
TdIF1-LSD1 Axis Regulates Epithelial—Mesenchymal Transition and Metastasis via Histone Demethylation of E-Cadherin Promoter in Lung Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
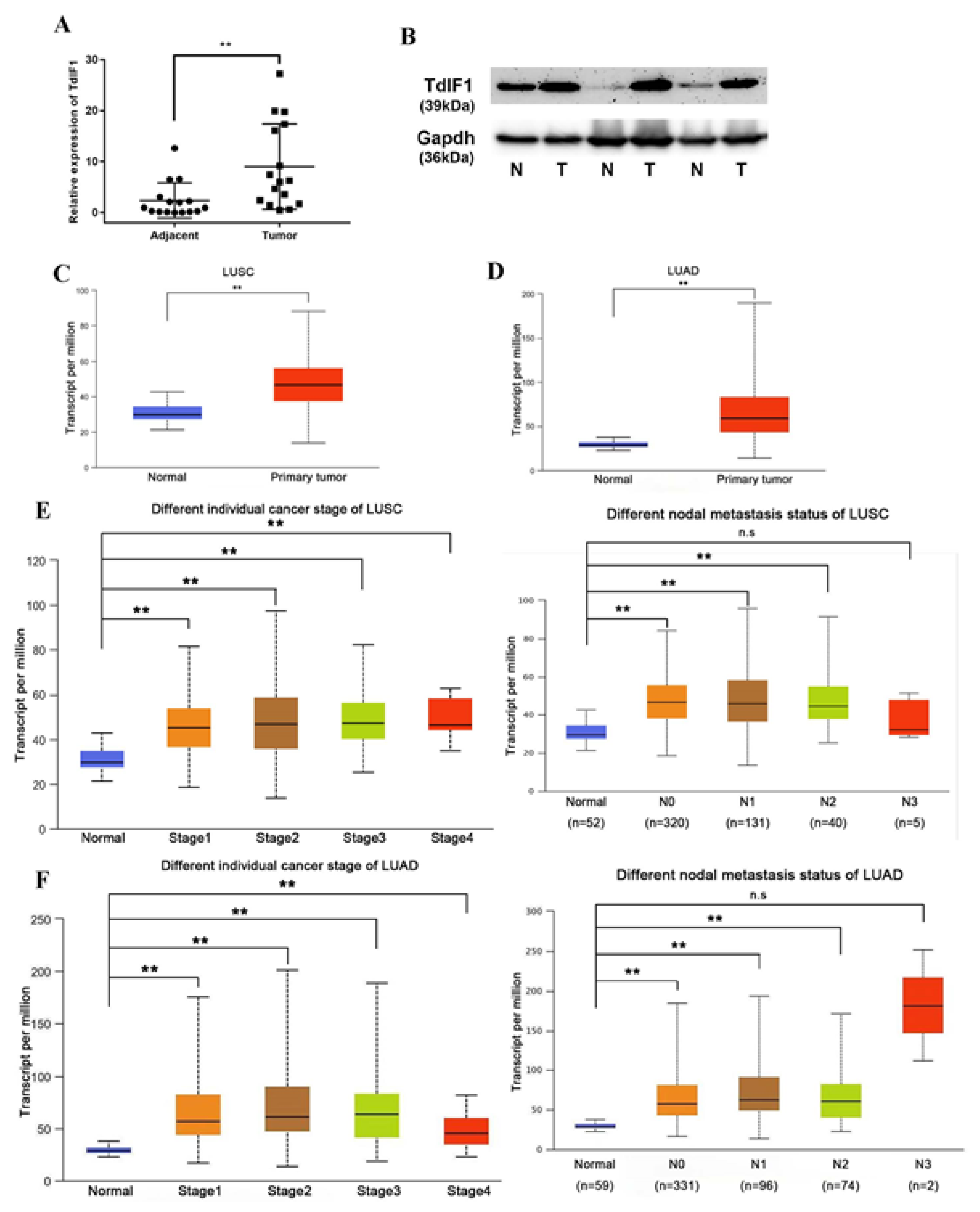
2.1. TdIF1 Is Highly Expressed in NSCLC and Positively Correlated with Metastasis
2.2. Knockdown TdIF1 Inhibits the Migration and Invasion of Lung Cancer Cells
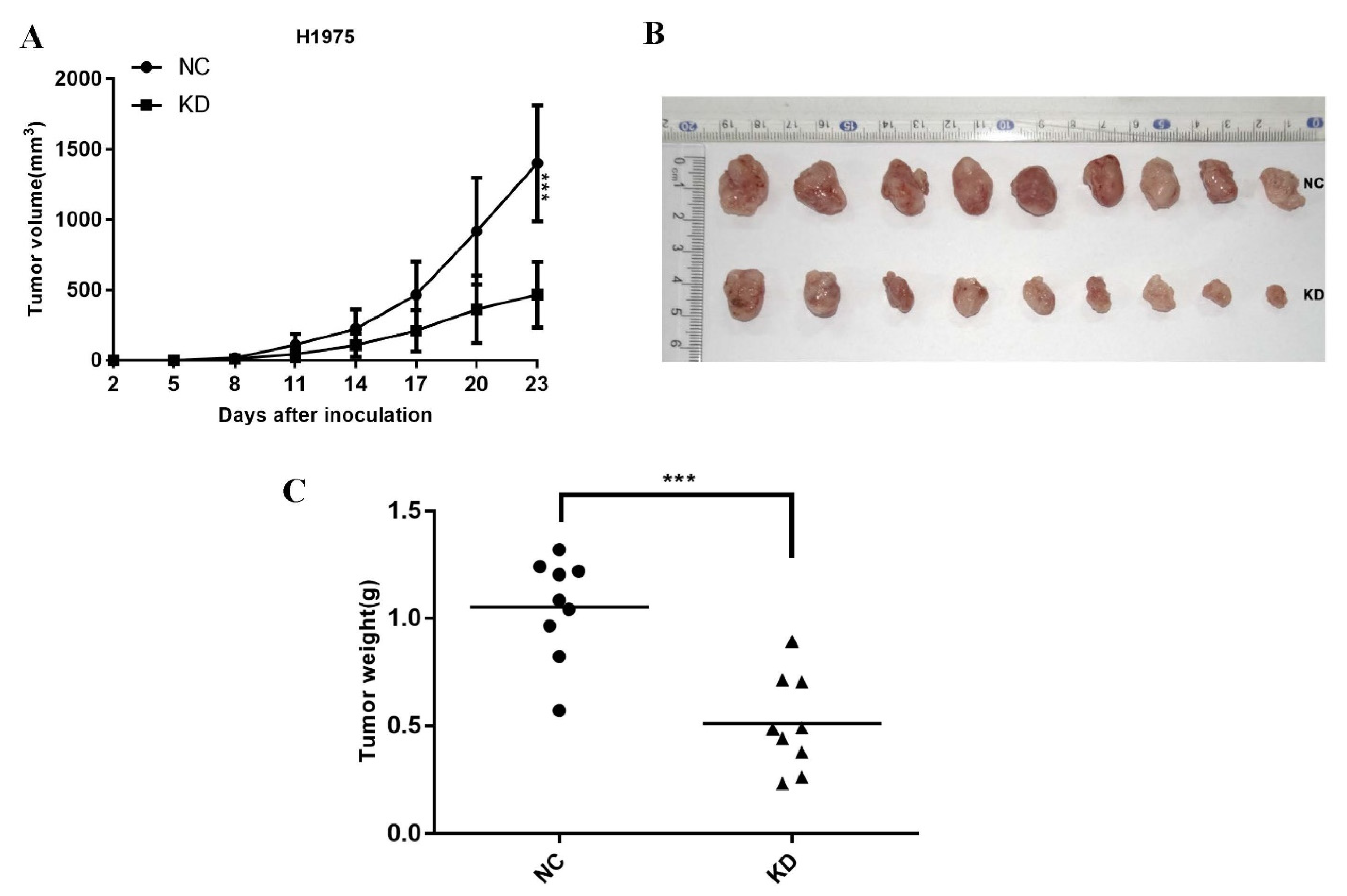
2.3. Knockdown TdIF1 Inhibits NSCLC Cells Tumorigenesis In Vivo
2.4. TdIF1 Interacts with LSD1 and Regulates EMT in NSCLC Cells
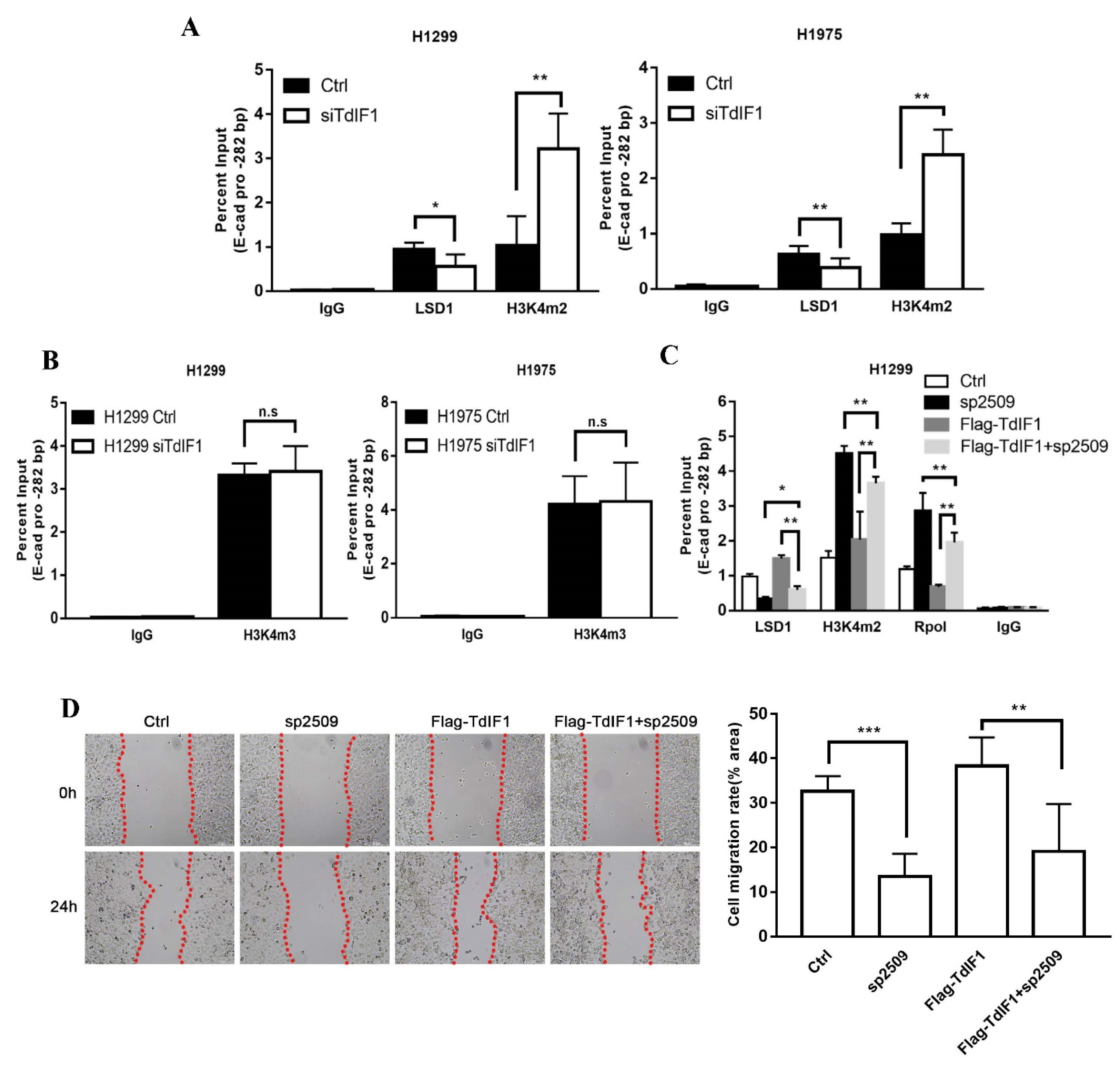
2.5. TdIF1 Regulates H3K4me2 Levels at the E-Cadherin Promoter by Recruiting LSD1
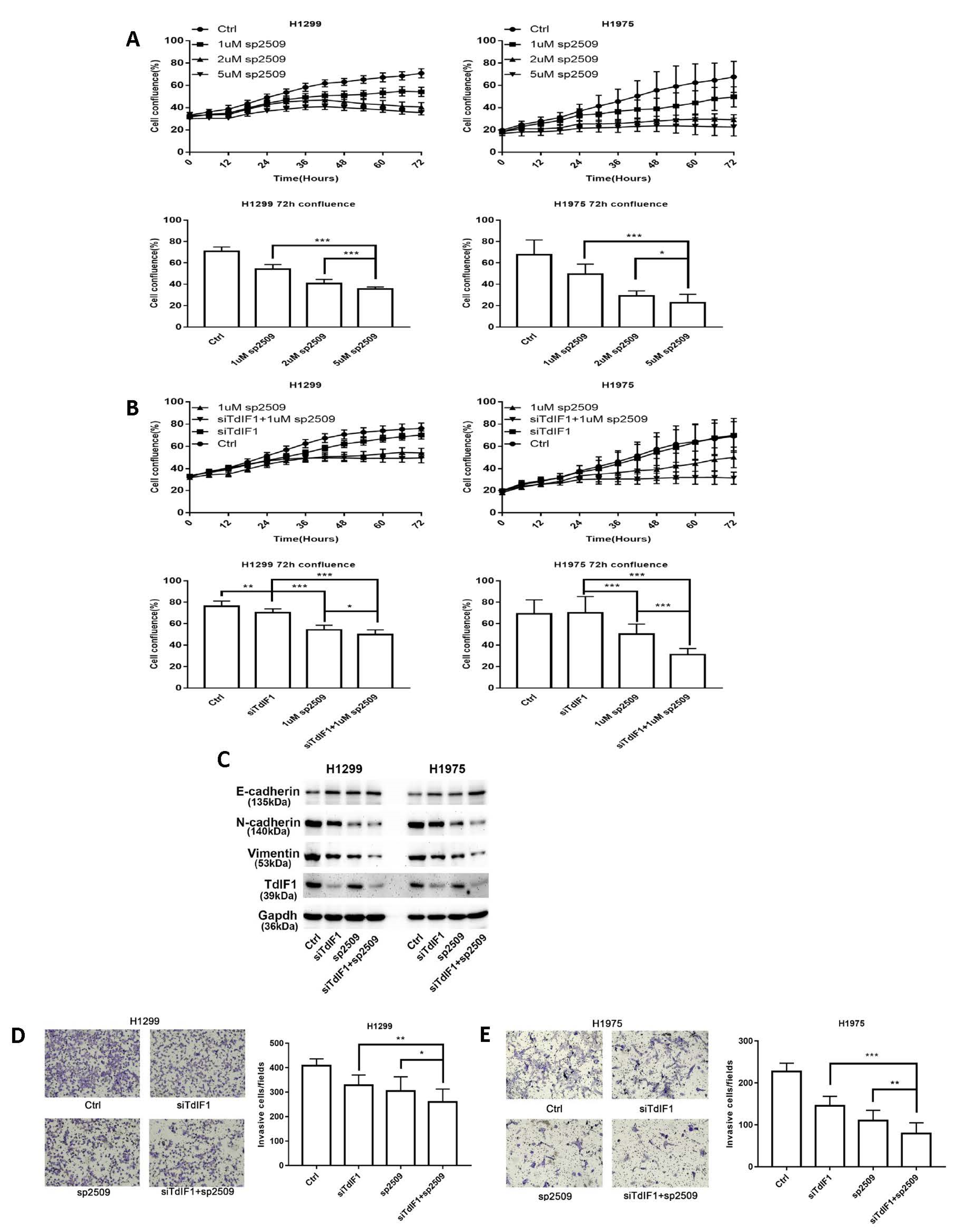
2.6. TdIF1 Knockdown Synergizes LSD1 Inhibitors in the Suppression of the Metastatic Cell Invasion of NSCLC
3. Discussion
4. Materials and Methods
4.1. Plasmid, siRNA, and LSD1 Inhibitor
4.2. Data Analysis Using the Cancer Genome Atlas (TCGA)
4.3. Animals
4.4. Cell Culture
4.5. Transfection and Interference Assay
4.6. Wound Healing Assay
4.7. Cell Migration and Invasion Assay
4.8. Incucyte Live-Cell Imaging System
4.9. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.10. Western Blot Analysis
4.11. Co-Immunoprecipitation (Co-IP)
4.12. Chromatin Immunoprecipitation Assay (ChIP)
4.13. Human Lung Cancer Xenograft Model
4.14. Human Tissue Samples
4.15. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung Cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Janku, F.; Stewart, D.J.; Kurzrock, R. Targeted therapy in non-small-cell lung cancer—Is it becoming a reality? Nat. Rev. Clin. Oncol. 2010, 7, 401–414. [Google Scholar] [CrossRef]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Novello, S.; Barlesi, F.; Califano, R.; Cufer, T.; Ekman, S.; Levra, M.G.; Kerr, K.; Popat, S.; Reck, M.; Senan, S.; et al. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v1–v27. [Google Scholar] [CrossRef]
- Yamashita, N.; Shimazaki, N.; Ibe, S.; Kaneko, R.; Tanabe, A.; Toyomoto, T.; Fujita, K.; Hasegawa, T.; Toji, S.; Tamai, K.; et al. Terminal deoxynucleotidyltransferase directly interacts with a novel nuclear protein that is homologous to p65. Genes Cells 2001, 6, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, S.; Sato, A.; Toyomoto, T.; Hayano, T.; Sugai, M.; Kubota, T.; Koiwai, O. Direct binding of TReP-132 with TdT results in reduction of TdT activity. Genes Cells 2006, 11, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Huang, Y.; Ying, M.; Wang, Y.; Xiong, J.; Liu, Q.; Cao, F.; Joshi, R.; Liu, Y.; et al. TdIF1: A putative oncogene in NSCLC tumor progression. Signal Transduct. Target. Ther. 2018, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Buonato, J.M.; Lazzara, M.J. ERK1/2 Blockade Prevents Epithelial–Mesenchymal Transition in Lung Cancer Cells and Promotes Their Sensitivity to EGFR Inhibition. Cancer Res. 2014, 74, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, I.; Greve, G.; Jung, M.; Lübbert, M. Epigenetic therapy approaches in non-small cell lung cancer: Update and perspectives. Epigenetics 2016, 11, 858–870. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Koiwai, O.; Hori, K.; Watanabe, N.; Koiwai, K. TdIF1 Recognizes a Specific DNA Sequence through Its Helix-Turn-Helix and AT-Hook Motifs to Regulate Gene Transcription. PLoS ONE 2013, 8, e66710. [Google Scholar] [CrossRef] [Green Version]
- Koiwai, K.; Kubota, T.; Watanabe, N.; Hori, K.; Koiwai, O.; Masai, H. Definition of the transcription factor TdIF1 consensus-binding sequence through genomewide mapping of its binding sites. Genes Cells 2015, 20, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Fleming-Waddell, J.N.; Wilson, L.M.; Olbricht, G.R.; Vuocolo, T.; Byrne, K.; Craig, B.A.; Tellam, R.L.; Cockett, N.E.; Bidwell, C.A. Analysis of gene expression during the onset of muscle hypertrophy in callipyge lambs. Anim. Genet. 2007, 38, 28–36. [Google Scholar] [CrossRef]
- Yu, H.; Waddell, J.N.; Kuang, S.; Tellam, R.L.; Cockett, N.E.; Bidwell, C.A. Identification of genes directly responding to DLK1 signaling in Callipyge sheep. BMC Genom. 2018, 19, 283. [Google Scholar] [CrossRef] [Green Version]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.-M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Pagliuca, F.; Collins, M.; Lichawska, A.; Zegerman, P.; Choudhary, J.S.; Pines, J. Quantitative Proteomics Reveals the Basis for the Biochemical Specificity of the Cell-Cycle Machinery. Mol. Cell 2011, 43, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, B.; Jin, H.; Kallappagoudar, S.; Sedkov, Y.; Martinez, T.; Sentmanat, M.F.; Poet, G.J.; Li, C.; Fan, Y.; Pruett-Miller, S.M.; et al. The histone deacetylase complex MiDAC regulates a neurodevelopmental gene expression program to control neurite outgrowth. eLife 2020, 9, e57519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat. Commun. 2020, 11, 3252. [Google Scholar] [CrossRef] [PubMed]
- Sawai, Y.; Kasamatsu, A.; Nakashima, D.; Fushimi, K.; Kasama, H.; Iyoda, M.; Kouzu, Y.; Shiiba, M.; Tanzawa, H.; Uzawa, K. Critical role of deoxynucleotidyl transferase terminal interacting protein 1 in oral cancer. Lab. Investig. 2018, 98, 980–988. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.; Mbalawa, C.G.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S.; et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. [Google Scholar] [CrossRef]
- Kerr, K.M.; Bubendorf, L.; Edelman, M.; Marchetti, A.; Mok, T.; Novello, S.; O’Byrne, K.; Stahel, R.; Peters, S.; Felip, E.; et al. Second ESMO consensus conference on lung cancer: Pathology and molecular biomarkers for non-small-cell lung cancer. Ann. Oncol. 2014, 25, 1681–1690. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef] [Green Version]
- Hayami, S.; Kelly, J.D.; Cho, H.-S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Kahl, P.; Gullotti, L.; Heukamp, L.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen Receptor Coactivators Lysine-Specific Histone Demethylase 1 and Four and a Half LIM Domain Protein 2 Predict Risk of Prostate Cancer Recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-K. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J. Gastroenterol. 2012, 18, 6651–6656. [Google Scholar] [CrossRef]
- Milano-Foster, J.; Ray, S.; Home, P.; Ganguly, A.; Bhattacharya, B.; Bajpai, S.; Pal, A.; Mason, C.W.; Paul, S. Regulation of human trophoblast syncytialization by histone demethylase LSD1. J. Biol. Chem. 2019, 294, 17301–17313. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Shenoy, A.K.; Li, X.; Jin, Y.; Jin, L.; Cai, Q.; Tang, M.; Liu, Y.; Chen, H.; Reisman, D.; et al. MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari-Amorotti, G.; Fragliasso, V.; Esteki, R.; Prudente, Z.; Soliera, A.R.; Cattelani, S.; Manzotti, G.; Grisendi, G.; Dominici, M.; Pieraccioli, M.; et al. Inhibiting Interactions of Lysine Demethylase LSD1 with Snail/Slug Blocks Cancer Cell Invasion. Cancer Res. 2013, 73, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Ferrari-Amorotti, G.; Chiodoni, C.; Shen, F.; Cattelani, S.; Soliera, A.R.; Manzotti, G.; Grisendi, G.; Dominici, M.; Rivasi, F.; Colombo, M.P.; et al. Suppression of Invasion and Metastasis of Triple-Negative Breast Cancer Lines by Pharmacological or Genetic Inhibition of Slug Activity. Neoplasia 2014, 16, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Chen, S.; Han, D.; Wang, Z.; Li, M.; Han, W.; Besschetnova, A.; Liu, M.; Zhou, F.; Barrett, D.; et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat. Genet. 2020, 52, 1011–1017. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 2017, 36, 6701–6711. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Ding, D.; Liu, X.; Guo, S.-W. Tranylcypromine, a lysine-specific demethylase 1 (LSD1) inhibitor, suppresses lesion growth and improves generalized hyperalgesia in mouse with induced endometriosis. Reprod. Biol. Endocrinol. 2016, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Jarroux, J.; Foretek, D.; Bertrand, C.; Gabriel, M.; Szachnowski, U.; Saci, Z.; Guo, S.; Londoño-Vallejo, A.; Pinskaya, M.; Morillon, A. HOTAIR lncRNA promotes epithelial–mesenchymal transition by redistributing LSD1 at regulatory chromatin regions. EMBO Rep. 2021, 22, e50193. [Google Scholar] [CrossRef]
- Sun, M.; Nie, F.; Wang, Y.; Zhang, Z.; Hou, J.; He, D.; Xie, M.; Xu, L.; De, W.; Wang, Z.; et al. LncRNA HOXA11-AS Promotes Proliferation and Invasion of Gastric Cancer by Scaffolding the Chromatin Modification Factors PRC2, LSD1, and DNMT1. Cancer Res. 2016, 76, 6299–6310. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; LaFleur, M.; Nguyen, T.; Chen, S.; Chakravarthy, A.; Conway, J.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019, 38, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, X.; Yang, Y.; Li, Y.; Wu, S. LSD1 silencing contributes to enhanced efficacy of anti-CD47/PD-L1 immunotherapy in cervical cancer. Cell Death Dis. 2021, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.; Burr, M.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Oeck, S.; Glazer, P.M. Hypoxia Promotes Resistance to EGFR Inhibition in NSCLC Cells via the Histone Demethylases, LSD1 and PLU-1. Mol. Cancer Res. 2018, 16, 1458–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Xiong, J.; Xu, D.; Hao, N.; Zhang, Y.; Sang, Y.; Wang, Z.; Zheng, X.; Min, J.; Diao, H.; et al. TdIF1-LSD1 Axis Regulates Epithelial—Mesenchymal Transition and Metastasis via Histone Demethylation of E-Cadherin Promoter in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 250. https://doi.org/10.3390/ijms23010250
Liu Q, Xiong J, Xu D, Hao N, Zhang Y, Sang Y, Wang Z, Zheng X, Min J, Diao H, et al. TdIF1-LSD1 Axis Regulates Epithelial—Mesenchymal Transition and Metastasis via Histone Demethylation of E-Cadherin Promoter in Lung Cancer. International Journal of Molecular Sciences. 2022; 23(1):250. https://doi.org/10.3390/ijms23010250
Chicago/Turabian StyleLiu, Qi, Juan Xiong, Derong Xu, Nan Hao, Yujuan Zhang, Yi Sang, Zhigang Wang, Xiufen Zheng, Jeffrey Min, Hong Diao, and et al. 2022. "TdIF1-LSD1 Axis Regulates Epithelial—Mesenchymal Transition and Metastasis via Histone Demethylation of E-Cadherin Promoter in Lung Cancer" International Journal of Molecular Sciences 23, no. 1: 250. https://doi.org/10.3390/ijms23010250
APA StyleLiu, Q., Xiong, J., Xu, D., Hao, N., Zhang, Y., Sang, Y., Wang, Z., Zheng, X., Min, J., Diao, H., Raphael, J., Vareki, S. M., Koropatnick, J., & Min, W. (2022). TdIF1-LSD1 Axis Regulates Epithelial—Mesenchymal Transition and Metastasis via Histone Demethylation of E-Cadherin Promoter in Lung Cancer. International Journal of Molecular Sciences, 23(1), 250. https://doi.org/10.3390/ijms23010250