: Mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 1997, 16, 211–220. [Google Scholar] [CrossRef]
- Kontrogianni-Konstantopoulos, A.; Ackermann, M.A.; Bowman, A.L.; Yap, S.V.; Bloch, R.J. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol. Rev. 2009, 89, 1217–1267. [Google Scholar] [CrossRef]
- Tonino, P.; Kiss, B.; Strom, J.; Methawasin, M.; Smith, J.E.; Kolb, J.; Labeit, S.; Granzier, H. The giant protein titin regulates the length of the striated muscle thick filament. Nat. Commun. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Prado, L.G.; Makarenko, I.; Andresen, C.; Krüger, M.; Opitz, C.A.; Linke, W.A. Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J. Gen. Physiol. 2005, 126, 461–480. [Google Scholar] [CrossRef]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity—Alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef]
- Greaser, M.L.; Pleitner, J.M. Titin isoform size is not correlated with thin filament length in rat skeletal muscle. Front. Physiol. 2014, 5, 35. [Google Scholar] [CrossRef]
- Greaser, M.L.; Warren, C.M.; Esbona, K.; Guo, W.; Duan, Y.; Parrish, A.M.; Krzesinski, P.R.; Norman, H.S.; Dunning, S.; Fitzsimons, D.P.; et al. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J. Mol. Cell. Cardiol. 2008, 44, 983–991. [Google Scholar] [CrossRef]
- Methawasin, M.; Hutchinson, K.R.; Lee, E.J.; Smith, J.E., 3rd; Saripalli, C.; Hidalgo, C.G.; Ottenheijm, C.A.; Granzier, H. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 2014, 129, 1924–1936. [Google Scholar] [CrossRef]
- Hooper, S.L.; Thuma, J.B. Invertebrate muscles: Muscle specific genes and proteins. Physiol. Rev. 2005, 85, 1001–1060. [Google Scholar] [CrossRef]
- Reedy, M.C.; Beall, C. Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev. Biol. 1993, 160, 443–465. [Google Scholar] [CrossRef]
- Chun, M.; Falkenthal, S. Ifm(2)2 is a myosin heavy chain allele that disrupts myofibrillar assembly only in the indirect flight muscle of Drosophila melanogaster. J. Cell Biol. 1988, 107, 2613–2621. [Google Scholar] [CrossRef]
- Bernstein, S.I.; O’Donnell, P.T.; Cripps, R.M. Molecular Genetic Analysis of Muscle Development, Structure, and Function in Drosophila. In International Review of Cytology; Jeon, K.W., Friedlander, M., Jarvik, J., Eds.; Academic Press: Cambridge, MA, USA, 1993; Volume 143, pp. 63–152. [Google Scholar]
- Vigoreaux, J.O.; Saide, J.D.; Valgeirsdottir, K.; Pardue, M.L. Flightin, a novel myofibrillar protein of Drosophila stretch-activated muscles. J. Cell Biol. 1993, 121, 587–598. [Google Scholar] [CrossRef]
- Ayer, G.; Vigoreaux, J.O. Flightin is a myosin rod binding protein. Cell Biochem. Biophys. 2003, 38, 41–54. [Google Scholar] [CrossRef]
- Reedy, M.C.; Bullard, B.; Vigoreaux, J.O. Flightin is essential for thick filament assembly and sarcomere stability in Drosophila flight muscles. J. Cell Biol. 2000, 151, 1483–1500. [Google Scholar] [CrossRef]
- Contompasis, J.L.; Nyland, L.R.; Maughan, D.W.; Vigoreaux, J.O. Flightin Is Necessary for Length Determination, Structural Integrity, and Large Bending Stiffness of Insect Flight Muscle Thick Filaments. J. Mol. Biol. 2010, 395, 340–348. [Google Scholar] [CrossRef]
- Gasek, N.S.; Nyland, L.R.; Vigoreaux, J.O. The Contributions of the Amino and Carboxy Terminal Domains of Flightin to the Biomechanical Properties of Drosophila Flight Muscle Thick Filaments. Biology 2016, 5, 16. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szikora, S.; Görög, P.; Mihály, J. The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles. Int. J. Mol. Sci. 2022, 23, 5306. https://doi.org/10.3390/ijms23105306
Szikora S, Görög P, Mihály J. The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles. International Journal of Molecular Sciences. 2022; 23(10):5306. https://doi.org/10.3390/ijms23105306
Chicago/Turabian StyleSzikora, Szilárd, Péter Görög, and József Mihály. 2022. "The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles" International Journal of Molecular Sciences 23, no. 10: 5306. https://doi.org/10.3390/ijms23105306
APA StyleSzikora, S., Görög, P., & Mihály, J. (2022). The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles. International Journal of Molecular Sciences, 23(10), 5306. https://doi.org/10.3390/ijms23105306