
The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
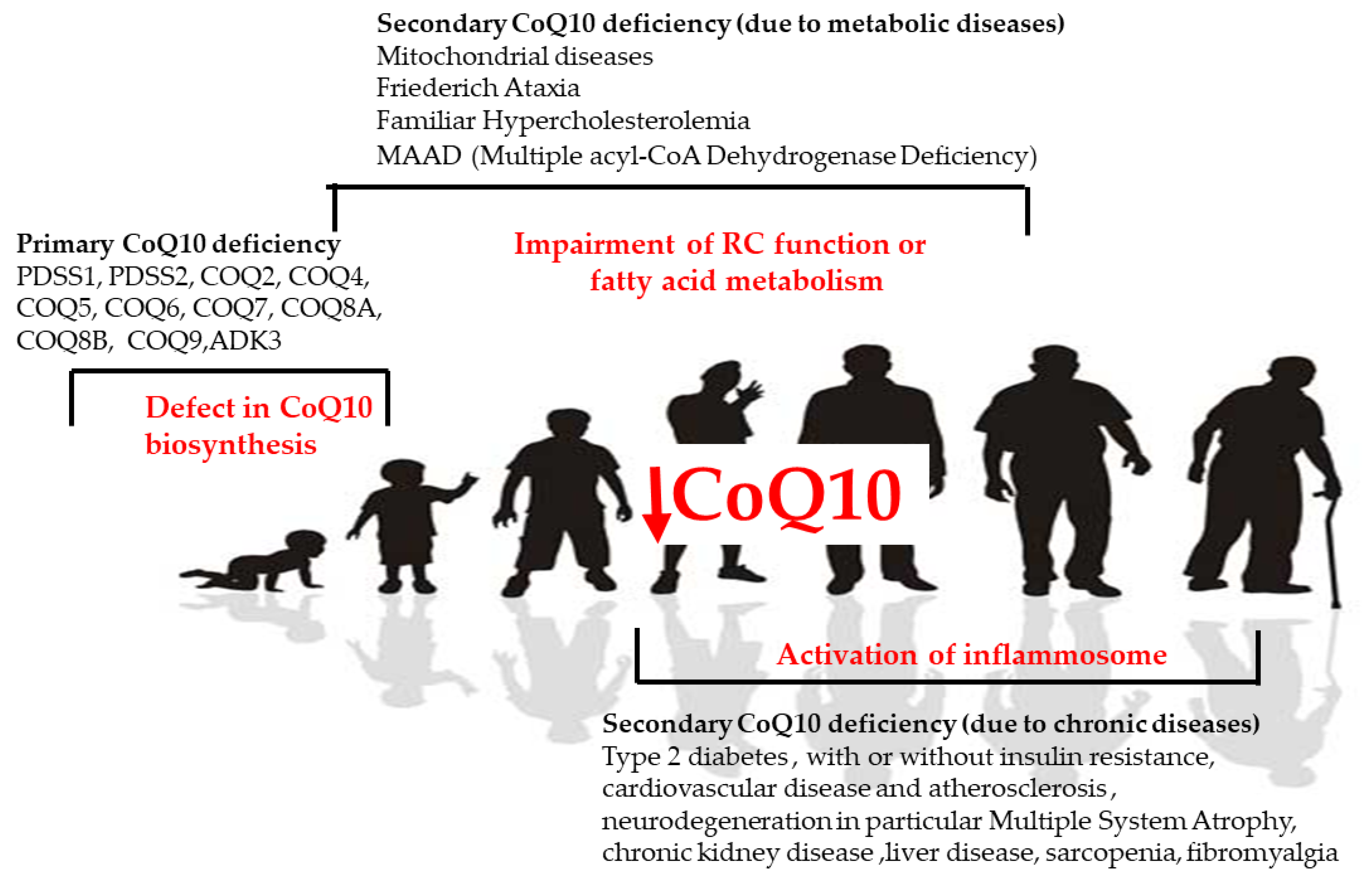
1. Introduction
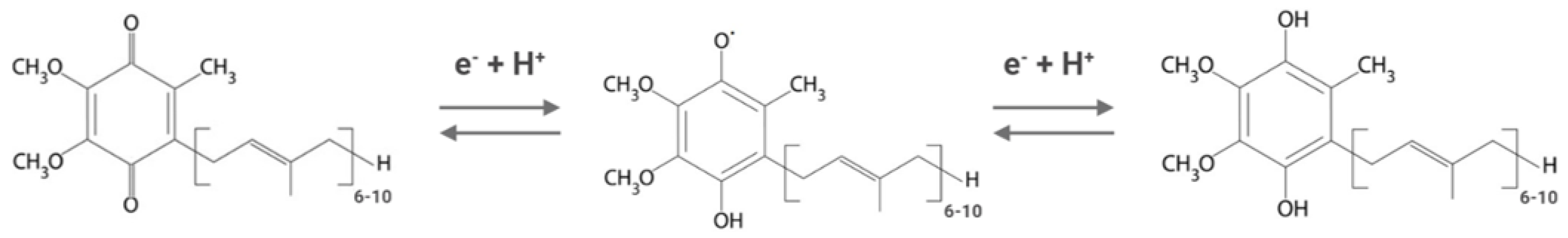
2. Ubiquinone/Coenzyme Q Biosynthesis
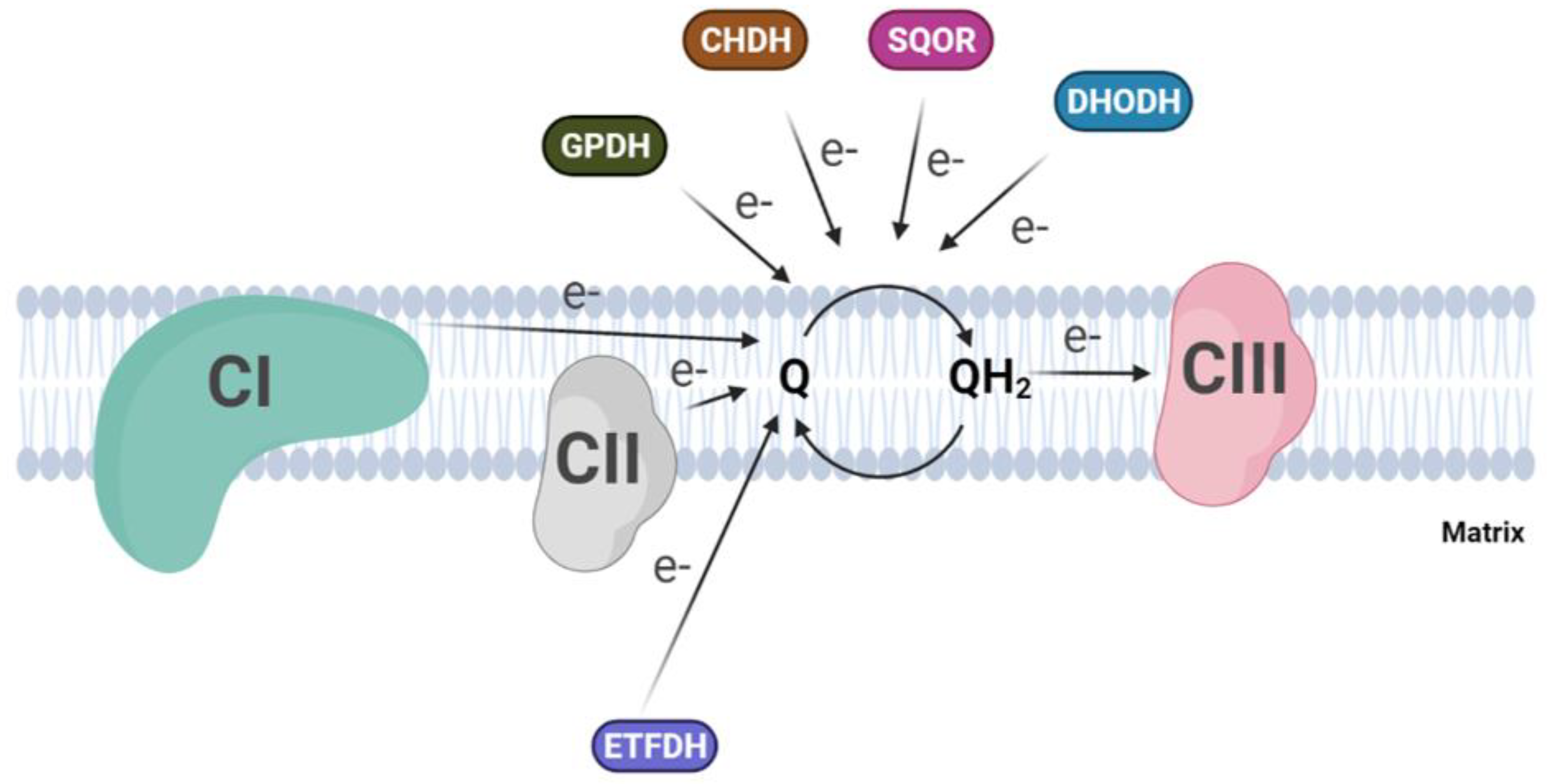
2.1. Ubiquinone in Mitochondrial Electron Transfer Chains
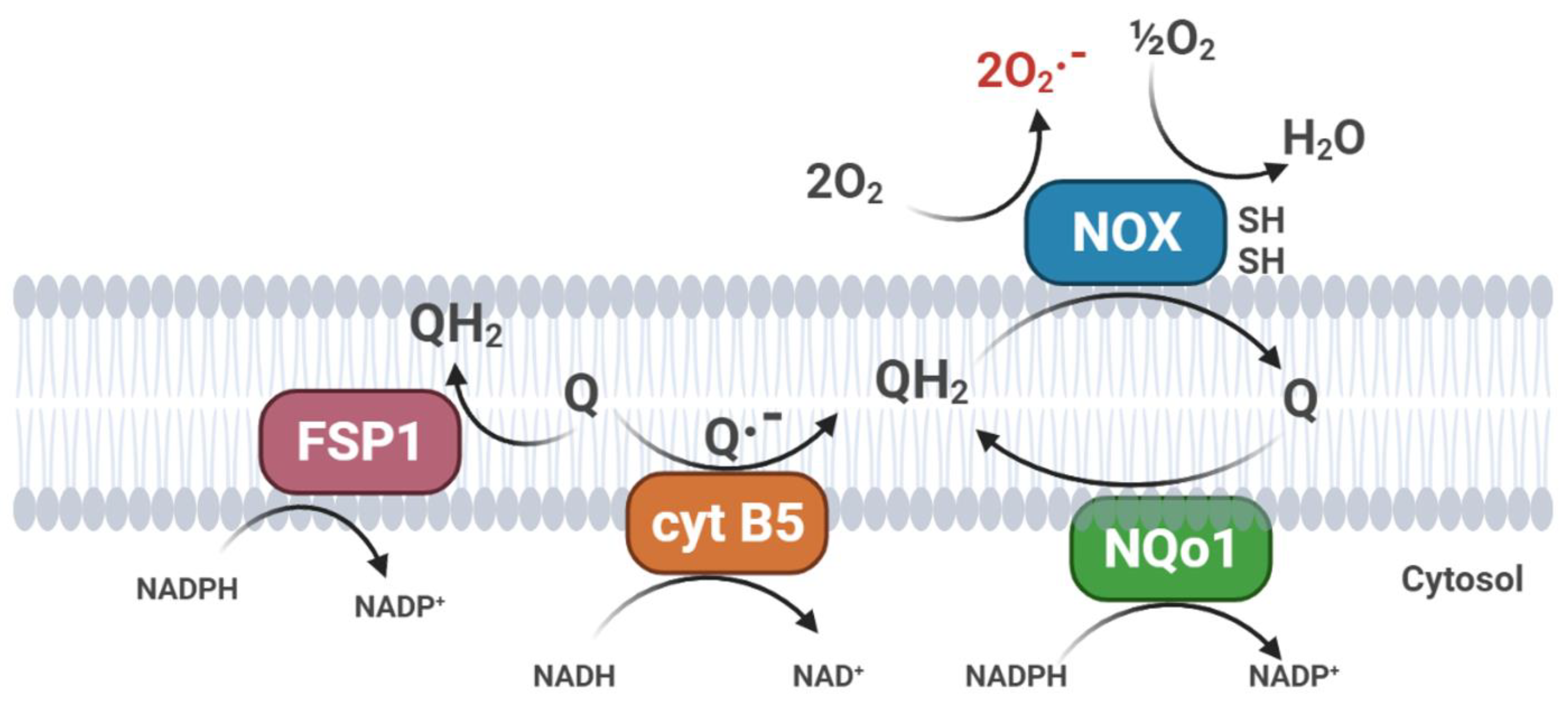
2.2. CoQ10 in Cell Membranes
2.3. CoQ as a Component in Low Density Lipoproteins (LDL)
2.4. Aging and Longevity: Role of CoQ10 Homeostasis
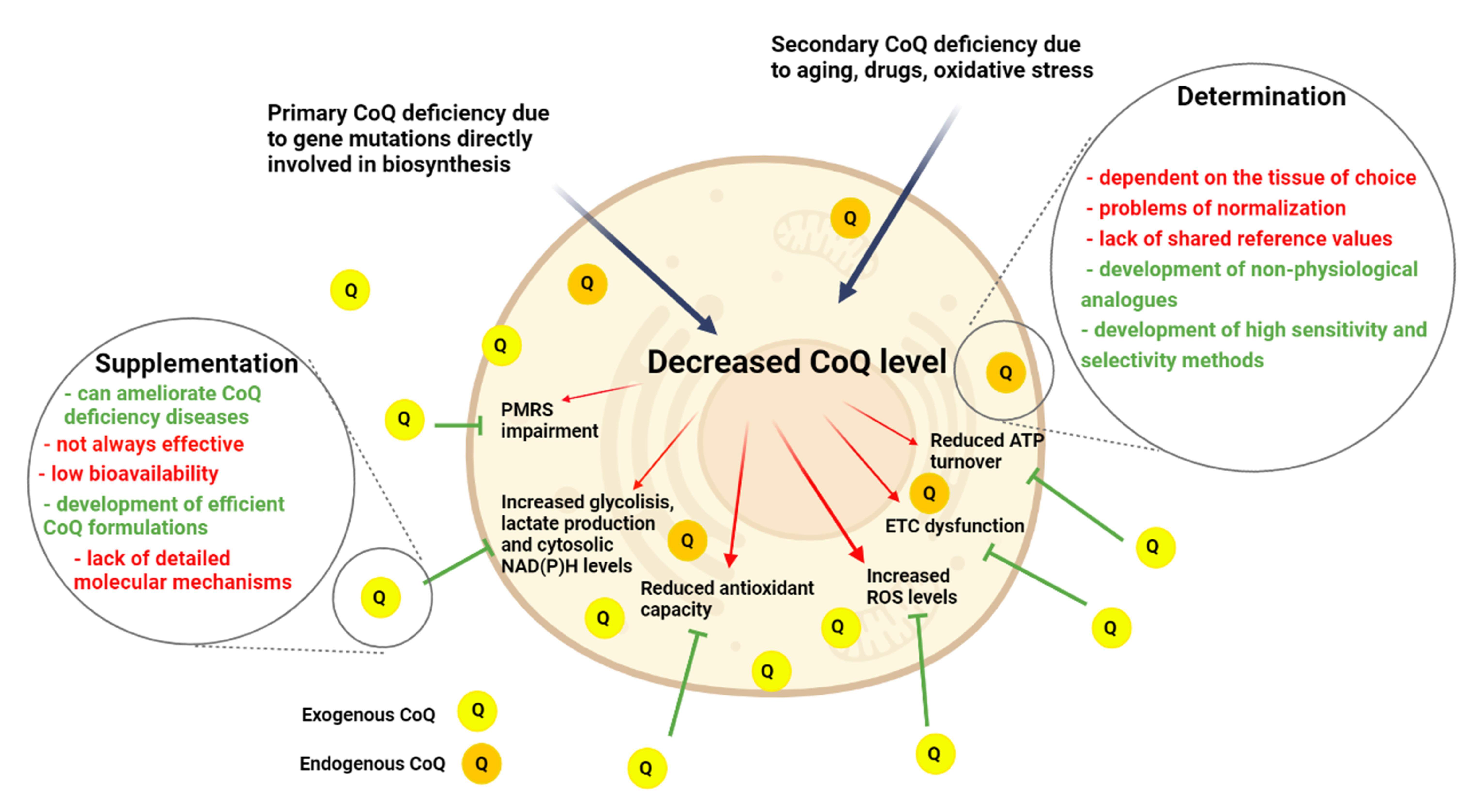
2.5. CoQ in Human Diseases
3. Coenzyme Q Determination in Biological Samples
3.1. CoQ Determination
3.2. Biological Samples
4. Coenzyme Q10 Supplementation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kofler, M. Ueber ein pflanzliches Chinon. In Festschrift Emil Christoph Barell; Hoffmann Laroche: Basel, Switzerland, 1946; pp. 199–212. [Google Scholar]
- Crane, F.L. Internal Distribution of Coenzyme Q in Higher Plants. Plant Physiol. 1959, 34, 128–131. [Google Scholar] [CrossRef]
- Crane, F.L. Isolation of Two Quinones with Coenzyme Q Activity from Alfalfa. Plant Physiol. 1959, 34, 546–551. [Google Scholar] [CrossRef]
- Crane, F.L. Quinones in electron transport. 1. Coenzymatic activity of plastoquinone, coenzyme Q and related natural quinones. Arch. Biochem. Biophys. 1960, 87, 198–202. [Google Scholar] [CrossRef]
- Lester, R.L.; Crane, F.L. The natural occurrence of coenzyme Q and related compounds. J. Biol. Chem. 1959, 234, 2169–2175. [Google Scholar] [CrossRef]
- Lester, R.L.; Ramasarma, T. Chromatography of the coenzyme Q family of compounds on silicone-impregnated paper. J. Biol. Chem. 1959, 234, 672–676. [Google Scholar] [CrossRef]
- Crane, F.L. Isolation and characterization of the coenzyme Q group and plastoquinone. In Ciba Foundation Symposium-Endocrinology of the Testis (Colloquia on Endocrinology); John Wiley & Sons, Ltd.: Chichester, UK, 1961; pp. 36–78. [Google Scholar]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Lane, N.; Allen, J.F.; Martin, W. How did LUCA make a living? Chemiosmosis in the origin of life. Bioessays 2010, 32, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ducluzeau, A.L.; Schoepp-Cothenet, B.; Baymann, F.; Russell, M.J.; Nitschke, W. Free energy conversion in the LUCA: Quo vadis? Biochim. Biophys. Acta 2014, 1837, 982–988. [Google Scholar] [CrossRef]
- Morré, D.J.; Morré, D.M. Non-mitochondrial coenzyme. Q. Biofactors. 2011, 37, 355–360. [Google Scholar] [CrossRef]
- Kishi, T.; Morré, D.M.; Morré, D.J. The plasma membrane NADH oxidase of HeLa cells has hydroquinone oxidase activity. Biochim. Biophys. Acta 1999, 1412, 66–77. [Google Scholar] [CrossRef][Green Version]
- Gómez-Díaz, C.; Rodríguez-Aguilera, J.C.; Barroso, M.P.; Villalba, J.M.; Navarro, F.; Crane, F.L.; Navas, P. Antioxi-dant ascorbate is stabilized by NADH-coenzyme Q10 reductase in the plasma membrane. J. Bioenerg. Biomembr. 1997, 29, 251–257. [Google Scholar] [CrossRef]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Or-landini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef]
- Döring, F.; Schmelzer, C.; Lindner, I.; Vock, C.; Fujii, K. Functional connections and pathways of coenzyme Q10-inducible genes: An in-silico study. IUBMB Life 2007, 59, 628–633. [Google Scholar] [CrossRef]
- Ernster, L. Lipid peroxidation in biological membranes: Mechanism and implications. In Active Oxygens, Lipid Peroxides, and Antioxidants; Yagi, K., Ed.; CRC Press: Tokyo, Japan, 1993; pp. 1–38. [Google Scholar]
- Ernster, L.; Forsmark-Andrée, P. Ubiquinol: An endogenous antioxidant in aerobic organisms. Clin. Investig. 1993, 71, S60–S65. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Cavazzoni, M.; Genova, M.L.; D’Aurelio, M.; Pich, M.M.; Pallotti, F.; Formiggini, G.; Marchetti, M.; Castelli, G.P.; Bovina, C. Oxidative stress, antioxidant defences and aging. BioFactors 1998, 8, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Kagan, T.; Davis, C.; Lin, L.; Zakeri, Z. Coenzyme Q10 can in some circumstances block apoptosis, and this effect is mediated through mitochondria. Ann. N. Y. Acad. Sci. 1999, 887, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta (BBA)-Biomembr. 2003, 1660, 171–199. [Google Scholar] [CrossRef]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q--biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Mikashinovich, Z.I.; Belousova, E.S.; Sarkisyan, O.G. Impairment of Energy-Dependent Processes in the Muscle Tissue as a Pathogenetic Mechanism of Statin-Induced Myopathy. Bull. Exp. Biol. Med. 2017, 162, 433–435. [Google Scholar] [CrossRef]
- Du Souich, P.; Roederer, G.; Dufour, R. Myotoxicity of statins: Mechanism of action. Pharmacol Ther. 2017, 175, 1–16. [Google Scholar] [CrossRef]
- Ramachandran, R.; Wierzbicki, A.S. Statins, Muscle Disease and Mitochondria. J. Clin. Med. 2017, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional conservation of coen-zyme Q biosynthetic genes among yeasts, plants, and humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in pa-ra-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Hernandez, M.A.R.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency ofCOQ4causes coenzyme Q10deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; De Lonlay, P.; De Baulny, H.O.; et al. CABC1 Gene Mutations Cause Ubiquinone Deficiency with Cerebellar Ataxia and Seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autoso-mal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Guo, W.; Han, X.; Dai, W.; Diao, Z.; Liu, W. Role of UBIAD1 in Intracellular Cholesterol Metabolism and Vascular Cell Calcification. PLoS ONE 2016, 11, e0149639. [Google Scholar] [CrossRef] [PubMed]
- Kemmerer, Z.A.; Robinson, K.P.; Schmitz, J.M.; Manicki, M.; Paulson, B.R.; Jochem, A.; Hutchins, P.D.; Coon, J.J.; Pagliarini, D.J. UbiB proteins regulate cellular CoQ distribution in Saccharomyces cerevisiae. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Green, D.E.; Tzagoloff, A. The mitochondrial electron transfer chain. Arch. Biochem. Biophys. 1966, 116, 293–304. [Google Scholar] [CrossRef]
- Kröger, A.; Klingenberg, M. The Kinetics of the Redox Reactions of Ubiquinone Related to the Electron-Transport Activity in the Respiratory Chain. JBIC J. Biol. Inorg. Chem. 1973, 34, 358–368. [Google Scholar] [CrossRef]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef]
- Schägger, H.; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexes I–V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: Random collisions vs. solid state electron channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Per-ales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex as-sembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C.; Butow, R.A.; Racker, E.; Chance, B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 1967, 242, 5169–5173. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Agüera, M.C.; Gao, L.; Gonzalez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Ashok, D.; Sarantos, M.R.; Shi, T.; Hughes, R.E.; Brand, M.D. Inhibitors of ROS production by the ubiqui-none-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 2013, 65, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sen-sor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef]
- Burger, N.; Logan, A.; Prime, T.A.; Mottahedin, A.; Caldwell, S.; Krieg, T.; Hartley, R.; James, A.M.; Murphy, M.P. A sensitive mass spectrometric assay for mitochondrial CoQ pool redox state in vivo. Free Radic. Biol. Med. 2019, 147, 37–47. [Google Scholar] [CrossRef]
- Rauchová, H.; Battino, M.; Fato, R.; Lenaz, G.; Drahota, Z. Coenzyme Q-pool function in glycerol-3-phosphate oxidation in hamster brown adipose tissue mitochondria. J. Bioenerg. Biomembr. 1992, 24, 235–241. [Google Scholar] [CrossRef]
- Moxley, M.A.; Tanner, J.J.; Becker, D.F. Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch. Biochem. Biophys. 2011, 516, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.C.; Dawson, A.P. The reaction of choline dehydrogenase with some electron acceptors. Biochem. J. 1975, 151, 677–683. [Google Scholar] [CrossRef] [PubMed]
- De Bari, L.; Moro, L.; Passarella, S. Prostate cancer cells metabolize d-lactate inside mitochondria via a D-lactate dehydrogenase which is more active and highly expressed than in normal cells. FEBS Lett. 2013, 587, 467–473. [Google Scholar] [CrossRef] [PubMed]
- De Bari, L.; Atlante, A.; Armeni, T.; Kalapos, M.P. Synthesis and metabolism of methylglyoxal, S-D-lactoylglutathione and D-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Ageing Res. Rev. 2019, 53, 100915. [Google Scholar] [CrossRef]
- Maurino, V.G.; Welchen, E.; García, L.; Schmitz, J.; Fuchs, P.; Wagner, S.; Wienstroer, J.; Schertl, P.; Braun, H.-P.; Schwarzländer, M.; et al. D-Lactate dehydrogenase links methylglyoxal degradation and electron transport through cytochrome C. Plant Physiol. 2016, 172, 901–912. [Google Scholar] [CrossRef]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic consequences from xenotopic expression of a tuni-cate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys Acta Bioenerg. 2020, 1861, 148137. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Villalba, J.; Navarro, F.; Gómez-Díaz, C.; Arroyo, A.; Bello, R.; Navas, P. Role of cytochrome b5 reductase on the antioxidant function of coenzyme Q in the plasma membrane. Mol. Asp. Med. 1997, 18, 7–13. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Aguilera, J.C.R.; López-Lluch, G.; Ocaña, C.S.; Villalba, J.; Gómez-Díaz, C.; Navas, P. Plasma membrane redox system protects cells against oxidative stress. Redox Rep. 2000, 5, 148–150. [Google Scholar] [CrossRef][Green Version]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q biosynthesis inhibition induces HIF-1α stabilization and metabolic switch toward glycolysis. FEBS J. 2020, 288, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Protonmotive redox mechanism of the cytochrome b-c1 complex in the respiratory chain: Pro-tonmotive ubiquinone cycle. FEBS Lett. 1975, 56, 1–6. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of fer-roptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to in-hibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Rizzardi, N.; Liparulo, I.; Antonelli, G.; Orsini, F.; Riva, A.; Bergamini, C.; Fato, R. Coenzyme Q10 Phytosome Formulation Improves CoQ10 Bioavailability and Mitochondrial Functionality in Cultured Cells. Antioxidants 2021, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G. Coenzyme Q homeostasis in aging: Response to non-genetic interventions. Free Radic. Biol. Med. 2021, 164, 285–302. [Google Scholar] [CrossRef]
- Stocker, R.; Bowry, V.W.; Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc. Natl. Acad. Sci. USA 1991, 88, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Tribble, D.L.; van den Berg, J.J.; Motchnik, P.A.; Ames, B.N.; Lewis, D.M.; Chait, A.; Krauss, R.M. Oxidative susceptibil-ity of low density lipoprotein subfractions is related to their ubiquinol-10 and alpha-tocopherol content. Proc. Natl. Acad. Sci. USA 1994, 91, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Mine, Y.; Okamoto, T. Extracellular coenzyme Q10 (CoQ10) is reduced to ubiquinol-10 by intact Hep G2 cells independent of intracellular CoQ10 reduction. Arch. Biochem. Biophys. 2019, 672, 108067. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Elstner, E.F. Coenzyme Q10, Vitamin E, and Dihydrothioctic Acid Cooperatively Prevent Diene Conjugation in Isolated Low-Density Lipoprotein. Antioxidants Redox Signal. 2000, 2, 327–333. [Google Scholar] [CrossRef] [PubMed]
- De Pinieux, G.; Chariot, P.; Ammi-Saïd, M.; Louarn, F.; Lejonc, J.L.; Astier, A.; Jacotot, B.; Gherardi, R. Lipid-lowering drugs and mitochondrial function: Effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br. J. Clin. Pharmacol. 1996, 42, 333–337. [Google Scholar] [CrossRef]
- SEARCH Collaborative Group; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- López-Lluch, G.; Hernández-Camacho, J.D.; Fernández-Ayala, D.J.M.; Navas, P. Mitochondrial dysfunction in metabolism and ageing: Shared mechanisms and outcomes? Biogerontology 2018, 19, 461–480. [Google Scholar] [CrossRef]
- Pagano, G.; Pallardó, F.V.; Lyakhovich, A.; Tiano, L.; Fittipaldi, M.R.; Toscanesi, M.; Trifuoggi, M. Aging-Related Disorders and Mitochondrial Dysfunction: A Critical Review for Prospect Mitoprotective Strategies Based on Mitochondrial Nutrient Mixtures. Int. J. Mol. Sci. 2020, 21, 7060. [Google Scholar] [CrossRef]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G.; Rodríguez-Aguilera, J.C.; Santos-Ocaña, C.; Navas, P. Is coenzyme Q a key factor in aging? Mech. Ageing Dev. 2010, 131, 225–235. [Google Scholar] [CrossRef]
- Maroz, A.; Anderson, R.F.; Smith, R.A.; Murphy, M.P. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: Implications for in vivo antioxidant activity. Free Radic. Biol. Med. 2009, 46, 105–109. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Kindermann, B.; Althammer, M.; Klapper, M.; Vormann, J.; Littarru, G.P.; Döring, F. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int. J. Biochem. Cell Biol. 2005, 37, 1208–1218. [Google Scholar] [CrossRef]
- Moreno Fernández-Ayala, D.J.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Hidalgo-Gutierrez, A.; Kleiner, G.; Lopez, L.C. The Role of Sulfide Oxidation Impairment in the Pathogenesis of Primary CoQ Deficiency. Front Physiol. 2017, 8, 525. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.-W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxidative Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lv, H.; Xu, X.; Ma, Y.; Li, Q. Clinical characteristics and gene mutation analysis of an adult patient with ETFDH-related multiple acyl-CoA dehydrogenase deficiency. Mol. Med. Rep. 2020, 22, 4396–4402. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Döring, F. Functions of coenzyme Q10 in inflammation and gene expression. Biofactors 2008, 32, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; De Miguel, M.; Moreno Fernández, A.M.; Carmona López, I.M.; Garrido Maraver, J.; Cotán, D.; Gómez Izquierdo, L.; Bonal, P.; Campa, F.; Bullon, P.; et al. Mitochondrial dysfunction and mitophagy activation in blood mononuclear cells of fibromyalgia patients: Implications in the patho-genesis of the disease. Arthritis Res. Ther. 2010, 12, R17. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Díaz-Parrado, E.; Carrión, A.M.; Alfonsi, S.; Sánchez-Alcazar, J.A.; Bullón, P.; Battino, M.; de Miguel, M. Is inflammation a mitochondrial dysfunction-dependent event in fibromyalgia? Antioxid Redox Signal. 2013, 18, 800–807. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mito-chondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7, e32111. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Miyazaki, T.; Takagi, A.; Sugita, Y.; Yatsu, S.; Murata, A.; Kato, T.; Suda, S.; Ouchi, S.; Aikawa, T.; et al. Low circulating coenzyme Q10 during acute phase is associated with inflammation, malnutrition, and in-hospital mortality in patients admitted to the coronary care unit. Hear. Vessel. 2016, 32, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, O.; Zhu, X.; Perry, G.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in neuro-degenerative disease. Sci. Aging Knowl. Environ. 2002, 2002, e16. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjakova, A.; Kucharska, J.; Sumbalova, Z.; Rausova, Z.; Chladekova, A.; Komlosi, M.; Szamosova, M.; Mojto, V. The importance of coenzyme Q10 and its ratio to cholesterol in the progress of chronic kidney diseases linked to non- -communicable diseases. Bratisl. Med. J. 2020, 121, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, X.; Xue, H.; Zhang, P.; Fang, W.; Chen, X.; Ling, W. Coenzyme Q10 attenuates high-fat di-et-induced non-alcoholic fatty liver disease through activation of the AMPK pathway. Food Funct. 2019, 10, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef] [PubMed]
- Åberg, F.; Appelkvist, E.-L.; Dallner, G.; Ernster, L. Distribution and redox state of ubiquinones in rat and human tissues. Arch. Biochem. Biophys. 1992, 295, 230–234. [Google Scholar] [CrossRef]
- Yuan, B.; Liu, C.; Xu, P.; Lin, L.; Pan, C.; Wang, L.; Xu, H. Validated HPLC method for the quantitative determination of CoQ10 in dog plasma and its application to a pharmacokinetic study. Biomed. Chromatogr. 2010, 25, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Onur, S.; Fischer, A.; Laudes, M.; Palussen, M.; Menke, T.; Döring, F. Coenzyme Q10 serum concentration and redox status in European adults: Influence of age, sex, and lipoprotein concentration. J. Clin. Biochem. Nutr. 2016, 58, 240–245. [Google Scholar] [CrossRef]
- Finckh, B.; Kontush, A.; Commentz, J.; Hübner, C.; Burdelski, M.; Kohlschütter, A. [31] High-performance liquid chromatography-coulometric electrochemical detection of ubiquinol 10, ubiquinone 10, carotenoids, and tocopherols in neonatal plasma. Methods Enzymol. 1999, 299, 341–348. [Google Scholar] [CrossRef]
- Tang, P.H.; Miles, M.V.; Degrauw, A.; Hershey, A.; Pesce, A. HPLC Analysis of Reduced and Oxidized Coenzyme Q10 in Human Plasma. Clin. Chem. 2001, 47, 256–265. [Google Scholar] [CrossRef]
- Barshop, B.A.; Gangoiti, J.A. Analysis of coenzyme Q in human blood and tissues. Mitochondrion 2007, 7, S89–S93. [Google Scholar] [CrossRef]
- Schou-Pedersen, A.M.V.; Schemeth, D.; Lykkesfeldt, J. Determination of Reduced and Oxidized Coenzyme Q10 in Canine Plasma and Heart Tissue by HPLC-ECD: Comparison with LC-MS/MS Quantification. Antioxidants 2019, 8, 253. [Google Scholar] [CrossRef]
- Molyneux, S.L.; Young, J.M.; Florkowski, C.M.; Lever, M.; George, P.M. Coenzyme Q10: Is There a Clini-cal Role and a Case for Measurement? Clin. Biochem. Rev. 2008, 29, 71–82. [Google Scholar]
- Kaikkonen, J.; Nyyssonen, K.; Salonen, J.; Kaikkonen, K.N.J. Measurement and stability of plasma reduced, oxidized and total coenzyme Q10 in humans. Scand. J. Clin. Lab. Investig. 1999, 59, 457–466. [Google Scholar] [CrossRef]
- Okamoto, T.; Fukunaga, Y.; Ida, Y.; Kishi, T. Determination of reduced and total ubiquinones in biological materials by liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1988, 430, 11–19. [Google Scholar] [CrossRef]
- Kaya, Y.; ÇEBİ, A.; Söylemez, N.; Demir, H.; Alp, H.H.; Bakan, E. Correlations between Oxidative DNA Damage, Oxidative Stress and Coenzyme Q10 in Patients with Coronary Artery Disease. Int. J. Med. Sci. 2012, 9, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Di Giovamberardino, G.; Petrillo, S.; Boenzi, S.; Bertini, E.; Dionisi-Vici, C.; Piemonte, F. Pediatric reference intervals for muscle coenzyme Q10. Biomarkers 2012, 17, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Bysted, A.; Hølmer, G. Coenzyme Q10 in the diet-daily intake and relative bioavailability. Mol. Asp. Med. 1997, 18, 251–254. [Google Scholar] [CrossRef]
- Vadhanavikit, S.; Sakamoto, N.; Ashida, N.; Kishi, T.; Folkers, K. Quantitative determination of coenzyme Q10 in human blood for clinical studies. Anal. Biochem. 1984, 142, 155–158. [Google Scholar] [CrossRef]
- Tang, P.H.; Miles, M.V.; Miles, L.; Quinlan, J.; Wong, B.; Wenisch, A.; Bove, K. Measurement of reduced and oxidized coenzyme Q9 and coenzyme Q10 levels in mouse tissues by HPLC with coulometric detection. Clin. Chim. Acta 2004, 341, 173–184. [Google Scholar] [CrossRef]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of Coenzyme Q10 Status in Blood Mononuclear Cells, Skeletal Muscle, and Plasma by HPLC with Di-Propoxy-Coenzyme Q10 as an Internal Standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef]
- Edlund, P.O. Determination of coenzyme Q10, α-tocopherol and cholesterol in biological samples by coupled-column liquid chromatography with coulometric and ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl. 1988, 425, 87–97. [Google Scholar] [CrossRef]
- Miles, M.V.; Horn, P.S.; Tang, P.H.; Morrison, J.A.; Miles, L.; DeGrauw, T.; Pesce, A.J. Age-related changes in plasma coenzyme Q10 concentrations and redox state in apparently healthy children and adults. Clin. Chim. Acta 2004, 347, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Bhagavan, H.N.; Chopra, R.K. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007, 7, S78–S88. [Google Scholar] [CrossRef]
- Yubero, D.; Allen, G.; Artuch, R.; Montero, R. The Value of Coenzyme Q10 Determination in Mitochondrial Patients. J. Clin. Med. 2017, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Kalén, A.; Norling, B.; Appelkvist, E.; Dallner, G. Ubiquinone biosynthesis by the microsomal fraction from rat liver. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1987, 926, 70–78. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. Int. J. Lab. Med. 2003, 40, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Diamant, B.; Edlund, P.O.; Lund, B.; Folkers, K.; Theorell, H. Plasma Ubiquinone, Al-pha-Tocopherol and Cholesterol in Man. Int. J. Vitam. Nutr. Res. 1992, 62, 160–164. [Google Scholar]
- McDonnell, M.; Archbold, G. Plasma ubiquinol/cholesterol ratios in patients with hyperlipidaemia, those with diabetes mellitus and in patients requiring dialysis. Clin. Chim. Acta 1996, 253, 117–126. [Google Scholar] [CrossRef]
- Niklowitz, P.; Scherer, J.; Döring, F.; Paulussen, M.; Menke, T. Oxidized proportion of muscle coenzyme Q10 increases with age in healthy children. Pediatr. Res. 2015, 78, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Menke, T.; Andler, W.; Okun, J.G. Simultaneous analysis of coenzyme Q10 in plasma, erythrocytes and platelets: Comparison of the antioxidant level in blood cells and their environment in healthy children and after oral supplementation in adults. Clin. Chim. Acta 2004, 342, 219–226. [Google Scholar] [CrossRef]
- Molyneux, S.L.; Florkowski, C.M.; Lever, M.; George, P.M. Biological Variation of Coenzyme Q10. Clin. Chem. 2005, 51, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Sebestianová, N.; Turiaková, J.; Kucera, I. Determination of coenzyme Q in human plasma. Physiol. Res. 1996, 45, 39–46. [Google Scholar]
- Wahlqvist, M.L.; Wattanapenpaiboon, N.; Savige, G.S.; Kannar, D. Bioavailability of Two Different For-mulations of Coenzyme Q10 in Healthy Subjects. Asia Pac. J. Clin. Nutr. 1998, 7, 37–40. [Google Scholar] [PubMed]
- Miles, M.V.; Horn, P.S.; Morrison, J.A.; Tang, P.H.; DeGrauw, T.; Pesce, A.J. Plasma coenzyme Q10 reference intervals, but not redox status, are affected by gender and race in self-reported healthy adults. Clin. Chim. Acta 2003, 332, 123–132. [Google Scholar] [CrossRef]
- Kontush, A.; Reich, A.; Baum, K.; Spranger, T.; Finckh, B.; Kohlschütter, A.; Beisiegel, U. Plasma ubiquinol-10 is decreased in patients with hyperlipidaemia. Atherosclerosis 1997, 129, 119–126. [Google Scholar] [CrossRef]
- Tomasetti, M.; Alleva, R.; Solenghi, M.D.; Littarru, G.P. Distribution of antioxidants among blood components and lipoproteins: Significance of lipids/CoQ10ratio as a possible marker of increased risk for atherosclerosis. BioFactors 1999, 9, 231–240. [Google Scholar] [CrossRef]
- Montero, R.; Yubero, D.; Salgado, M.C.; González, M.J.; Campistol, J.; O’Callaghan, M.D.M.; Pineda, M.; Delgadillo, V.; Maynou, J.; Fernandez, G.; et al. Plasma coenzyme Q10 status is impaired in selected genetic conditions. Sci. Rep. 2019, 9, 793. [Google Scholar] [CrossRef]
- Artuch, R.; Vilaseca, M.-A.; Moreno, J.; Lambruschini, N.; Cambra, F.J.; Campistol, J. Decreased serum ubiquinone-10 concentrations in phenylketonuria. Am. J. Clin. Nutr. 1999, 70, 892–895. [Google Scholar] [CrossRef]
- Cooper, J.M.; Korlipara, L.V.P.; Hart, P.E.; Bradley, J.L.; Schapira, A.H.V. Coenzyme Q10and vitamin E deficiency in Friedreich’s ataxia: Predictor of efficacy of vitamin E and coenzyme Q10therapy. Eur. J. Neurol. 2008, 15, 1371–1379. [Google Scholar] [CrossRef]
- Colomé, C.; Artuch, R.; Vilaseca, M.A.; Sierra, C.; Brandi, N.; Cambra, F.J.; Lambruschini, N.; Campistol, J. Ubiquinone-10 content in lymphocytes of phenylketonuric patients. Clin. Biochem. 2002, 35, 81–84. [Google Scholar] [CrossRef]
- Steen, G.; Axelsson, H.; Bowallius, M.; Holthuis, N.; Molander, B.-M. Isoprenoid biosynthesis in multiple sclerosis, II. A possible role of NADPH. Acta Neurol. Scand. 1987, 76, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.V.; Tang, P.H.; Miles, L.; Steele, P.E.; Moye, M.J.; Horn, P.S. Validation and application of an HPLC-EC method for analysis of coenzyme Q10 in blood platelets. Biomed. Chromatogr. 2008, 22, 1403–1408. [Google Scholar] [CrossRef]
- Mortensen, S.; Heidt, P.; Sehested, J. Clinical Perspectives in Treatment of Cardiovascular Diseases with Coenzyme Q 10. In Highlights in Ubiquinone Research; Taylor and Francis: London, UK, 1998; pp. 226–227. [Google Scholar]
- Crane, F.L. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 2007, 7, S2–S7. [Google Scholar] [CrossRef]
- Ogasahara, S.; Engel, A.G.; Frens, D.; Mack, D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Artuch, R.; Briones, P.; Nascimento, A.; García-Cazorla, A.; Vilaseca, M.; Śnchez-Alćzar, J.; Navas, P.; Montoya, J.; Pineda, M. Muscle coenzyme Q10 concentrations in patients with probable and definite diagnosis of respiratory chain disorders. BioFactors 2005, 25, 109–115. [Google Scholar] [CrossRef]
- Miles, M.V.; Miles, L.; Tang, P.H.; Horn, P.S.; Steele, P.E.; DeGrauw, A.J.; Wong, B.L.; Bove, K.E. Systematic evaluation of muscle coenzyme Q10 content in children with mitochondrial respiratory chain enzyme deficiencies. Mitochondrion 2008, 8, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Louw, R.; Smuts, I.; Wilsenach, K.-L.; Jonck, L.-M.; Schoonen, M.; van der Westhuizen, F. The dilemma of diagnosing coenzyme Q10 deficiency in muscle. Mol. Genet. Metab. 2018, 125, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.F.; de la Mata, M.; Pavon, A.D.; Alcocer-Gomez, E.; Calero, C.P.; Paz, M.V.; Alanis, M.; de Lavera, I. Clinical applications of coenzyme Q10. Front Biosci 2014, 19, 619–633. [Google Scholar]
- Neergheen, V.; Chalasani, A.; Wainwright, L.; Yubero, D.; Montero, R.; Artuch, R.; Hargreaves, I. Coenzyme Q10 in the Treatment of Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Larriva, A.P.A.-D.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 Supplementation for the Reduction of Oxidative Stress: Clinical Implications in the Treatment of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed]
- Pravst, I.; Rodriguez Aguilera, J.C.; Cortes Rodriguez, A.B.; Jazbar, J.; Locatelli, I.; Hristov, H.; Žmitek, K. Comparative Bioavailability of Different Coenzyme Q10 Formulations in Healthy Elderly Individuals. Nutrients 2020, 12, 784. [Google Scholar] [CrossRef]
- Li, H.; Chen, F. Preparation and quality evaluation of coenzyme Q10 long-circulating liposomes. Saudi J. Biol. Sci. 2015, 24, 797–802. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, G.; Zhang, J.; Sun, N.; Duan, M.; Yan, Z.; Xia, Q. Novel Lipid-Free Nanoformulation for Improving Oral Bioavailability of Coenzyme Q10. BioMed Res. Int. 2014, 2014, 793879. [Google Scholar] [CrossRef] [PubMed]
- Masotta, N.E.; Martinefski, M.R.; Lucangioli, S.; Rojas, A.M.; Tripodi, V.P. High-dose coenzyme Q10-loaded oleogels for oral therapeutic supplementation. Int. J. Pharm. 2018, 556, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Song, J. Inclusion of coenzyme Q10 with beta-cyclodextrin studied by polarography. Yao Xue Xue Bao 2006, 41, 671–674. [Google Scholar]
- Manzar, H.; Abdulhussein, D.; Yap, T.; Cordeiro, M. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef]
- Löw, H.; Crane, F.L.; Morré, D.J. Putting together a plasma membrane NADH oxidase: A tale of three labor-atories. Int. J. Biochem. Cell Biol. 2012, 44, 1834–1838. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2022, 23, 128. https://doi.org/10.3390/ijms23010128
Pallotti F, Bergamini C, Lamperti C, Fato R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. International Journal of Molecular Sciences. 2022; 23(1):128. https://doi.org/10.3390/ijms23010128
Chicago/Turabian StylePallotti, Francesco, Christian Bergamini, Costanza Lamperti, and Romana Fato. 2022. "The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions" International Journal of Molecular Sciences 23, no. 1: 128. https://doi.org/10.3390/ijms23010128
APA StylePallotti, F., Bergamini, C., Lamperti, C., & Fato, R. (2022). The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. International Journal of Molecular Sciences, 23(1), 128. https://doi.org/10.3390/ijms23010128