
Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors



Abstract
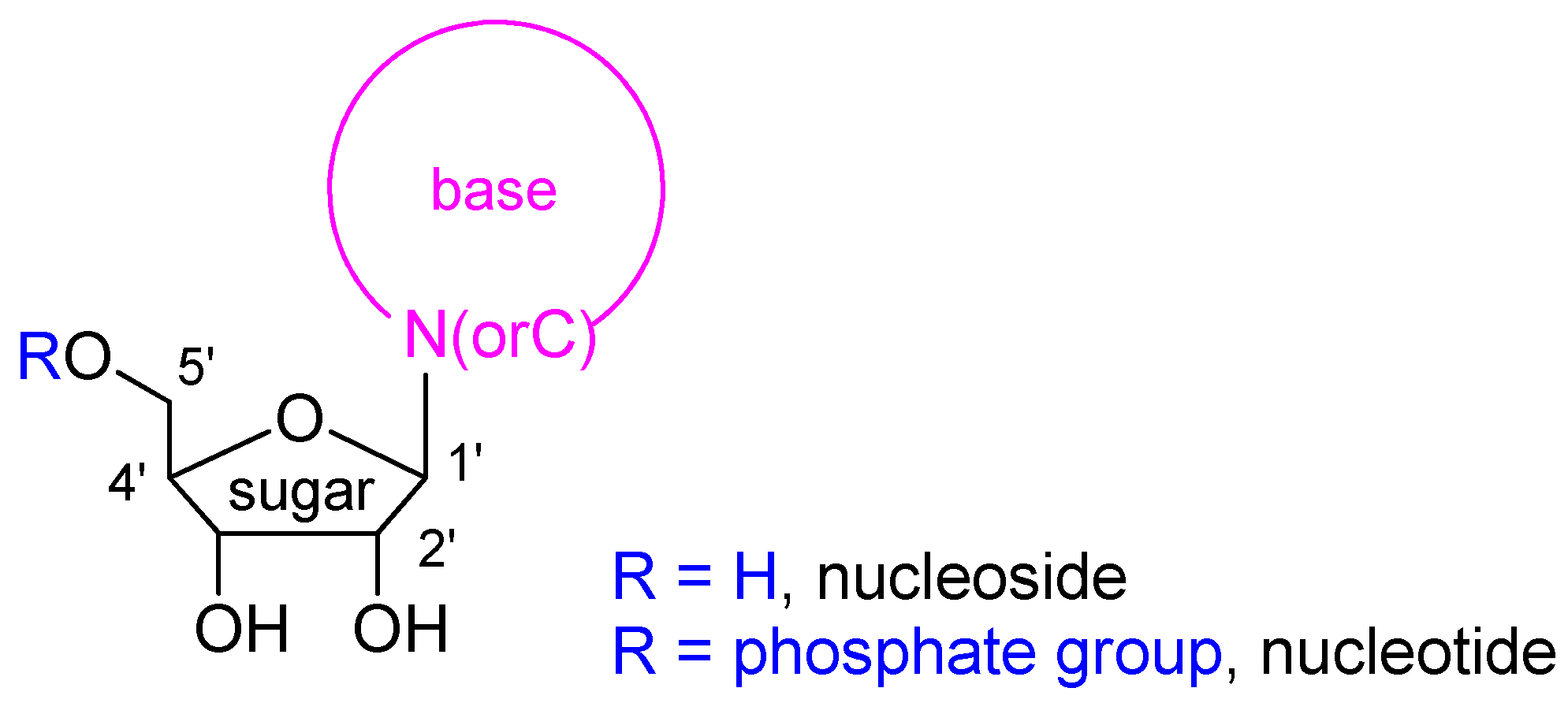
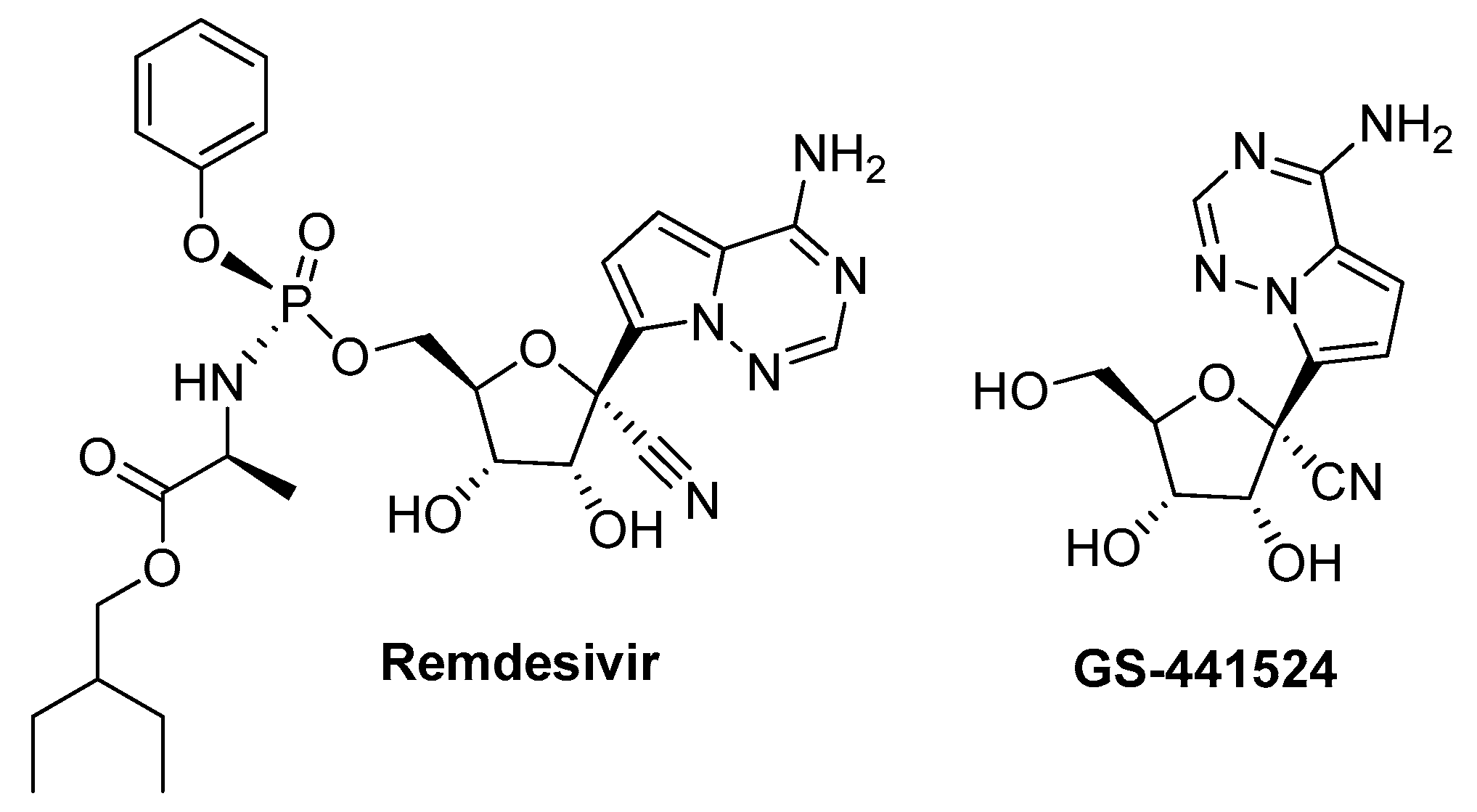
:1. Introduction
2. Results and Discussion
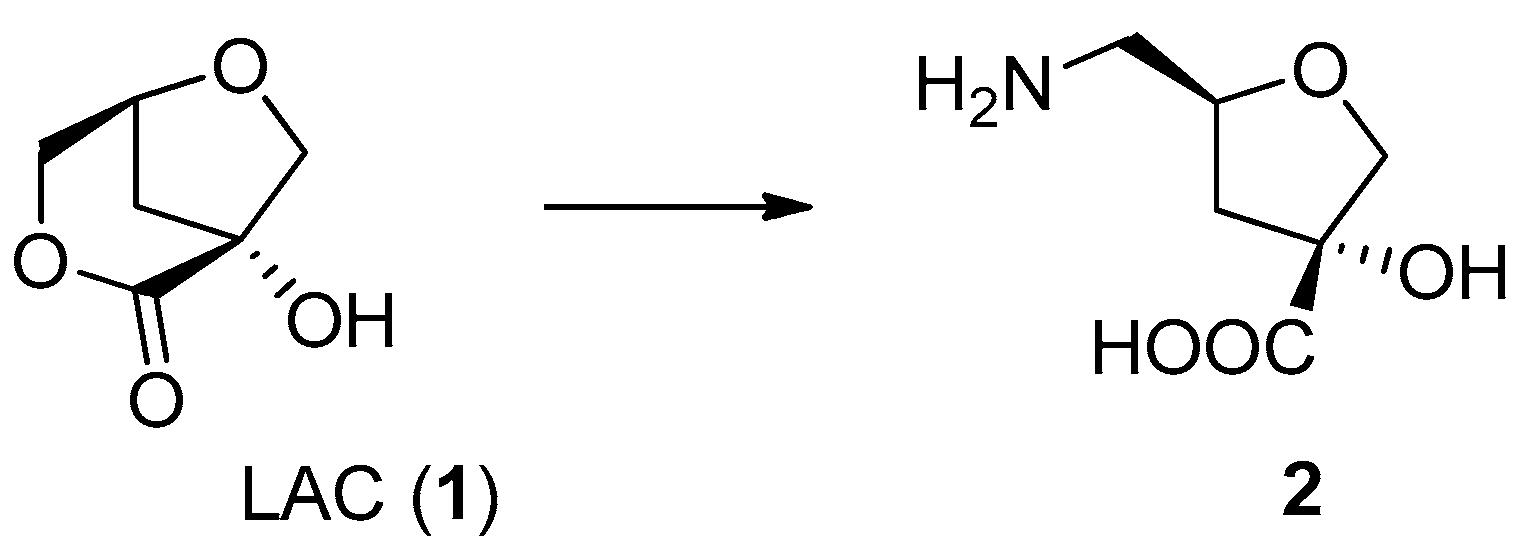
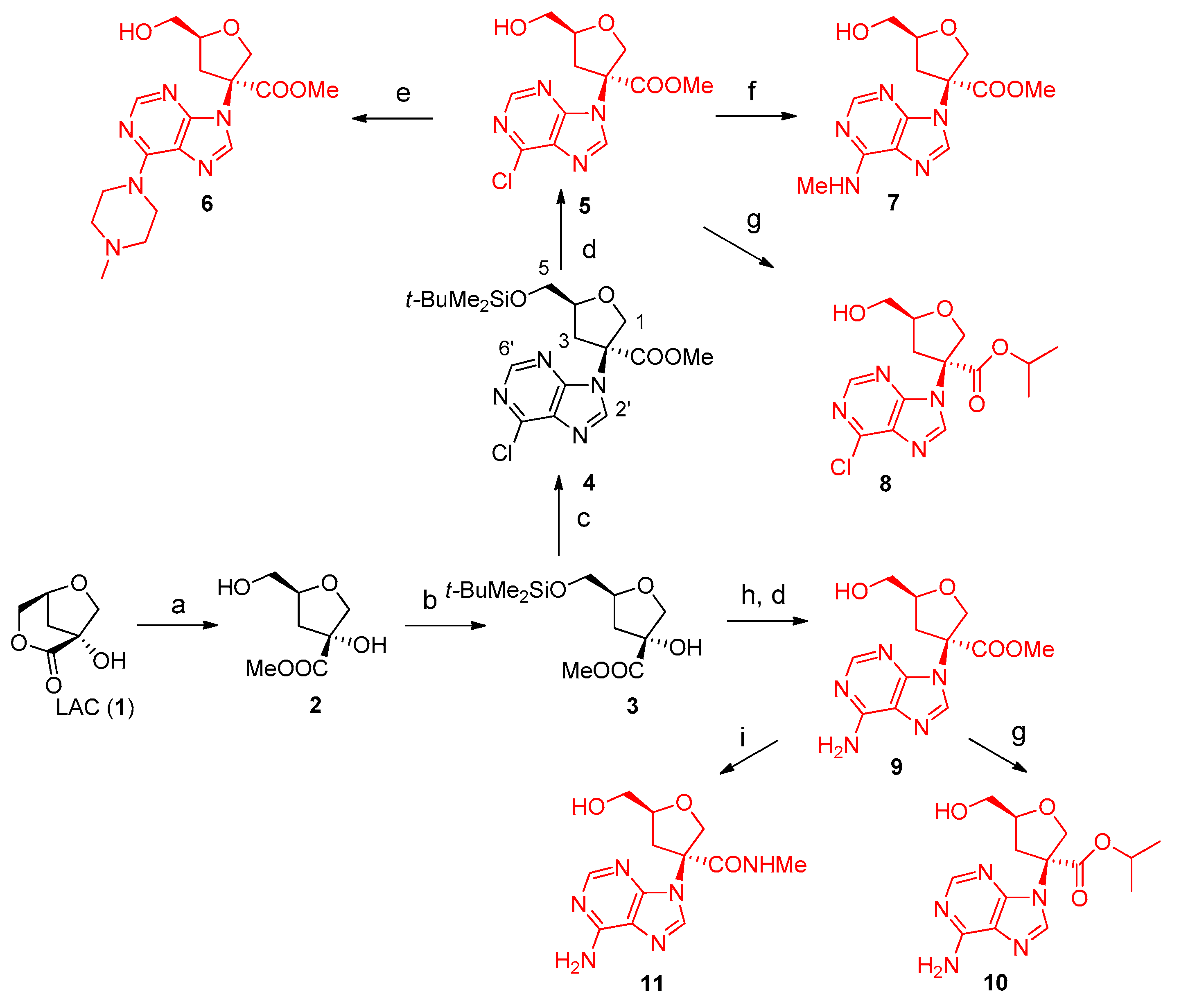
2.1. Synthesis of Nucleosides 5–11
2.2. Pharmacokinetics Studies
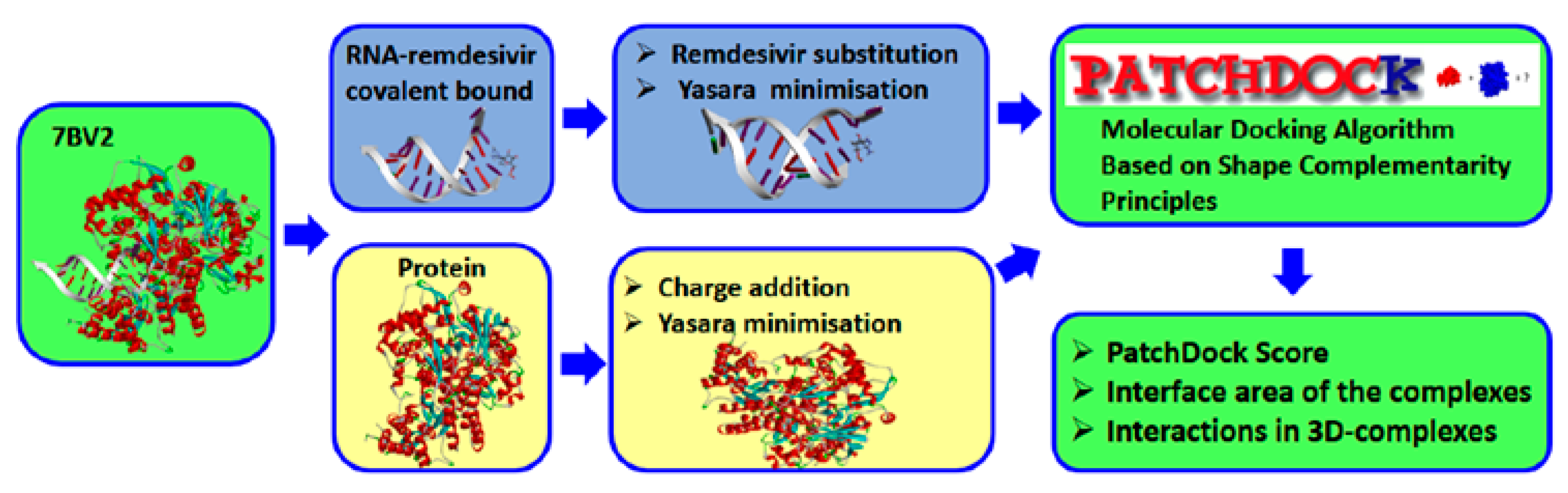
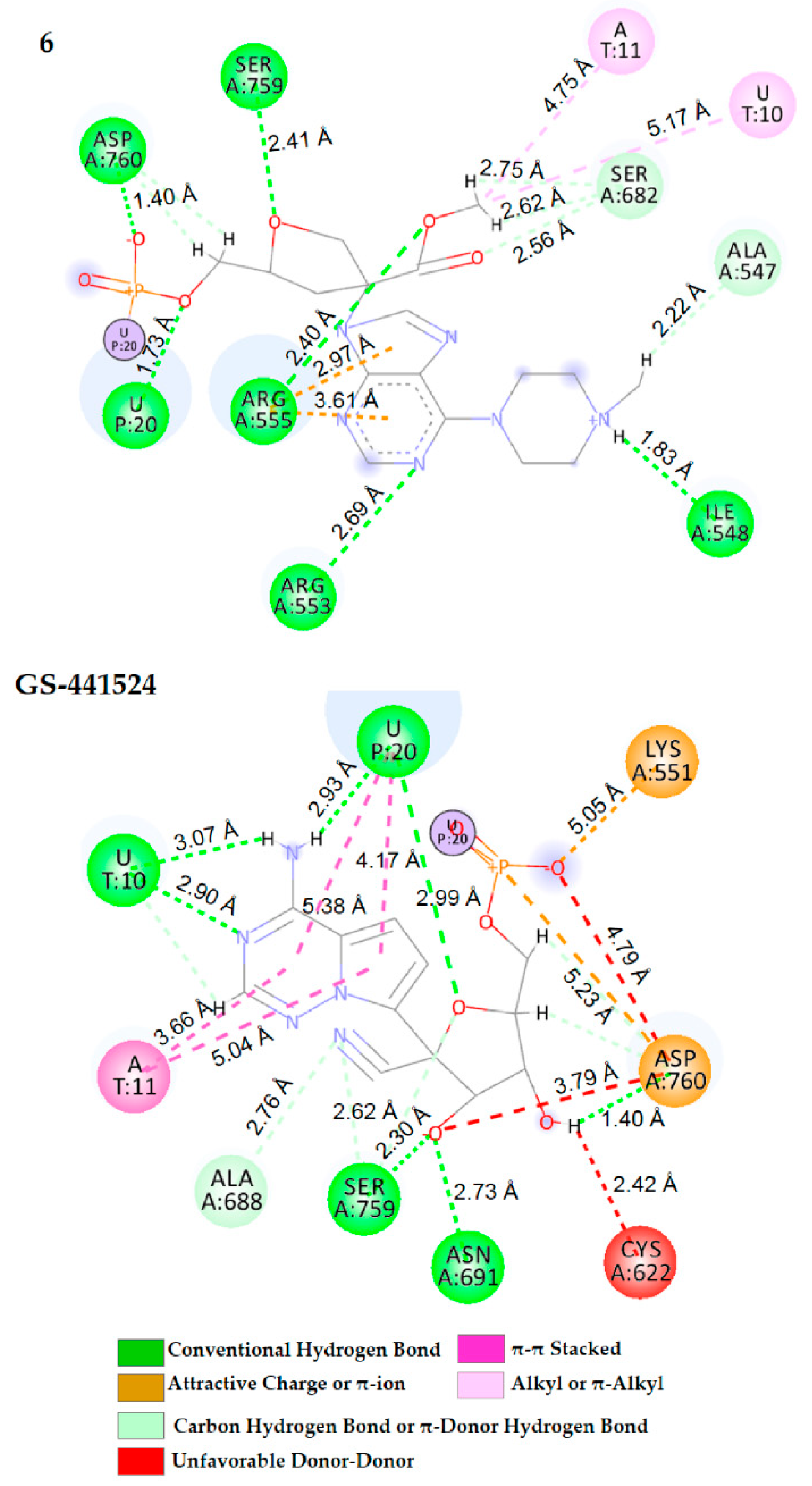
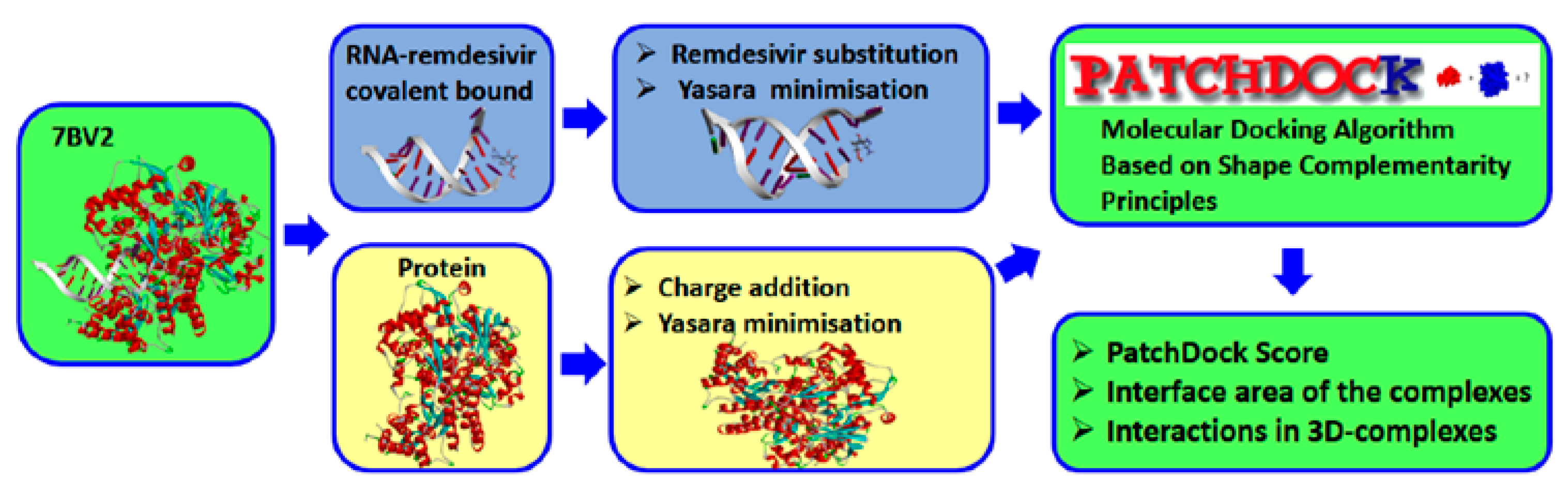
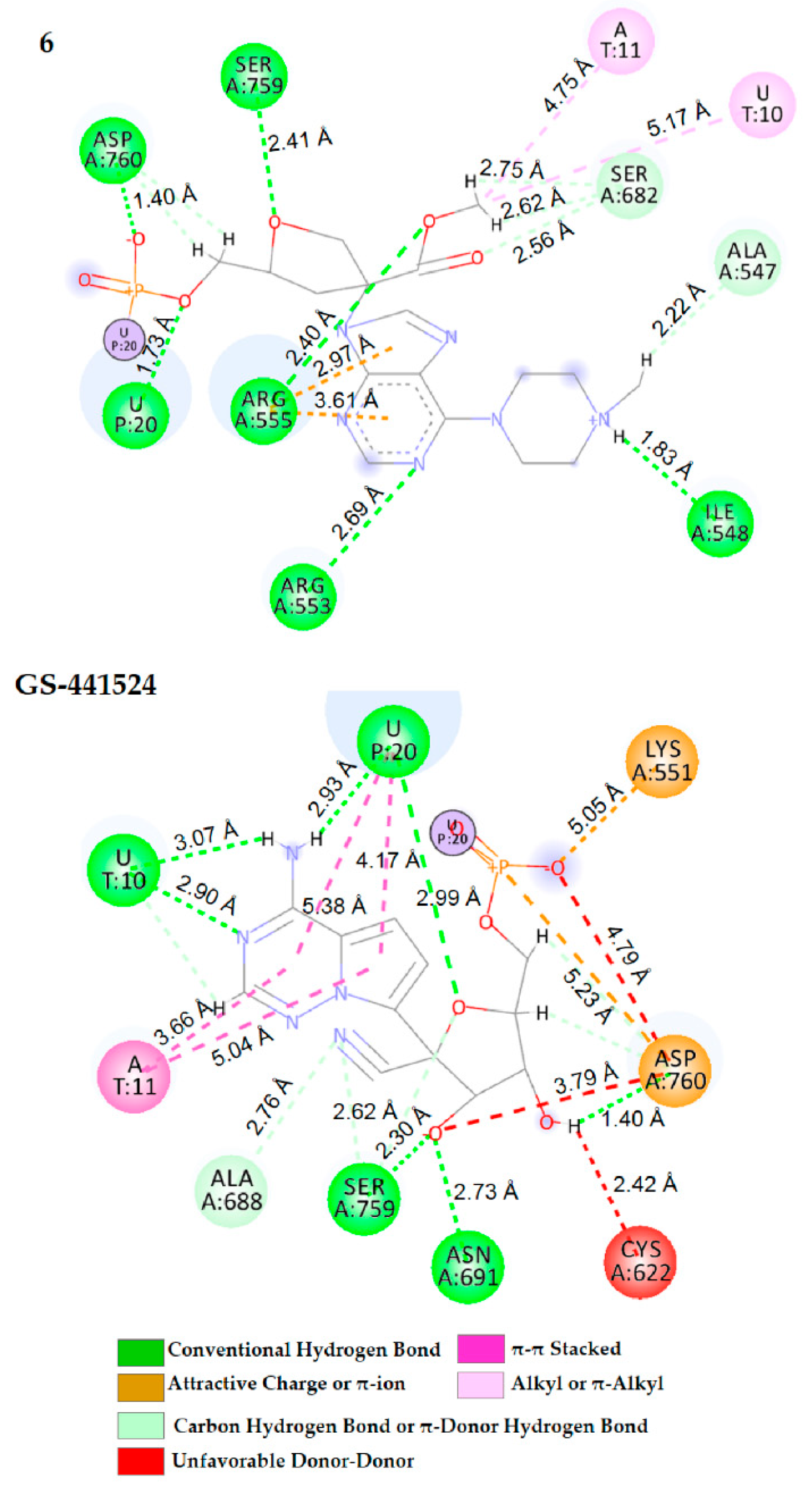
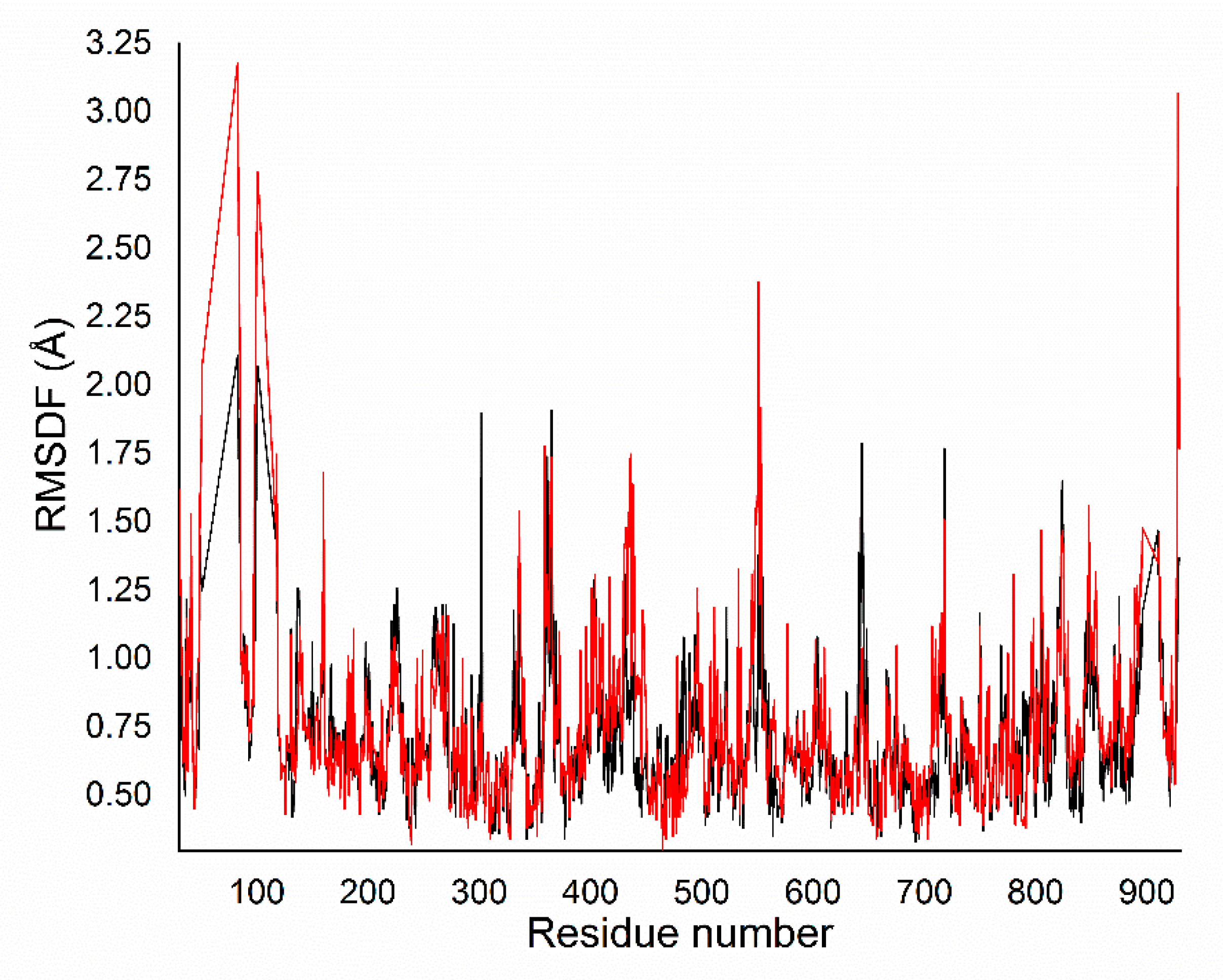
2.3. Docking Calculation and Molecular Dynamics
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis and Structural Characterization of Compounds 3–11
- (3R,5S)-Methyl 3-hydroxy-5-(hydroxymethyl)tetrahydrofuran-3-carboxylate (2). The compound was obtained following the procedure reported and structural assignment is in agreement with known data [3].
- (3R,5S)-Methyl 5-(((tert-butyldimethylsilyl)oxy)methyl)-3-hydroxytetrahydrofuran-3-carboxylate (3). A solution of compound 2 (0.998 g, 5.67 mmol) in dichloromethane (20 mL) was stirred with tert-butylchlorodimethylsilane (1.20 g, 7.94 mmol) and imidazole (0.965 g, 14.18 mmol) at room temperature for 2 h, monitoring the total conversion by TLC. The reaction mixture was concentrated in vacuo and the residue subjected to FC on silica gel, using hexane/ethyl acetate gradient elution as eluent, obtaining pure 3 as a colorless oil. TLC (hexane/EtOAc = 40:60 v/v): Rf = 0.88. Yield: 78%. FT-IR (cm−1): 2952 (w), 1727 (s), 1463 (w), 1238 (s), 1099 (s), 837 (vs), 779 (s). 1H-NMR (400 MHz, CDCl3) δ 4.25 (m, 1H, H4), 4.13 (d, J = 9.5 Hz, 1H, Ha1) 3.79 (d, J = 9.5 Hz, 1H, Hb1), 3.78 (s, 3H, OMe), 3.73 (dd, J = 11.0, 3.9 Hz, 1H, Ha 5), 3.67 (dd, J = 11.0, 4.3 Hz, 1H, Hb 5), 2.30 (dd, J = 12.5, 9.4 Hz, 1H, Ha 3), 2.02 (dd, J = 12.5, 5.5 Hz, 1H, Hb 3), 0.85 (s, 9H, tBu), 0.03 (s, 6H, SiMe2).13C-NMR (100 MHz, CDCl3) δ 181.3 (COO), 80.9, 79.7, 77.5 (C1), 67.9 (C5), 57.9 (OMe), 37.5 (C3), 28.3 (tBu), 1.5 (SiMe2). EI-MS: m/z 290 (M+, 1), 233 (24), 159 (8), 73 (100).
- (3S,5S)-Methyl 5-(((tert-butyldimethylsilyl)oxy)methyl)-3-(6-chloro-9H-purin-9-yl)tetrahydrofuran-3-carboxylate (4). Diethyl azodicarboxylate (DEAD, 40% in toluene, 1.442 g, 8.27 mmol) was slowly added to triphenylphosphine (2.168 g, 8.27 mmol) in THF (8 mL) under N2 atmosphere at 0 °C. After stirring for 30 min, 6-chloropurine (0.478 g, 3.10 mmol) and compound 3 (0.600 g, 2.068 mmol) in THF (8 mL) were added and the reaction mixture refluxed for 24 h. The solvent was evaporated and the crude residue was purified by column chromatography using hexane/EtOAc gradient elution to recover 230 mg of 3 and pure 4 (252 mg) as a light yellow oil TLC (hexane/EtOAc = 70:30 v/v): Rf = 0.20. Yield 48%. [α]D20= +20.0 (25.1 mg/mL, acetone). FT-IR (cm−1): 2954 (m), 2930 (m), 2858 (w), 1747 (s), 1589 (s), 1562 (s), 1486 (m), 1472 (m), 1437 (m), 1400 (m), 1343 (m), 1297 (w), 1257 (s), 1220 (s), 1149 (s), 1092 (m), 1006 (m), 936 (m), 837 (s), 779 (m). 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 1H, H6′), 8.27 (s, 1H, H2′), 4.73 (d, J = 12 Hz, 1H, Ha1) 4.47 (d, J = 12 Hz, 1H, Hb1), 4.37 (m, 1H, H4), 3.71 (s, 3H, OMe), 3.69 (m,1H, Ha5), 3.60 (dd, J = 11.2, 3.9 Hz, 1H, Hb5), 3.16 (dd, J = 13.6, 7.9 Hz, 1H, Ha3), 2.69 (dd, J = 13.6, 7.3 Hz, 1H, Hb3), 0.74 (s, 9H, tBu), −0.07 (s, 6H, SiMe2). 13C-NMR (100 MHz, CDCl3) δ 169.5 (COO), 152.0 (C6′), 151.6, 143.5 (C2′), 131.9, 80.2 (C4), 74.7(C1), 70.3, 63.7 (C5), 53.8 (OMe), 37.5 (C3), 25.1 (tBu), −6.1 (SiMe2). ESI(+)-MS: m/z 449 [M+Na]+, 427 [M+H]+. HREI-MS: m/z 411.12448 ± 0.0010, (M+˙− Me, calcd. for C17H2435ClN4O4Si: 411.12554), m/z 369.07799 ± 0.0010, (M+˙− tBu, calcd for C14H1835ClN4O4Si: 369.07859).
- (3S,5S)-Methyl 3-(6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3-carboxylate (5). Compound 4 (240 mg, 0.56 mmol) and tetrabutylammonium fluoride (TBAF, 2 M solution in THF, 1.35 mL, 0.676 mmol) were stirred for 2 h at room temperature. The crude product obtained by evaporation of the solvent was purified by FC using hexane/ethyl acetate gradient elution, to give pure 5. TLC (dichloromethane/MeOH = 90:10): Rf 0.18. Yield: 87%. HPLC analysis (column CN, hexane/i-PrOH/MeOH 20:75:5) under UV detection at 254 nm showed a single peak at Rt = 18.2 min. [α]D20 = +18.2 (14.4 mg/mL, MeOH). FT-IR: 3400 (w, broad), 2900 (w broad), 1742 (s), 1590 (s), 1563 (s), 1436 (m), 1398 (m), 1343 (m), 1220 (s), 1050 (m), 934 (m), 834 (m), 636 (m). 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H, H6′), 8.40 (s, 1H, H2′), 4.78 (d, J = 10.3 Hz, 1H, Ha1) 4.46 (d, J = 10.3 Hz, 1H, Hb1), 4.39 (m, 1H, H4), 3.68 (s, 3H, OMe), 3.81 (broad d, J = 12.1 Hz, 1H, Ha 5), 3.51 (dd, J = 12.1, 3.0 Hz, 1H, Hb 5), 3.10 (dd, J = 13.4, 8.0 Hz, 1H, Ha 3), 2.76 (dd, J = 13.4, 7.0 Hz, 1H, Hb 3). 13C NMR (100 MHz, CDCl3) δ 169.5 (COO), 151.9, 151.8 (C6′), 151.1, 144.2 (C2′), 132.0, 80.2 (C4), 74.5 (C2), 70.3 (C1), 62.7 (C5), 53.8 (OMe), 37.5 (C3). ESI(+)-MS: m/z 336 [M+Na]+, 313 [M+H]+. HRESI(+)-MS: m/z 313.07031 ± 0.0005, calcd. for C12H1435ClN4O4: 313.06981.
- (3S,5S)-Methyl 5-(hydroxymethyl)-3-(6-(4-methylpiperazin-1-yl)-9H-purin-9-yl)tetrahydrofuran-3-carboxylate (6). Compound 5 (10 mg, 0.024 mmol) and N-methyl piperazine (0.008 mL, 0.072 mmol) in MeOH (1 mL) were microwave irradiated for 1.5 h at 70 °C. The crude mixture was purified by preparative TLC, using dichlorometane/MeOH = 85:15 v/v added of few drops of trimethylamine. TLC: (dichloromethane/MeOH = 80:20 v/v): Rf 0.63. Yield: 75%. HPLC analysis (column CN, hexane/i-PrOH/MeOH 20:75:5) under UV detection at 254 nm showed a single peak at Rt = 26.2 min. [α]D20 = +21.2 (6.66 mg/mL in MeOH). FT-IR: 3351 (w, broad), 2923 (w, broad), 1742 (m), 1693 (w), 1586 (s), 1453 (m), 1250 (m), 1046 (m), 793 (m), 646 (m). 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, H6′), 7.90 (s, 1H, H2′), 4.70 (d, J = 10.3 Hz, 1H, Ha1) 4.53 (d, J = 10.3 Hz, 1H, Hb1), 4.39 (m, 1H, H4), 4.31 (m, 4H), 3.70 (s, 3H, OMe), 3.80 (dd, J = 12.0, 2.6 Hz, 1H, Ha 5), 3.52 (dd, J = 12.0, 4.5 Hz, 1H, Hb 5), 3.09 (dd, J = 13.8, 7.5 Hz, 1H, Ha 3), 2.67 (dd, J = 13.8, 7.9 Hz, 1H, Hb 3), 2.54 (pseudo t, J = 4.6 Hz, 4H), 2.34 (s, 3H, NMe). 13C NMR (100 MHz, CDCl3) δ 170.2 (COO), 153.7, 152.3 (C6′), 151.2, 149.5, 136.5 (C2′), 79.9, 69.5, 63.2, 54.7, 53.4, 45.8, 45.6, 37.4 (C3). ESI(+)-MS: m/z 399 [M+Na]+, 377 [M+H]+; MS/MS (377): m/z 320, 162. HRESI(+)-MS: m/z 377.19344 ± 0.0003, [M+H+], calcd. for C17H25N6O4: 377.19318).
- (3S,5S)-Methyl 5-(hydroxymethyl)-3-(6-(methylamino)-9H-purin-9-yl)tetrahydrofuran-3-carboxylate (7). Compound 5 (25 mg, 0.081 mmol) and methylamine (2M in THF, 0.230 mL, 0.46 mmol) in MeOH (0.5 mL) were microwave irradiated for 20 min at 70 °C. The crude mixture was purified by preparative TLC was purified by preparative TLC, using dichloromethane/MeOH = 90:10 v/v added of few drops of trimethylamine, obtaining pure 7 as a light-yellow solid. TLC (dichloromethane/MeOH = 90:10 v/v). Rf 0.25. Yield 92%. [α]D20= +16.9 (12.5 mg/mL in MeOH). FT-IR (cm−1): 3364 broad w, 2925 m, 1740 m, 1623 s, 1456 w, 1376 w, 1297 w, 1241 m, 1045 m, 963 w, 645 s broad. 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H, H6′), 7.97 (s, 1H, H2′), 6.28 (br s, 1H), 4.85 (d, J = 10.3 Hz, 1H, Ha1) 4.43 (d, J = 10.3 Hz, 1H, Hb1), 4.37 (m, 1H, H4), 3.65 (s, 3H, OMe), 3.83 (brd, J = 11.7 Hz, 1H, Ha 5), 3.51 (dd, J = 11.7, 3.6 Hz, 1H, Hb 5), 3.26 (m, 1H), 3.05 (br s, 3H, NMe), 2.98 (dd, J = 13.6, 8.1 Hz, 1H, Ha 3), 2.83 (dd, J = 13.6, 6.5 Hz, 1H, Hb 3), 3.05 (br s, 3H, NMe). 13C NMR (100 MHz, CDCl3) δ 170.3 (COO), 155.1, 153.2 (C6′), 148.8, 138.4 (C2′), 119.7, 80.5, 74.5, 69.8 (C1), 62.6 (C5), 53.4 (OMe), 37.2 (C3), 27.2 (NMe). ESI(+)-MS: m/z 308 [M+H]+, MS/MS (308): m/z 290, 248; HRESI(+)-MS: m/z 308.13566 ± 0.0004, [M+H]+, calcd. for C13H18N5O4: 308.13533.
- (3S,5S)-Isopropyl 3-(6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3-carboxylate (8). Compound 5 (30 mg, 0.08 mmol,) and triazabicyclodecene (TBD, 3 mg, 0.024 mmol) in i-PrOH (1 mL) were microwave irradiated for 30 min at 50 °C. The crude mixture was purified using CN-column chromatography using dichloromethane/MeOH gradient elution to obtain pure 8 as light-yellow solid. TLC: CN phase, dichloromethane/MeOH 99:1 v/v. Rf = 0.56. Yield 53%. [α]D20 = +26.1 (17.4 mg/mL in MeOH). FT-IR (cm−1): 3400 (m, broad), 2925 (m), 1720 (s), 1688 (s), 1589 (m), 1453 (m), 1376 (m), 1228 (s), 1100 (s), 1039 (s), 827 (m), 603 (s). 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H, H6′), 8.26 (s, 1H, H2′), 4.76 (d, J = 10.3 Hz, 1H, Ha1) 4.42 (d, J = 10.3 Hz, 1H, Hb1), 5.00 (sept, J = 6.4 Hz, 1H, OiPr), 4.37 (m, 1H, H4), 3.80 (dd, J = 12.5, 2.5 Hz, 1H, Ha 5), 3.50 (dd, J = 12.5, 3.8 Hz, 1H, Hb 5), 3.07 (dd, J = 13.6, 8.0 Hz, 1H, Ha 3), 2.72 (dd, J = 13.6, 7.2 Hz, 1H, Hb 3), 2.97 (br s, 1H), 1.06 (pseudo t, J = 6.4Hz, 6H, i-Pr). 13C NMR (100 MHz, CDCl3) δ 168.2 (COO), 151.9, 151.0, 151.6 (C6′), 144.1 (C2′), 131.8, 80.2 (C4), 74.4 (C2), 71.2 (C1), 70.6 (i-PrO), 62.7 (C5), 37.4 (C3), 21.4 (i-PrO). ESI(+)-MS: m/z 341 [M+H]+, 323 [M+H-H2O]+. HRESI(+)-MS: m/z 341.10181 ± 0.0007, calcd. for C14H1835ClN4O4: 341.10111.
- (3S,5S)-Methyl 3-(6-amino-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3-carboxylate (9). Diethylazodicarboxylate (DEAD, 40% in toluene, 721 mg, 4.14 mmol) was slowly added to triphenylphosphine (1084 mg, 4.14 mmol) under N2 atmosphere at 0 °C. After 30 min, 3 (300 mg, 1.03 mmol) and adenine in THF were added, stirring under reflux for 10 h, monitoring by TLC (hexane/EtOAc 6:4 v/v with few drops of trimethylamine). The reaction mixture was concentrated and the residue subjected to FC using hexane/AcOEt added of 1% triethylamine gradient elution. The recovered product was then treated with TBAF in THF stirring 2 h at room temperature as described in the procedure for obtaining 5. The deprotected product was purified using CN-column chromatography with hexane/AcOEt gradient elution obtaining pure 9 as light yellow solid TLC: dichloromethane/MeOH 85:15 v/v. Rf = 0.32. Yield 52%. [α]D20 = +16.8 (8.57 mg/mL in MeOH). FT-IR (cm−1) 3193 (m, broad), 2928 (m), 1704 (m), 1642 (m), 1581 (s), 1437 (s), 1250 (s), 1119 (vs), 837 (m). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H, H6′), 8.03 (s, 1H, H2′), 6.06 (br s, 2H), 4.84 (d, J = 10.3 Hz, 1H, Ha1) 4.45 (d, J = 10.3 Hz, 1H, Hb1), 4.41 (m, 1H, H4), 3.69 (s, 3H, OMe), 3.87 (dd, J = 12.6, 2.6 Hz, 1H, Ha 5), 3.53 (dd, J = 12.6, 3.6 Hz, 1H, Hb 5), 3.05 (dd, J = 13.8, 8.1 Hz, 1H, Ha 3), 2.84 (dd, J = 13.8, 7.2 Hz, 1H, Hb3). 13C NMR (100 MHz, CDCl3) δ 170.0 (COO), 155.3, 152.7 (C6′), 150.0, 139.0 (C2′), 119.6, 80.4, 74.6, 69.8 (C1), 62.6 (C5), 53.5 (OMe), 37.3 (C3). EI-MS: m/z 293 [M+.]; HRESI(+)-MS: m/z 294.11985 ± 0.0003, [M+H] +, calcd. for C12H15N5O4: 294.11968.
- (3S,5S)-Isopropyl 3-(6-amino-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3-carboxylate (10). Compound 9 (30 mg, 0.10 mmol) and TBD (4.3 mg, 0.03 mmol) i-PrOH (1 mL) were microwave irradiated for 30 min at 50 °C. The crude product was purified by FC using dichloromethane/MeOH gradient elution, to obtain 10 as a pure white solid. TLC: dichloromethane/MeOH 90:10 v/v, +1% triethylamine. Rf = 0.57. Yield: 51%. [α]D20 = +13.0 (9.4 mg/mL in MeOH). FT-IR (cm−1) 3333 (m, broad), 2926 (m), 2360 (w), 1734 (m), 1641 (s), 1600 (s), 1473 (m), 1292 (m), 1226 (s), 1102 (s), 1042 (m), 799 (m), 647 (m). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.03 (s, 1H), 5.93 (br s, 2H, NH2), 4.81 (d, J = 10.2 Hz, 1H, Ha1) 4.45 (d, J = 10.2 Hz, 1H, Hb1), 5.03 (sept, J = 6.1 Hz, 1H, i-PrO), 4.40 (m, 1H, H4), 3.87 (brd, J = 12.5 Hz, 1H, Ha5), 3.54 (dd, J = 12.5, 3.9 Hz, 1H, Hb5), 3.04 (dd, J = 13.7, 8.1 Hz, 1H, Ha3), 2.80 (dd, J = 13.7, 7.6 Hz, 1H, Hb3), 1.08 ( d, J = 6.1Hz, 6H, i-Pr). 13C NMR (100 MHz, CDCl3) δ 168.9 (COO), 155.3, 152.6, 150.1, 139.0, 119.5, 80.4, 74.5, 70.7, 70.0, 62.7, 37.1, 21.3. ESI(+)-MS: m/z 344 [M+Na]+, 322 [M+H]+; HRESI(+)-MS: m/z 322.15094 ± 0.0003, [M+H]+, calcd. for C14H20N5O4: 322.15098.
- (3S,5S)-3-(6-Amino-9H-purin-9-yl)-5-(hydroxymethyl)-N-methyltetrahydrofuran-3-carboxamide (11). Methylamine (2M in THF, 0.273 mmol, 0.137 mL) was slowly added to a solution of 9 (20 mg, 0.68 mmol) in MeOH (1 mL) at 0 °C. The reaction was stirred overnight at r.t., then concentrated in vacuo and the residue subjected to FC, using dichloromethane/MeOH gradient elution, to obtain pure 11 as white solid. TLC: dichloromethane/MeOH 85:15 v/v. Rf: 0.23. Yield: 60%. [α]D20= +21.4 (7.8 g/mL in MeOH). FT-IR (cm−1): 3326 m broad, 2926 w, 1639 s, 1601 m, 1573 m, 1475 m, 1414 m, 1376 m, 1339 m, 1260 m, 1226 m, 1042 m, 644 s broad. 1H NMR (400 MHz, acetone-d6) δ 8.16 (s, 1H), 8.14 (s, 1H), 6.65 (br s, 2H, NH2), 4.65 (d, J = 9.8 Hz, 1H, Ha1) 4.56 (d, J = 9.8 Hz, 1H, Hb1), 4.29 (m, 1H, H4), 3.50 (dd, J = 11.6, 4.2 Hz, 1H, Ha 5), 3.46 (dd, J = 11.6, 4.8 Hz, 1H, Hb 5), 3.14 (dd, J = 13.4, 7.8 Hz, 1H, Ha 3), 2.67 (dd, J = 13.4, 7.6 Hz, 1H, Hb3), 2.66 (d, J = 4.6 Hz, 3H, NMe). 13C NMR (100 MHz, acetone-d6) δ 169.4 (COO), 156.2, 152.5 (C6′), 150.2, 139.6 (C2′), 120.1, 79.8, 73.9, 70.3 (C1), 63.3 (C5), 37.5 (C3), 25.7 (NMe). ESI(+)-MS m/z 293 [M+H]+; MS/MS(293): m/z 234; HRESI(+)-MS: m/z 293.13586±0.0003, [M+H]+, calcd. for C12H17N6O3: 293.13566.
3.2. Computational Analysis
3.2.1. ADME Predictions
3.2.2. Docking Calculation
3.2.3. Molecular Dynamics Procedure
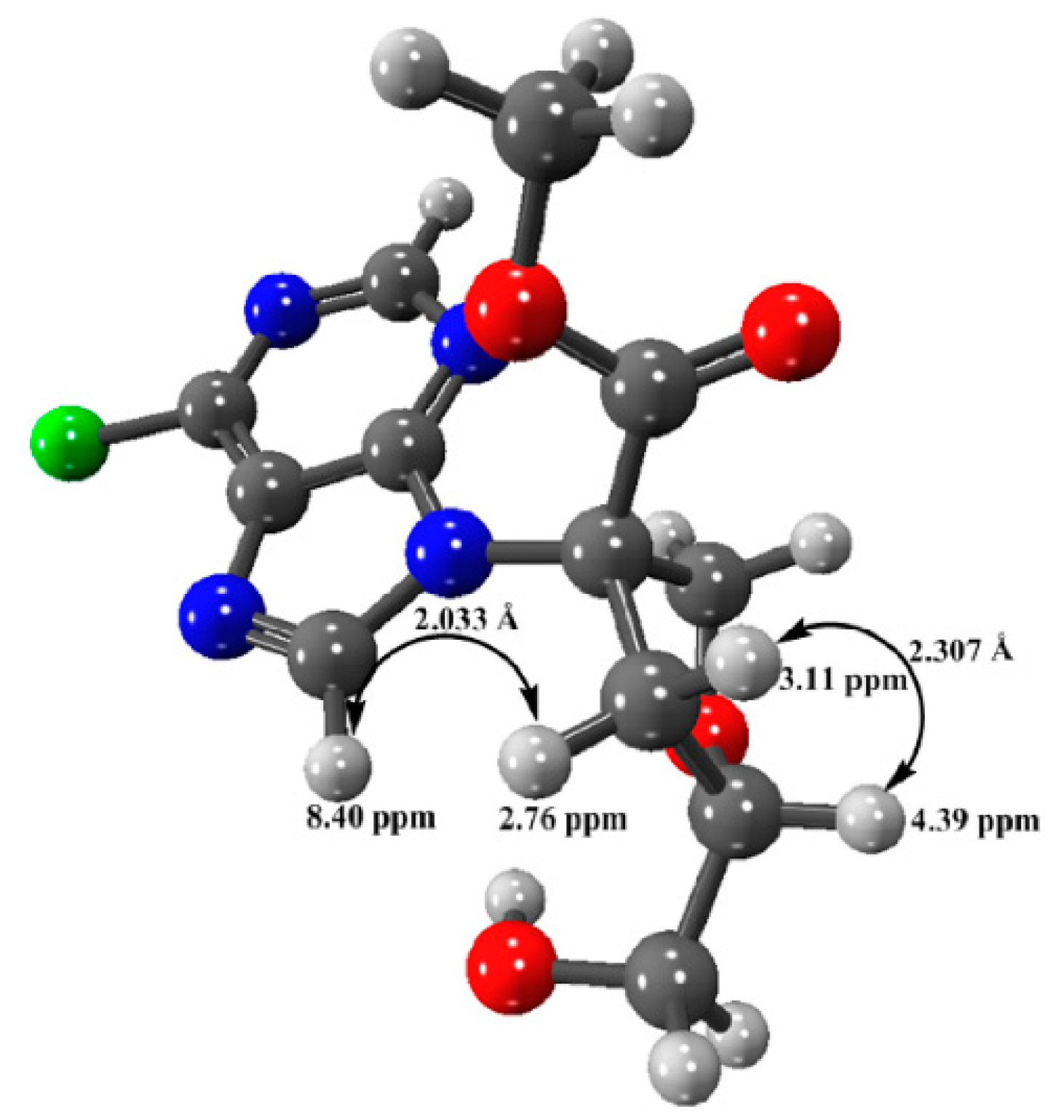
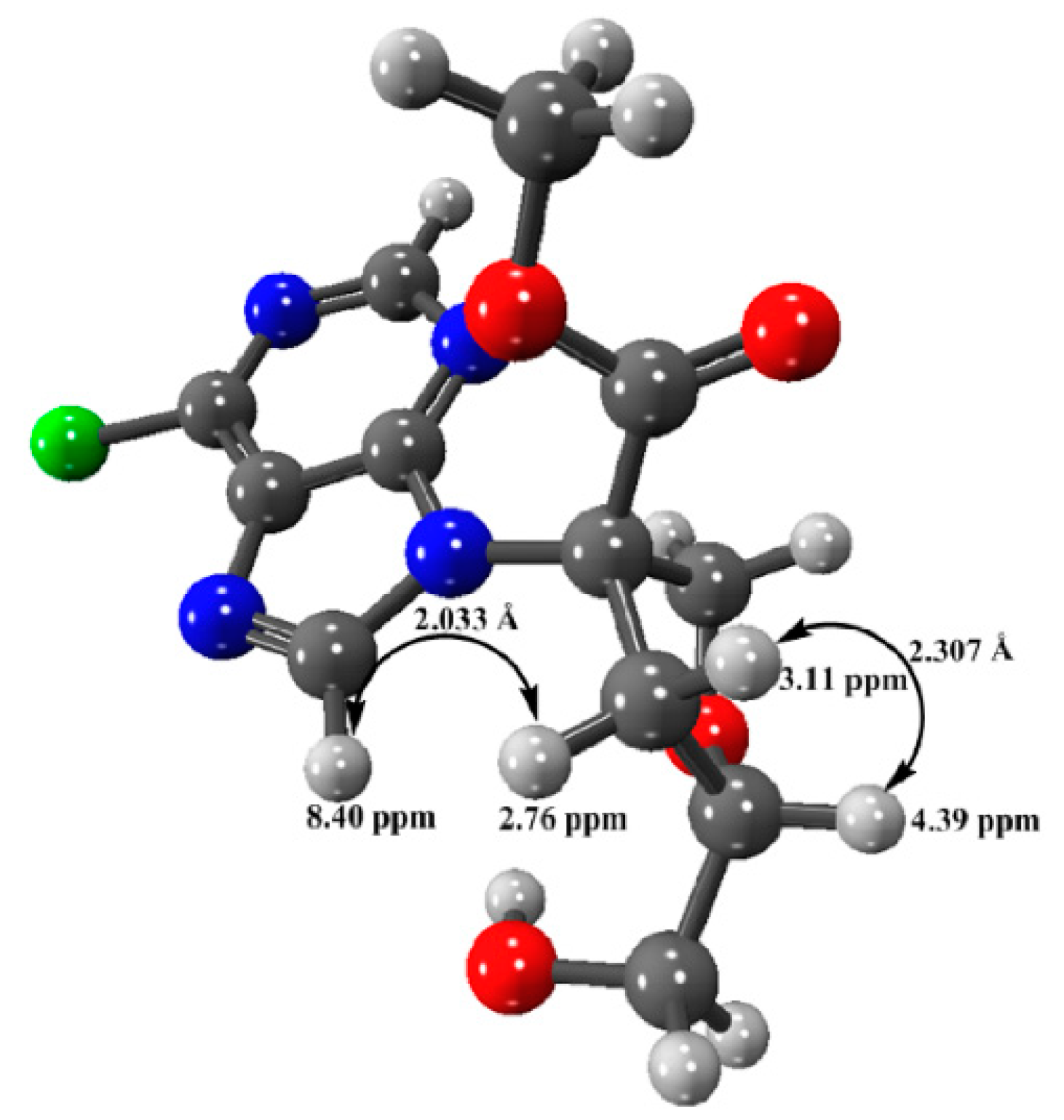
3.2.4. Minimized Structure of Compound 5 by DFT Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabbri, D.; Torri, C.; Mancini, I. Pyrolysis of cellulose catalysed by nanopowder metal oxides: Production and characterisation of a chiral hydroxylactone and its role as building block. Green Chem. 2007, 9, 1374–1379. [Google Scholar] [CrossRef]
- Mancini, I.; Dosi, F.; Defant, A.; Crea, F.; Miotello, A. Upgraded production of (1R,5S)-1-hydroxy-3,6-dioxa-bicyclo[3.2.1] octan-2-one from cellulose catalytic pyrolysis and its detection in bio-oils by spectroscopic methods. J. Anal. Appl. Pyrolysis 2014, 110, 285–290. [Google Scholar] [CrossRef]
- Defant, A.; Mancini, I.; Torri, C.; Malferrari, D.; Fabbri, D. An efficient route towards a new branched tetrahydrofurane δ-sugar amino acid from a pyrolysis product of cellulose. Amino Acids 2010, 40, 633–640. [Google Scholar] [CrossRef]
- Defant, A.; Mancini, I.; Matucci, R.; Bellucci, C.; Dosi, F.; Malferrari, D.; Fabbri, D. Muscarine-like compounds derived from a pyrolysis product of cellulose. Org. Biomol. Chem. 2015, 13, 6291–6298. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.; Le, N.T.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793–104800. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral drug discovery: Preparing for the next pandemic. Chem. Soc. Rev. 2021, 50, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Yan, V.C.; Muller, F.L. Advantages of the Parent Nucleoside GS-441524 over Remdesivir for COVID-19 Treatment. ACS Med. Chem. Lett. 2020, 11, 1361–1366. [Google Scholar] [CrossRef]
- Roy, A.; Schneller, S.W. 4’- and 1’-Methyl-substituted 5’-norcarbanucleosides. J. Org. Chem. 2003, 68, 9269–9273. [Google Scholar] [CrossRef]
- Martin, C.Y. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Wakchaure, P.D.; Ghosh, S.; Bishwajit Ganguly, B. Revealing the Inhibition Mechanism of RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2 by Remdesivir and Nucleotide Analogues: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2020, 124, 10641–10652. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules. In Proceedings of the 2nd Workshop on Algorithms in Bioinformatics (WABI), Rome, Italy, 17–21 September 2002; Lecture Notes in Computer Science 2452. Springer: New York, NY, USA, 2002; pp. 185–200. [Google Scholar]
- Milanović, Ž.B.; Antonijević, M.R.; Amić, A.D.; Avdović, E.H.; Dimić, D.S.; Milenković, D.A.; Marković, Z.S. Inhibitory activity of quercetin, its metabolite, and standard antiviral drugs towards enzymes essential for SARS-CoV-2: The role of acid-base equilibria. RSC Adv. 2021, 11, 2838–2847. [Google Scholar] [CrossRef]
- Nemaysh, V.; Luthra, P.M. Computational analysis revealing that K634 and T681 mutations modulate the 3D-structure of PDGFR- and lead to sunitinib resistance. RSC Adv. 2017, 7, 37612–37626. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.T.; Iyer, S.; Christy, J.P.; Siva, R.; Tayubi, I.A.; Priya Doss, C.G.; Zayed, H. A comparative computational approach toward pharmacological chaperones (NN-DNJ and ambroxol) on N370S and L444P mutations causing Gaucher’s disease. Adv. Protein Chem. Struct. Biol. 2019, 114, 315–339. [Google Scholar] [CrossRef]
- Hannan, M.; Dash, R.; Sohag, A.A.M.; Moon, I.S. Deciphering Molecular Mechanism of the Neuropharmacological Action of Fucosterol through Integrated System Pharmacology and in silico Analysis. Mar. Drugs 2019, 17, 639. [Google Scholar] [CrossRef] [Green Version]
- Swiss ADME. Available online: http://www.swissadme.ch/ (accessed on 21 October 2021).
- PatchDock Server. Available online: https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php (accessed on 1 September 2021).
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids. Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering; Springer: Berlin, Germany, 2018; pp. 43–67. [Google Scholar]
- Stewart, J.J. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, effcient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Gr. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Lipparini, F.; Mennucci, B. Perspective: Polarizable continuum models for quantum-mechanical descriptions. J. Chem. Phys. 2016, 144, 160901–160909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A. Left-Right correlation energy. J. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.J.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scalmani, G.; Scuseria, G.E. Can short-range hybrids describe long-range-dependent properties? J. Chem. Phys. 2009, 131, 044108. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Remdesivir | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|
| Score | 16,264 | 17,496 | 19,052 | 18,336 | 17,460 | 18,538 | 18,118 | 17,918 |
| Area (Å2) | 1924 | 2477 | 2362 | 2257 | 2302 | 2257 | 2093 | 2295 |
| Compound | H-Bond | π-π π-Alkyl | vdW | ch-ch π-ion | Unf. |
|---|---|---|---|---|---|
| GS-441524 | U10 (3.07, 2.90) U20 (2.93, 2.99) Asn691 (2.73) Ser759 (2.30) Asp760 (1.40) | A11 (3.66, 5.04) U20 (4.17, 5.38) | Ala688 (2.76) Ser759 (2.62) | Lys551 (5.05), Asp760 (4.79) | Cys622 (2.42) Asp760 (3.79) |
| 5 | Arg555 (1.79, 2.77) Ser759 (1.71) Asp760 (1.10) | U10 (4.13, 5.94) | U10 (4.11) Lys545 (4.55) Val557 (4.41) Ser682 (2.55, 2.77) | Asp760 (3.21) | - |
| 6 | U20 (1.73) Ile548 (1.83) Arg553(2.69) Arg555 (2.40) Ser759 (2.41) Asp760 (1.40) | - | U10 (5.17), A11 (4.75) Ala547 (2.22), Ser682 (2.62) | Arg555 (2.97, 3.61) | - |
| 7 | Arg555 (1.77) Thr556 (2.07) Asp760 (1.14) | Ser681 (3.69, 4.69) | Thr556 (2.76) Thr687 (2.27) Ser759 (2.71) Asp760 (2.81) | Ser682 (4.77), Asp760 (3.15) | - |
| 8 | Lys551 (1.77) Ser759 (1.61) | U10 (4.80) A11 (4.75) | U20 (2.39) Ala688 (3.81) Asp760 (2.67) Ser814 (2.89) | Lys551 (1.67) | Lys551 (3.03) |
| 9 | U20 (1.59) Arg553 (1.83) Arg555 (1.88) Asp760 (1.10) | - | Thr556 (2.47) Val557 (4.59) Cys622 (5.21) Asp62 (2.45) Ser682 (2.79) Asp760 (2.32) | Asp760 (3.09) | - |
| 10 | U20 (2.22) Arg555 (1.93, 2.21) | - | A11 (4.91), Leu758 (2.66) Asp760 (2.29) | Asp760 (4.82) Asp761(4.43) | Asp760 (4.99) |
| 11 | A11(2.56) U20 (1.68) Arg553 (2.24) Asp760 (1.10) | U20 (5.42) | Arg555 (5.31) Asp760 (2.53) Asp763 (2.97) Ser682 (2.37, 2.51) | Arg555 (4.92, 4.96) Asp760 (3.11) | Cys622 (2.11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Defant, A.; Dosi, F.; Innocenti, N.; Mancini, I. Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors. Int. J. Mol. Sci. 2022, 23, 518. https://doi.org/10.3390/ijms23010518
Defant A, Dosi F, Innocenti N, Mancini I. Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors. International Journal of Molecular Sciences. 2022; 23(1):518. https://doi.org/10.3390/ijms23010518
Chicago/Turabian StyleDefant, Andrea, Federico Dosi, Nicole Innocenti, and Ines Mancini. 2022. "Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors" International Journal of Molecular Sciences 23, no. 1: 518. https://doi.org/10.3390/ijms23010518
APA StyleDefant, A., Dosi, F., Innocenti, N., & Mancini, I. (2022). Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors. International Journal of Molecular Sciences, 23(1), 518. https://doi.org/10.3390/ijms23010518