
Resolvin D2 and Resolvin D1 Differentially Activate Protein Kinases to Counter-Regulate Histamine-Induced [Ca2+]i Increase and Mucin Secretion in Conjunctival Goblet Cells


Abstract
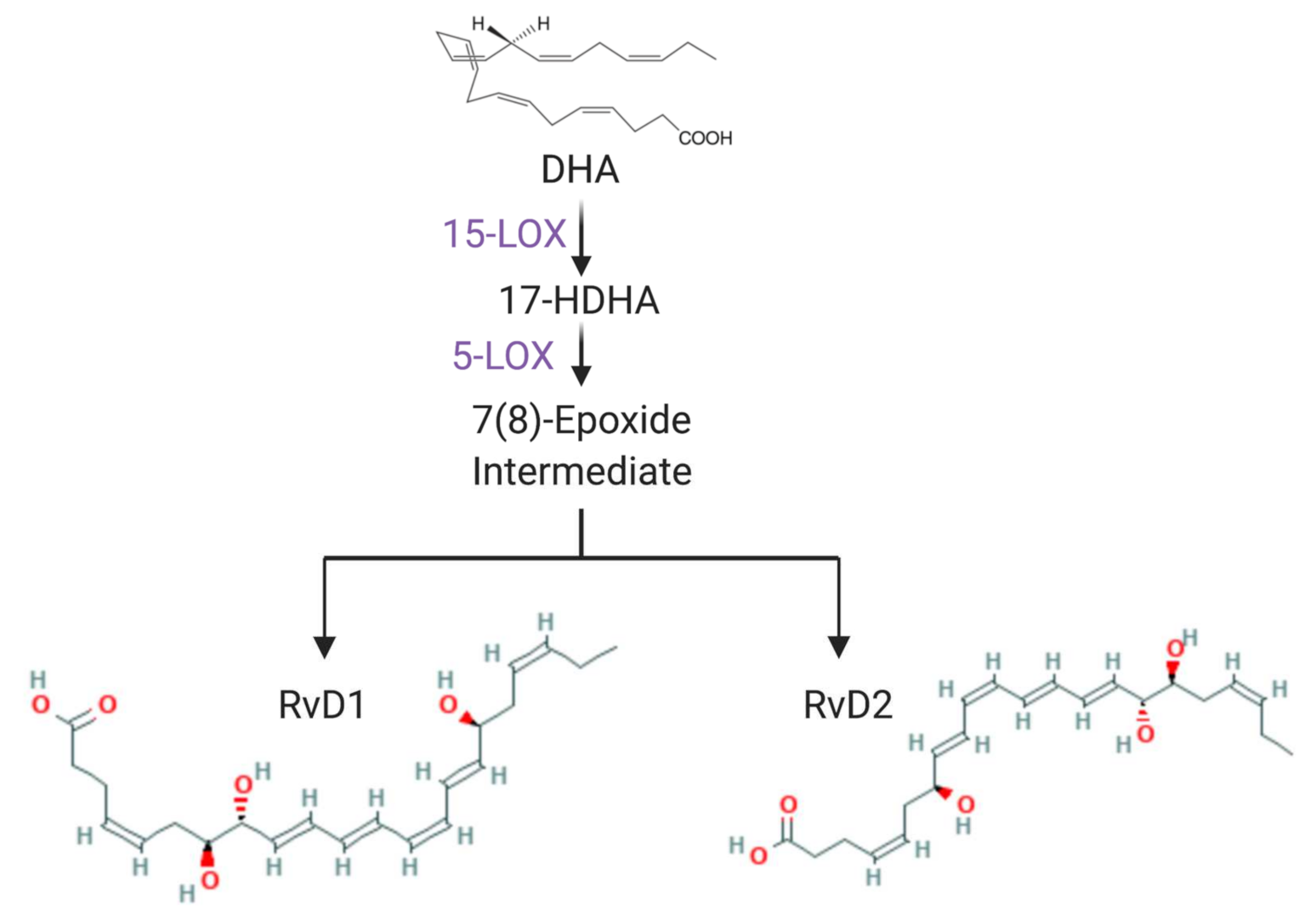
:1. Introduction
2. Results
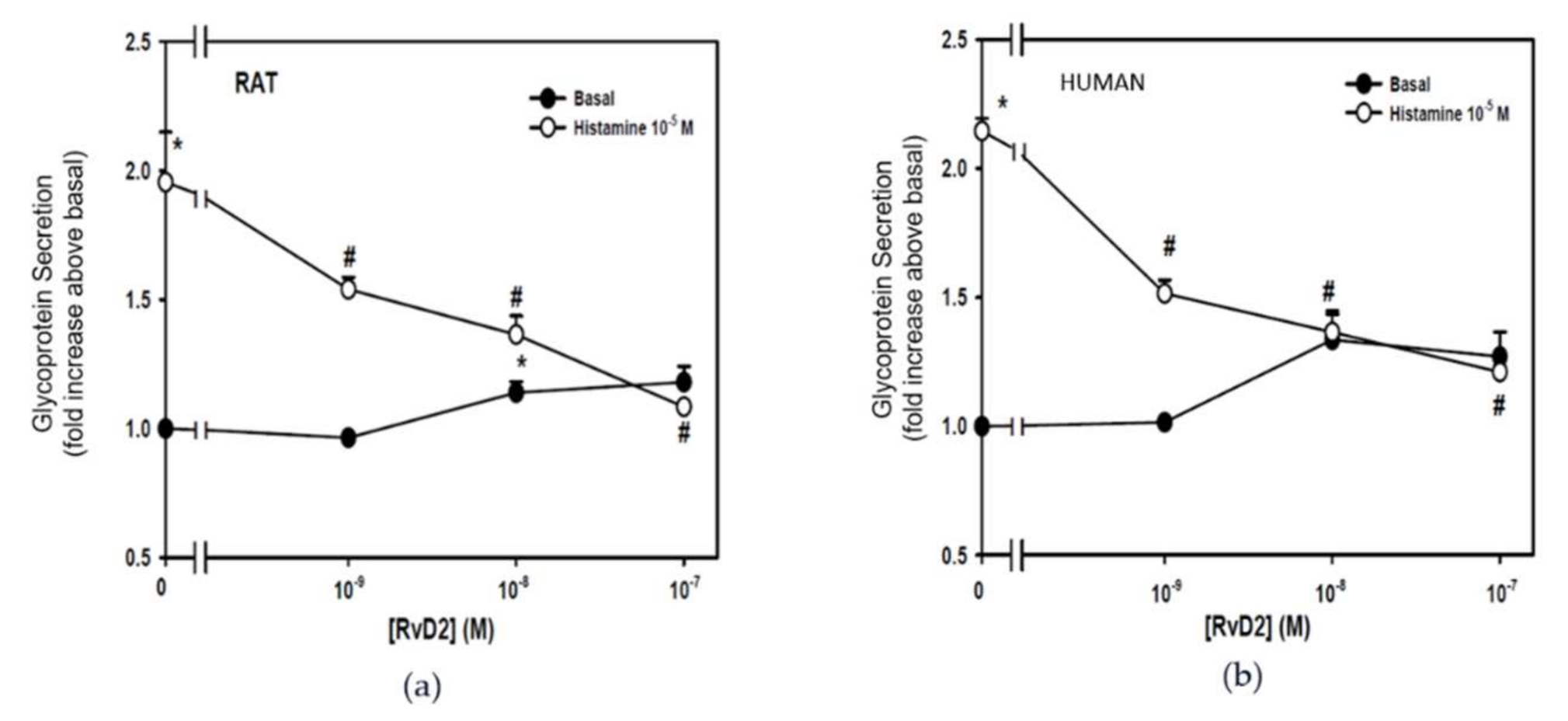
2.1. RvD2 Counter-Regulates Histamine-Stimulated Secretion from Rat- and Human-Derived Cultured Conjunctival Goblet Cells
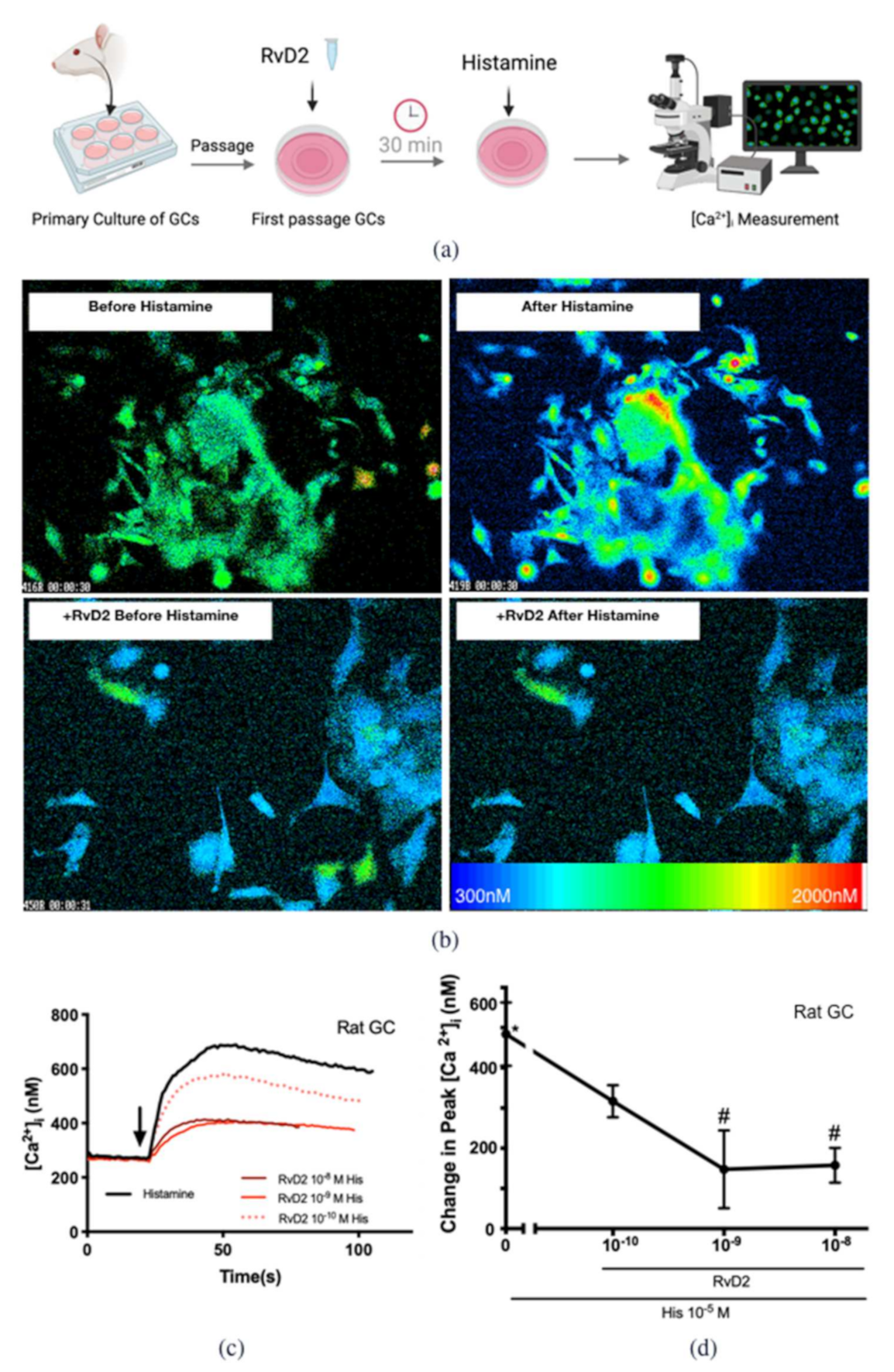
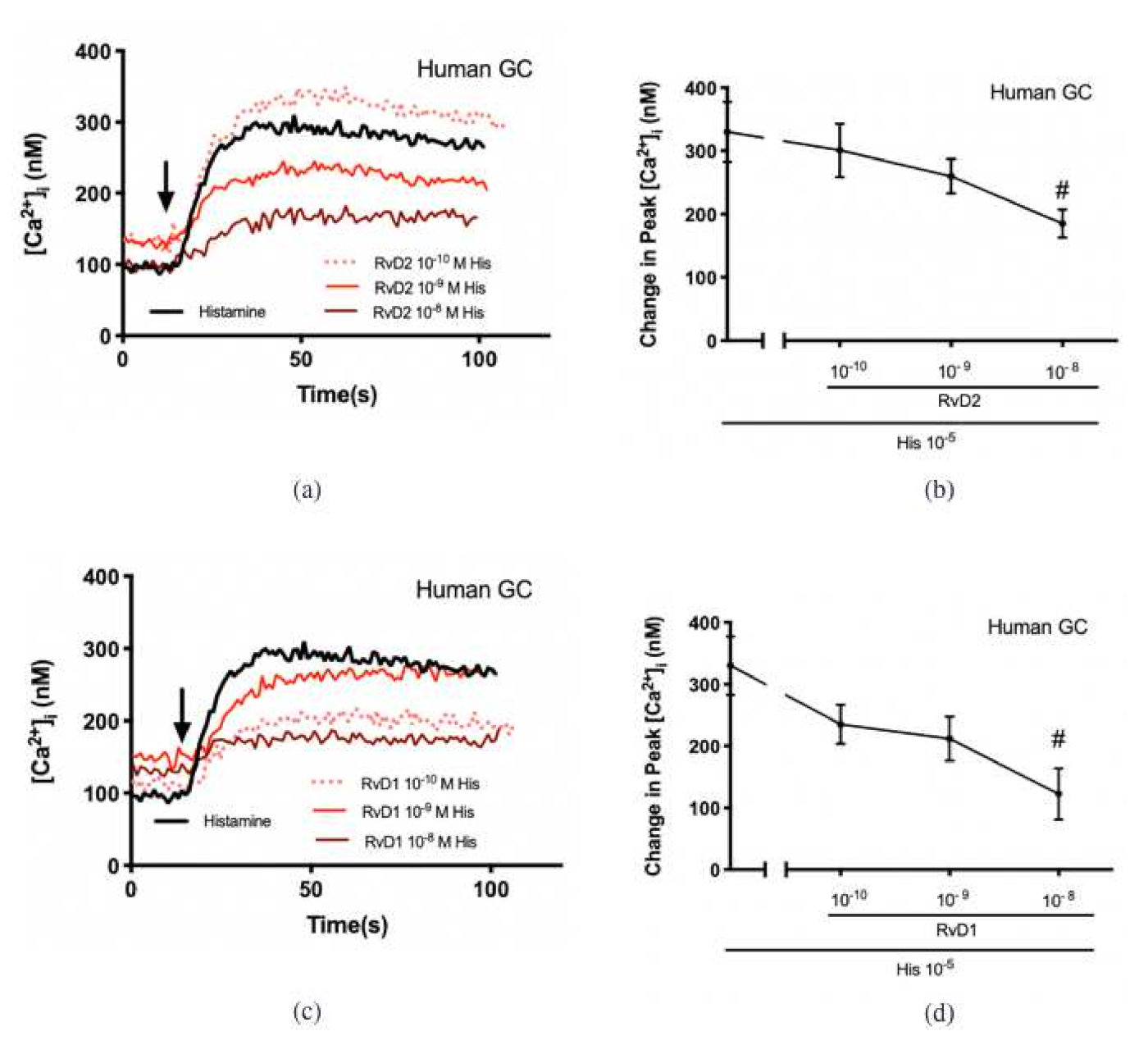
2.2. RvD2 Counter-Regulates Histamine-Stimulated Increase in [Ca2+]i in Cultured Conjunctival Goblet Cells Derived from Rats and Humans
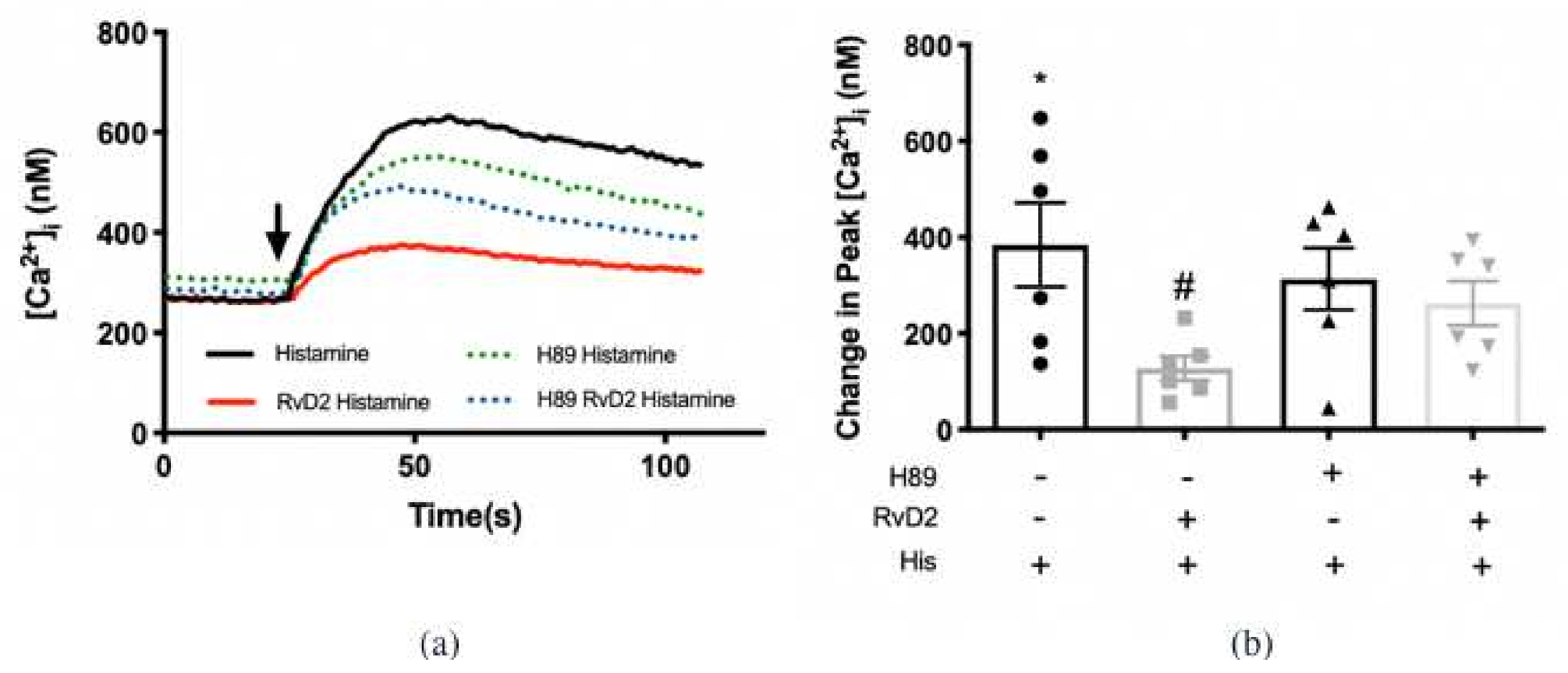
2.3. RvD2 Uses PKA to Counter-Regulate Histamine-Stimulated Increase in [Ca2+]i in Cultured Conjunctival Goblet Cells Derived from Rats
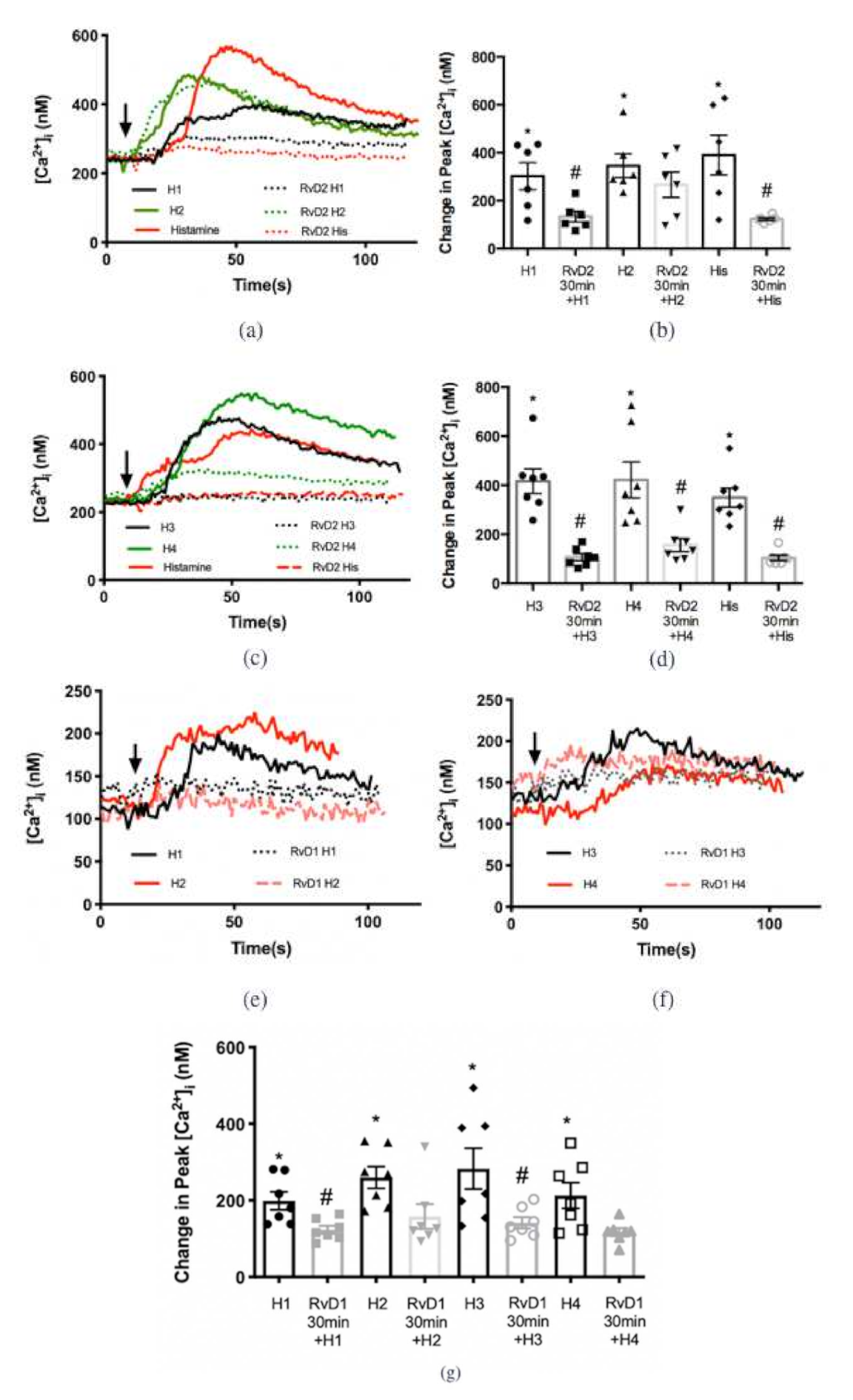
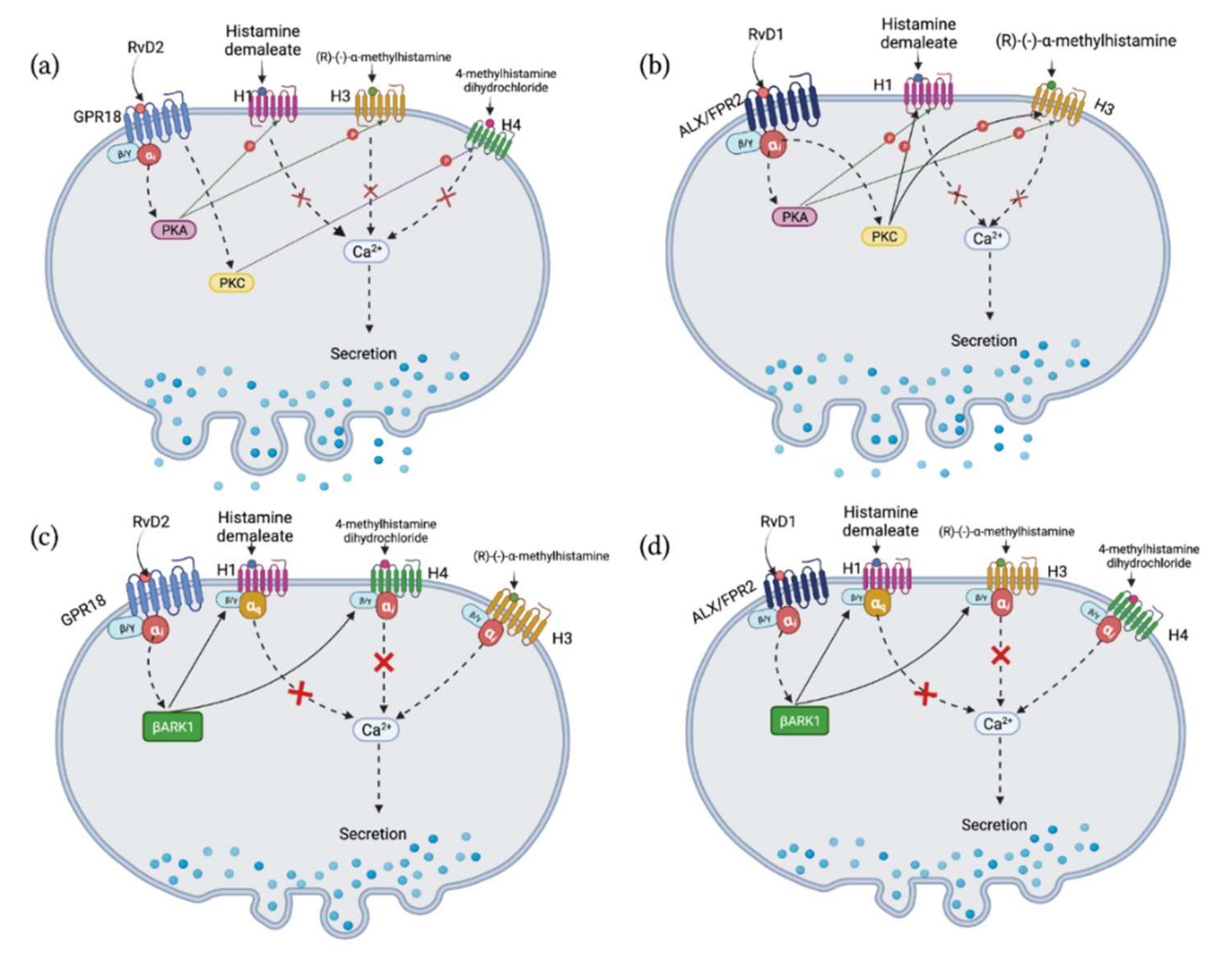
2.4. RvD2 Counter-Regulates Increase in [Ca2+]i Stimulated by Histamine Receptor 1, 3, and 4 Specific Agonists, while RvD1 Counter-Regulates Response to Histamine Receptor 1 and 3 Specific Agonists in Cultured Conjunctival Goblet Cells Derived from Rats
2.5. RvD2 Uses β-ARK1 to Counter-Regulate Increase in [Ca2+]i Induced by H1 and H4, but Not H3 Receptor Subtype Agonists in Cultured Conjunctival Goblet Cells Derived from Rats
2.6. RvD1 Uses β-ARK1 to Counter-Regulate the Increase in [Ca2+]i Induced by H3 Receptor Subtype in Cultured Conjunctival Goblet Cells Derived from Rats
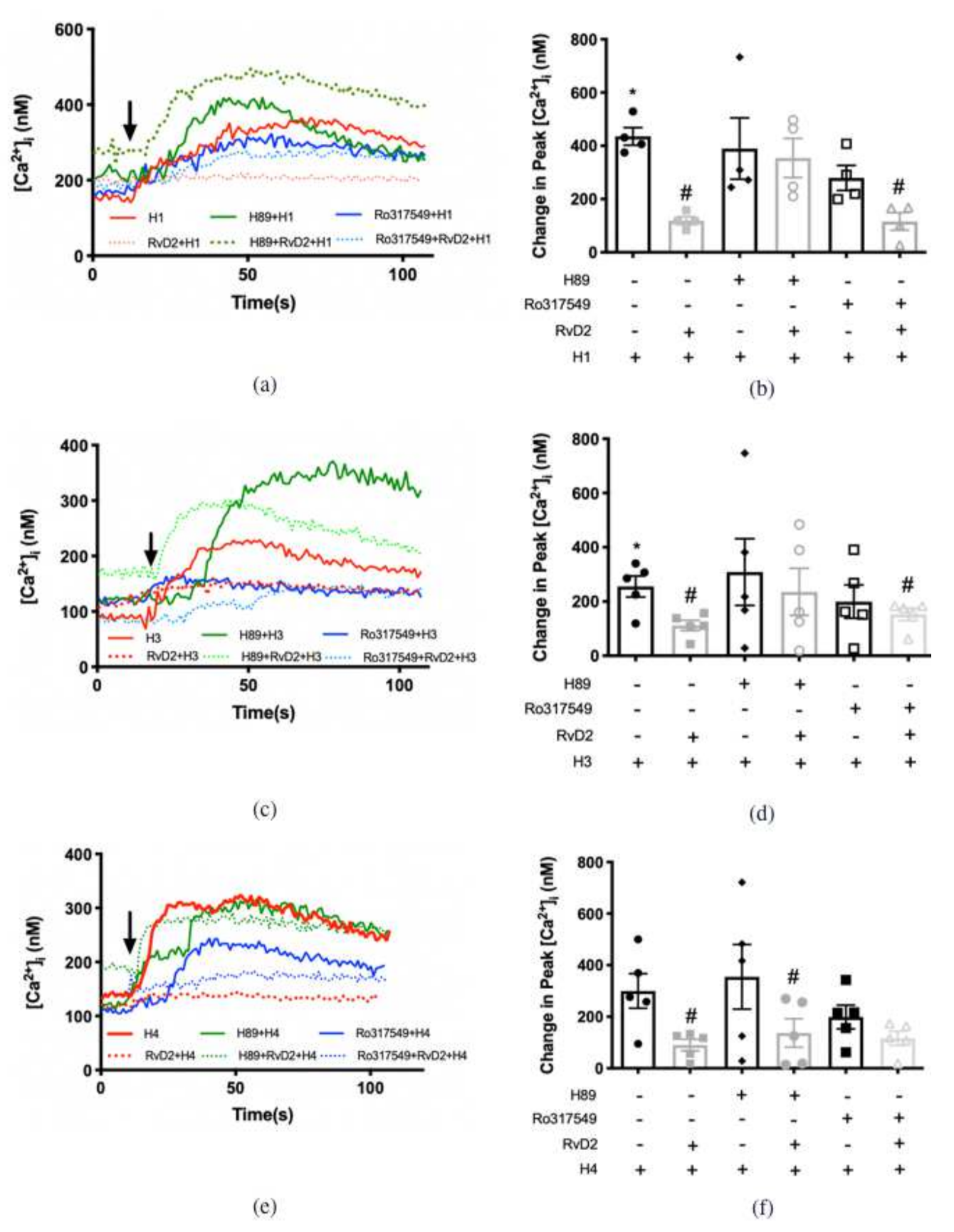
2.7. RvD2 Uses PKA to Counter-Regulate the Increase in [Ca2+]i Induced by H1 and H3 Receptor Subtypes Agonists, but Uses PKC for the Action of H4 Receptor Subtype Agonist in Cultured Conjunctival Goblet Cells Derived from Rats
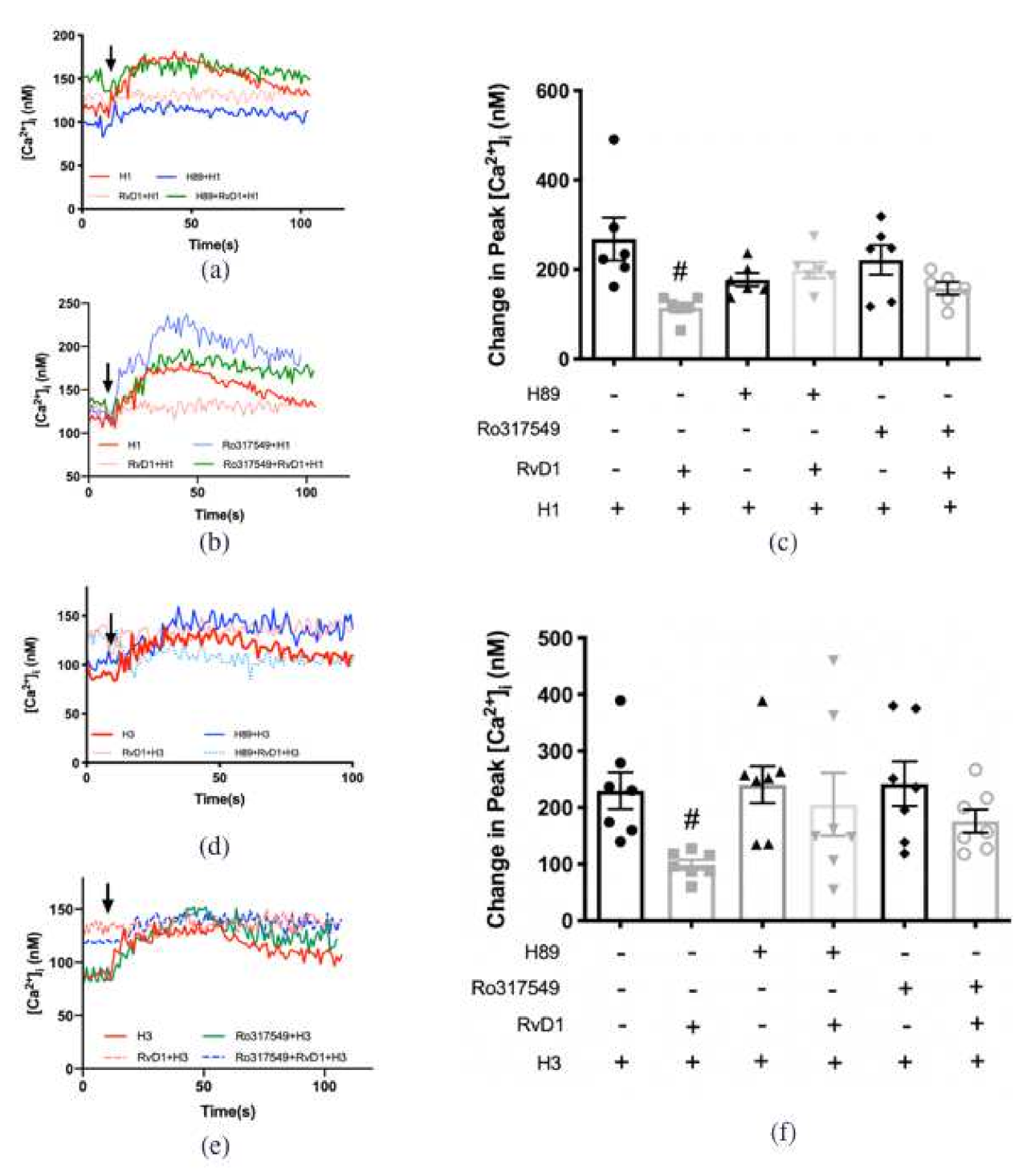
2.8. RvD1 Uses Both PKA and PKC to Counter-Regulate the Increase in [Ca2+]i Induced by H1 and H3 Agonists in Cultured Conjunctival Goblet Cells Derived from Rats
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Human Tissue
4.4. Cell Culture
4.5. High-Molecular Weight Glycoprotein Secretion Measurements
4.6. [Ca2+]i Measurements
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ono, S.J.; Abelson, M.B. Allergic conjunctivitis: Update on pathophysiology and prospects for future treatment. J. Allergy Clin. Immunol. 2005, 115, 118–122. [Google Scholar] [CrossRef]
- Bielory, L.; Delgado, L.; Katelaris, C.H.; Leonardi, A.; Rosario, N.; Vichyanoud, P. ICON: Diagnosis and management of allergic conjunctivitis. Ann. Allergy Asthma Immunol. 2020, 124, 118–134. [Google Scholar] [CrossRef] [Green Version]
- Brozek, G.; Lawson, J.; Szumilas, D.; Zejda, J. Increasing prevalence of asthma, respiratory symptoms, and allergic diseases: Four repeated surveys from 1993-2014. Respir. Med. 2015, 109, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Axelrod, S.; Bielory, L. The epidemiology of ocular and nasal allergy in the United States, 1988–1994. J. Allergy Clin. Immunol. 2010, 126, 778–783.e6. [Google Scholar] [CrossRef]
- Leonardi, A. Role of histamine in allergic conjunctivitis. Acta Ophthalmol. Scand. 2000, 78, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, A.A.; Smith, L.M.; Fregona, I.A.; Salmaso, M.; Secchi, A.G. Tear histamine and histaminase during the early (EPR) and late (LPR) phases of the allergic reaction and the effects of lodoxamide. Eur. J. Ophthalmol. 1996, 6, 106–112. [Google Scholar] [CrossRef]
- Leonardi, A.; Battista, M.C.; Gismondi, M.; Fregona, I.A.; Secchi, A.G. Antigen sensitivity evaluated by tear-specific and serum-specific IgE, skin tests, and conjunctival and nasal provocation tests in patients with ocular allergic disease. Eye 1993, 7 Pt 3, 461–464. [Google Scholar] [CrossRef]
- Azouz, N.P.; Fukuda, M.; Rothenberg, M.E.; Sagi-Eisenberg, R. Investigating mast cell secretory granules; from biosynthesis to exocytosis. J. Vis. Exp. 2015, 95, e52505. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, D.; Li, D.; Hayashi, C.; Shatos, M.; Hodges, R.R.; Dartt, D.A. Role of histamine and its receptor subtypes in stimulation of conjunctival goblet cell secretion. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2993–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inada, N.; Shoji, J.; Shiraki, Y.; Aso, H.; Yamagami, S. Histamine H1 and H4 receptor expression on the ocular surface of patients with chronic allergic conjunctival diseases. Allergol. Int. 2017, 66, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Shatos, M.A.; Ríos, J.D.; Horikawa, Y.; Hodges, R.R.; Chang, E.L.; Bernardino, C.R.; Rubin, P.A.D.; Dartt, D.A. Isolation and characterization of cultured human conjunctival goblet cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Carozza, R.B.; Shatos, M.A.; Hodges, R.R.; Dartt, D.A. Effect of histamine on Ca2+-dependent signaling pathways in rat conjunctival goblet cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6928–6938. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Ota, H.; Sugiyama, A.; Maruta, F.; Ikezawa, T.; Hotta, K.; Ishihara, K. Effects of a novel histamine H2-receptor antagonist, lafutidine, on the mucus barrier of human gastric mucosa. J. Gastroenterol. Hepatol. 2007, 22, 1800–1805. [Google Scholar] [CrossRef]
- Leurs, R.; Bakker, R.A.; Timmerman, H.; de Esch, I.J. The histamine H3 receptor: From gene cloning to H3 receptor drugs. Nat. Rev. Drug Discov. 2005, 4, 107–120. [Google Scholar] [CrossRef]
- Guyonnet Duperat, V.; Audie, J.P.; Debailleul, V.; Laine, A.; Buisine, M.P.; Galiegue-Zouitina, S.; Pigny, P.; Degand, P.; Aubert, J.P.; Porchet, N. Characterization of the human mucin gene MUC5AC: A consensus cysteine-rich domain for 11p15 mucin genes? Biochem. J. 1995, 305 Pt 1, 211–219. [Google Scholar] [CrossRef]
- Morse, K.L.; Behan, J.; Laz, T.M.; West, R.E., Jr.; Greenfeder, S.A.; Anthes, J.C.; Umland, S.; Wan, Y.; Hipkin, R.W.; Gonsiorek, W.; et al. Cloning and characterization of a novel human histamine receptor. J. Pharmacol. Exp. Ther. 2001, 296, 1058–1066. [Google Scholar]
- Ling, P.; Ngo, K.; Nguyen, S.; Thurmond, R.L.; Edwards, J.P.; Karlsson, L.; Fung-Leung, W.-P. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br. J. Pharmacol. 2004, 142, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, J.T.; Norris, P.C.; Hodges, R.R.; Dartt, D.A.; Serhan, C.N. Identification and Profiling of Specialized Pro-Resolving Mediators in Human Tears by Lipid Mediator Metabolomics. Prostaglandins Leukot. Essent. Fat. Acids 2017, 117, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Lippestad, M.; Hodges, R.R.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Resolvin D1 Increases Mucin Secretion in Cultured Rat Conjunctival Goblet Cells via Multiple Signaling Pathways. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4530–4544. [Google Scholar] [CrossRef] [PubMed]
- Botten, N.; Hodges, R.R.; Li, D.; Bair, J.A.; Shatos, M.A.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Resolvin D2 elevates cAMP to increase intracellular [Ca2+] and stimulate secretion from conjunctival goblet cells. FASEB J. 2019, 33, 8468–8478. [Google Scholar] [CrossRef]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Hodges, R.R.; Jiao, J.; Carozza, R.B.; Shatos, M.A.; Chiang, N.; Serhan, C.N.; Dartt, D.A. Resolvin D1 and aspirin-triggered resolvin D1 regulate histamine-stimulated conjunctival goblet cell secretion. Mucosal Immunol. 2013, 6, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Saban, D.R.; Hodges, R.R.; Mathew, R.; Reyes, N.J.; Yu, C.; Kaye, R.; Swift, W.; Botten, N.; Serhan, C.N.; Dartt, D.A. Resolvin D1 treatment on goblet cell mucin and immune responses in the chronic allergic eye disease (AED) model. Mucosal Immunol. 2019, 12, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, C.; Penela, P.; Murga, C.; Salcedo, A.; Garcia-Hoz, C.; Jurado-Pueyo, M.; Aymerich, I.; Mayor, F., Jr. The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta 2007, 1768, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Hodges, R.R.; Li, D.; Shatos, M.A.; Bair, J.A.; Lippestad, M.; Serhan, C.N.; Dartt, D.A. Lipoxin A4 activates ALX/FPR2 receptor to regulate conjunctival goblet cell secretion. Mucosal Immunol. 2017, 10, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Hofstra, C.L.; Desai, P.J.; Thurmond, R.L.; Fung-Leung, W.-P. Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J. Pharmacol. Exp. Ther. 2003, 305, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, A.; Leifer, C.A.; Mullen, G.E.D.; Kennedy, M.N.; Klinman, D.M.; Segal, D.M. Cutting edge: Histamine inhibits IFN-alpha release from plasmacytoid dendritic cells. J. Immunol. 2003, 170, 2269–2273. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, A.; Young, H.A.; Spitzer, J.H.; Visintin, A.; Segal, D.M. Histamine regulates cytokine production in maturing dendritic cells, resulting in altered T cell polarization. J. Clin. Investig. 2001, 108, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Mommert, S.; Gregor, K.; Rossbach, K.; Schaper, K.; Witte, T.; Gutzmer, R.; Werfel, T. Histamine H2 receptor stimulation upregulates TH2 chemokine CCL17 production in human M2a macrophages. J. Allergy Clin. Immunol. 2018, 141, 782–785.e5. [Google Scholar] [CrossRef] [Green Version]
- Bissonnette, E.Y. Histamine inhibits tumor necrosis factor alpha release by mast cells through H2 and H3 receptors. Am. J. Respir. Cell Mol. Biol. 1996, 14, 620–626. [Google Scholar] [CrossRef]
- Clark, R.A.; Sandler, J.A.; Gallin, J.I.; Kaplan, A.P. Histamine modulation of eosinophil migration. J. Immunol. 1977, 118, 137–145. [Google Scholar] [PubMed]
- Kim, C.M.; Dion, S.B.; Benovic, J.L. Mechanism of beta-adrenergic receptor kinase activation by G proteins. J. Biol. Chem. 1993, 268, 15412–15418. [Google Scholar] [CrossRef]
- Koch, W.J.; Inglese, J.; Stone, W.C.; Lefkowitz, R.J. The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J. Biol. Chem. 1993, 268, 8256–8260. [Google Scholar] [CrossRef]
- Iwata, K.; Luo, J.; Penn, R.B.; Benovic, J.L. Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J. Biol. Chem. 2005, 280, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, N.; Gottardo, F.L.; Alonso, M.N.; Monczor, F.; Shayo, C.; Davio, C. Roles of phosphorylation-dependent and -independent mechanisms in the regulation of histamine H2 receptor by G protein-coupled receptor kinase 2. J. Biol. Chem. 2011, 286, 28697–28706. [Google Scholar] [CrossRef] [Green Version]
- Verweij, E.W.E.; Al Araaj, B.; Prabhata, W.R.; Prihandoko, R.; Nijmeijer, S.; Tobin, A.B.; Leurs, R.; Vischer, H.F. Differential Role of Serines and Threonines in Intracellular Loop 3 and C-Terminal Tail of the Histamine H4 Receptor in beta-Arrestin and G Protein-Coupled Receptor Kinase Interaction, Internalization, and Signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Pope, N.H.; Salmon, M.; Davis, J.P.; Chatterjee, A.; Su, G.; Conte, M.S.; Ailawadi, G.; Upchurch, G.R., Jr. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. Faseb. J. 2016, 30, 4192–4201. [Google Scholar] [CrossRef] [Green Version]
- Jannaway, M.; Torrens, C.; Warner, J.A.; Sampson, A.P. Resolvin E1, resolvin D1 and resolvin D2 inhibit constriction of rat thoracic aorta and human pulmonary artery induced by the thromboxane mimetic U46619. Br. J. Pharmacol. 2018, 175, 1100–1108. [Google Scholar] [CrossRef]
- Park, C.-K.; Xu, Z.-Z.; Liu, T.; Lü, N.; Serhan, C.N.; Ji, R.-R. Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: Distinct roles of resolvin D1, D2, and E1. J. Neurosci. 2011, 31, 18433–18438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Puro, D.G. Role of ion channels in the functional response of conjunctival goblet cells to dry eye. Am. J. Physiol. Cell Physiol. 2018, 315, C236–C246. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RvD2 | RvD1 | ||||||
|---|---|---|---|---|---|---|---|
| H Receptors | Pathway | β-ARK1 | PKA | PKC | β-ARK1 | PKA | PKC |
| H1 | Gq-PKC-IP3 | + | + | – | + | + | + |
| H2 | Gαs—cAMP | Not inhibited by RvD2 | Not inhibited by RvD1 | ||||
| H3 | Gαi/o | – | + | – | + | + | + |
| H4 | Gαi/o | + | – | + | Not inhibited by RvD1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Botten, N.; Hodges, R.; Bair, J.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Resolvin D2 and Resolvin D1 Differentially Activate Protein Kinases to Counter-Regulate Histamine-Induced [Ca2+]i Increase and Mucin Secretion in Conjunctival Goblet Cells. Int. J. Mol. Sci. 2022, 23, 141. https://doi.org/10.3390/ijms23010141
Yang M, Botten N, Hodges R, Bair J, Utheim TP, Serhan CN, Dartt DA. Resolvin D2 and Resolvin D1 Differentially Activate Protein Kinases to Counter-Regulate Histamine-Induced [Ca2+]i Increase and Mucin Secretion in Conjunctival Goblet Cells. International Journal of Molecular Sciences. 2022; 23(1):141. https://doi.org/10.3390/ijms23010141
Chicago/Turabian StyleYang, Menglu, Nora Botten, Robin Hodges, Jeffrey Bair, Tor P. Utheim, Charles N. Serhan, and Darlene A. Dartt. 2022. "Resolvin D2 and Resolvin D1 Differentially Activate Protein Kinases to Counter-Regulate Histamine-Induced [Ca2+]i Increase and Mucin Secretion in Conjunctival Goblet Cells" International Journal of Molecular Sciences 23, no. 1: 141. https://doi.org/10.3390/ijms23010141
APA StyleYang, M., Botten, N., Hodges, R., Bair, J., Utheim, T. P., Serhan, C. N., & Dartt, D. A. (2022). Resolvin D2 and Resolvin D1 Differentially Activate Protein Kinases to Counter-Regulate Histamine-Induced [Ca2+]i Increase and Mucin Secretion in Conjunctival Goblet Cells. International Journal of Molecular Sciences, 23(1), 141. https://doi.org/10.3390/ijms23010141