
Anaphylaxis: Focus on Transcription Factor Activity



Abstract
1. Anaphylaxis, a General Overview: Definition, Effector Cells and Mechanisms
2. Proinflammatory Mediators Involved in Anaphylaxis
2.1. Cysteinyl Leukotrienes (CysLTs) and Prostaglandins
2.2. Cytokines and Chemokines
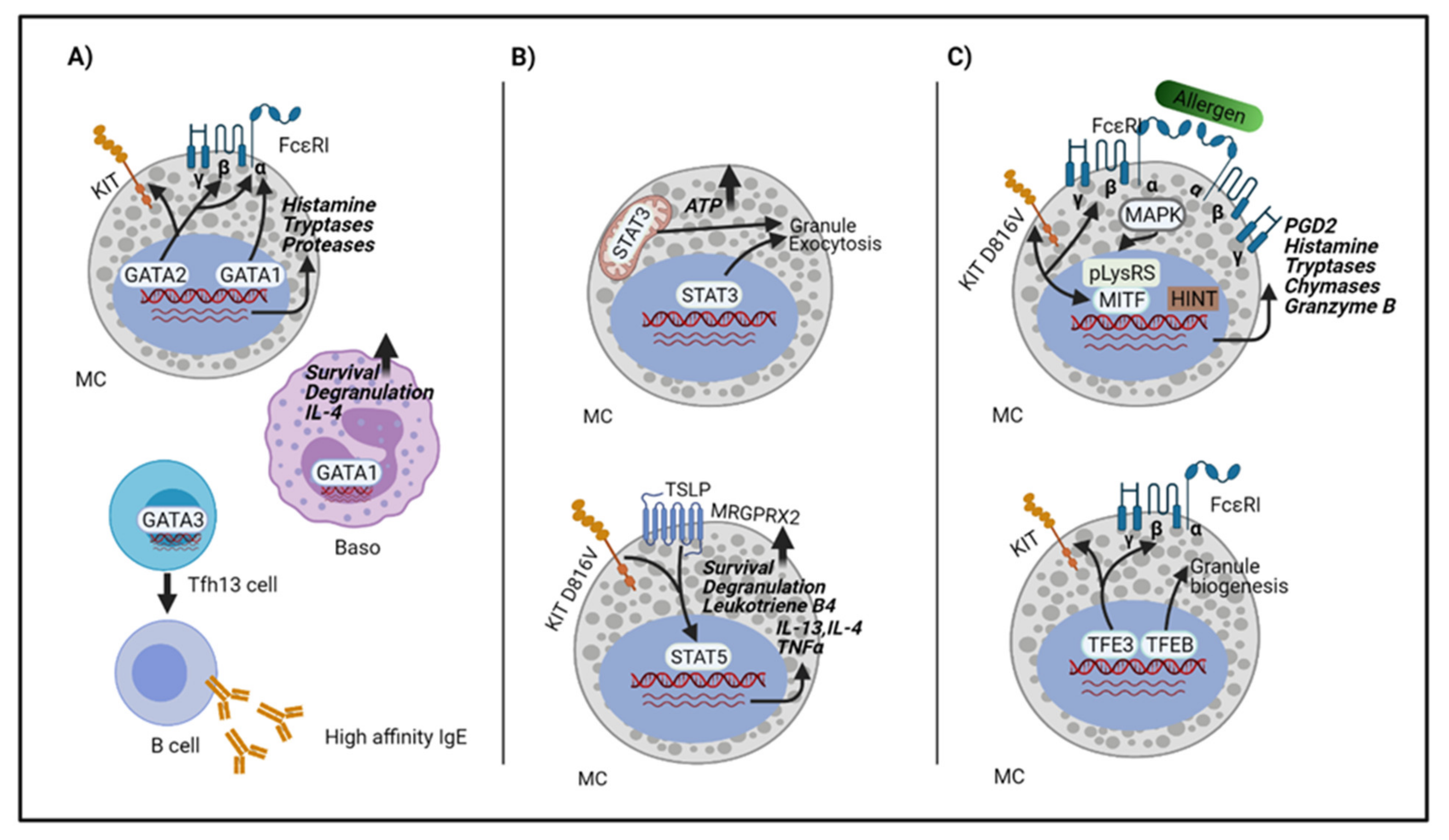
3. Transcriptions Factors Involved in Anaphylaxis
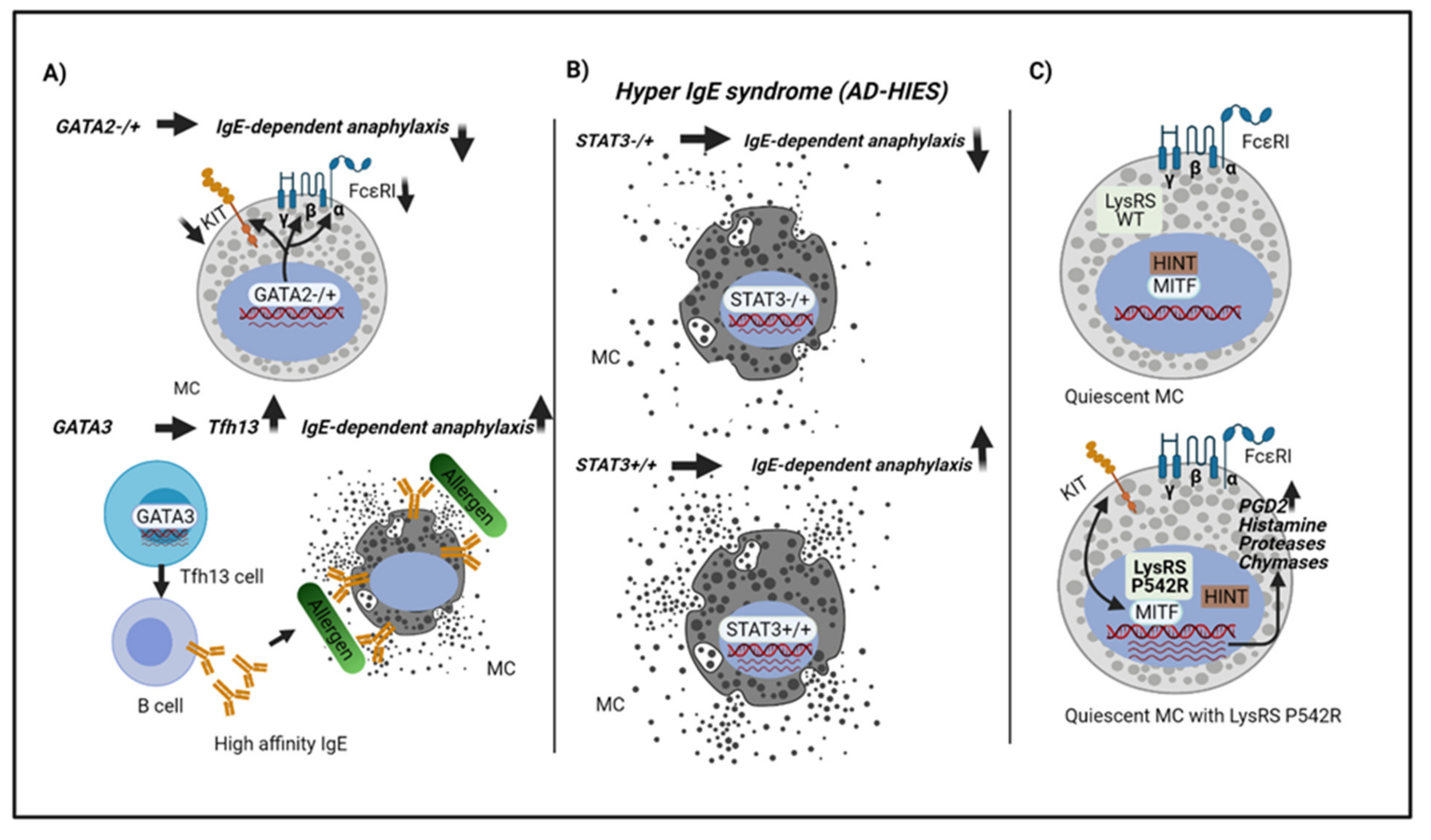
3.1. The GATA Family
3.2. The STAT Family
3.3. The MiTF/TFE Family
3.4. Other Transcription Factors
3.5. Transcription Factors in Mast Cell Activation
4. Host Genetic Factors or Mutations Related to Anaphylaxis
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castells, M. Diagnosis and management of anaphylaxis in precision medicine. J. Allergy Clin. Immunol. 2017, 140, 321–333. [Google Scholar] [CrossRef]
- Regateiro, F.S.; Marques, M.L.; Gomes, E.R. Drug-Induced Anaphylaxis: An Update on Epidemiology and Risk Factors. Int. Arch. Allergy Immunol. 2020, 181, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zink, A.; Schuster, B.; Winkler, J.; Eyerich, K.; Darsow, U.; Brockow, K.; Eberlein, B.; Biedermann, T. Allergy and sensitization to Hymenoptera venoms in unreferred adults with a high risk of sting exposure. World Allergy Organ. J. 2019, 12, 100039. [Google Scholar] [CrossRef]
- Muraro, A.; Mendoza Hernandez, D.A. Managing food allergy and anaphylaxis: A new model for an integrated approach. Allergol. Int. 2020, 69, 19–27. [Google Scholar] [CrossRef]
- Reber, L.L.; Hernandez, J.D.; Galli, S.J. The pathophysiology of anaphylaxis. J. Allergy Clin. Immunol. 2017, 140, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, F.D.; Khodoun, M.V.; Strait, R. Human IgE-independent systemic anaphylaxis. J. Allergy Clin. Immunol. 2016, 137, 1674–1680. [Google Scholar] [CrossRef]
- van der Linden, P.W.; Hack, C.E.; Poortman, J.; Vivié-Kipp, Y.C.; Struyvenberg, A.; van der Zwan, J.K. Insect-sting challenge in 138 patients: Relation between clinical severity of anaphylaxis and mast cell activation. J. Allergy Clin. Immunol. 1992, 90, 110–118. [Google Scholar] [CrossRef]
- Schuch, A.; Brockow, K. Mastocytosis and Anaphylaxis. Immunol. Allergy Clin. North. Am. 2017, 37, 153–164. [Google Scholar] [CrossRef]
- Beutier, H.; Gillis, C.M.; Iannascoli, B.; Godon, O.; England, P.; Sibilano, R.; Reber, L.L.; Galli, S.J.; Cragg, M.S.; Van Rooijen, N.; et al. IgG subclasses determine pathways of anaphylaxis in mice. J. Allergy Clin. Immunol. 2017, 139, 269–280.e7. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Ali, H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 2010, 128, 36. [Google Scholar] [CrossRef]
- Brown, S.G.A.; Stone, S.F.; Fatovich, D.M.; Burrows, S.A.; Holdgate, A.; Celenza, A.; Coulson, A.; Hartnett, L.; Nagree, Y.; Cotterell, C.; et al. Anaphylaxis: Clinical patterns, mediator release, and severity. J. Allergy Clin. Immunol. 2013, 132, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guhl, S.; Franke, K.; Artuc, M.; Zuberbier, T.; Babina, M. IL-33 and MRGPRX2-Triggered Activation of Human Skin Mast Cells—Elimination of Receptor Expression on Chronic Exposure, but Reinforced Degranulation on Acute Priming. Cells 2019, 8, 341. [Google Scholar] [CrossRef] [PubMed]
- Wedi, B.; Gehring, M.; Kapp, A. The pseudoallergen receptor MRGPRX2 on peripheral blood basophils and eosinophils: Expression and function. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- Porebski, G.; Kwiecien, K.; Pawica, M.; Kwitniewski, M. Mas-Related G Protein-Coupled Receptor-X2 (MRGPRX2) in Drug Hypersensitivity Reactions. Front. Immunol. 2018, 9, 3027. [Google Scholar] [CrossRef]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Investig. 2016, 126, 3981–3998. [Google Scholar] [CrossRef]
- An, J.; Lee, J.H.; Won, H.K.; Kang, Y.; Song, W.J.; Kwon, H.S.; Cho, Y.S.; Moon, H.B.; Kim, T.B. Clinical significance of serum MRGPRX2 as a new biomarker in allergic asthma. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 959–962. [Google Scholar] [CrossRef]
- Fujisawa, D.; Kashiwakura, J.; Kita, H.; Kikukawa, Y.; Fujitani, Y.; Sasaki-Sakamoto, T.; Kuroda, K.; Nunomura, S.; Hayama, K.; Terui, T.; et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J. Allergy Clin. Immunol. 2014, 134, 622–633.e9. [Google Scholar] [CrossRef]
- Quan, P.L.; Sabaté-Brescó, M.; Guo, Y.; Martín, M.; Gastaminza, G. The Multifaceted Mas-Related G Protein-Coupled Receptor Member X2 in Allergic Diseases and Beyond. Int. J. Mol. Sci. 2021, 22, 4421. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Tkaczyk, C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006, 6, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Kraft, S.; Kinet, J.-P.P. New developments in FcεRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.F.; Cotterell, C.; Isbister, G.K.; Holdgate, A.; Brown, S.G.A. Elevated serum cytokines during human anaphylaxis: Identification of potential mediators of acute allergic reactions. J. Allergy Clin. Immunol. 2009, 124, 786–792.e4. [Google Scholar] [CrossRef]
- Vadas, P.; Perelman, B.; Liss, G. Platelet-activating factor, histamine, and tryptase levels in human anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.H. Tryptase genetics and anaphylaxis. J. Allergy Clin. Immunol. 2006, 117, 1411–1414. [Google Scholar] [CrossRef]
- Ohneda, K.; Ohmori, S.; Yamamoto, M. Mouse tryptase gene expression is coordinately regulated by GATA1 and GATA2 in bone marrow-derived mast cells. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Lyons, J.J.; Chovanec, J.; O’Connell, M.P.; Liu, Y.; Šelb, J.; Zanotti, R.; Bai, Y.; Kim, J.; Le, Q.T.; DiMaggio, T.; et al. Heritable risk for severe anaphylaxis associated with increased α-tryptase–encoding germline copy number at TPSAB1. J. Allergy Clin. Immunol. 2021, 147, 622–632. [Google Scholar] [CrossRef]
- Vadas, P.; Gold, M.; Perelman, B.; Liss, G.M.; Lack, G.; Blyth, T.; Simons, F.E.R.; Simons, K.J.; Cass, D.; Yeung, J. Platelet-Activating Factor, PAF Acetylhydrolase, and Severe Anaphylaxis. N. Engl. J. Med. 2008, 358, 28–35. [Google Scholar] [CrossRef]
- Finkelman, F.D. Anaphylaxis: Lessons from mouse models. J. Allergy Clin. Immunol. 2007, 120, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Ono, E.; Taniguchi, M.; Mita, H.; Fukutomi, Y.; Higashi, N.; Miyazaki, E.; Kumamoto, T.; Akiyama, K. Increased production of cysteinyl leukotrienes and prostaglandin D2 during human anaphylaxis. Clin. Exp. Allergy 2009, 39, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Vantur, R.; Rihar, M.; Koren, A.; Rijavec, M.; Kopac, P.; Bidovec-Stojkovic, U.; Erzen, R.; Korosec, P. Chemokines during anaphylaxis: The importance of CCL2 and CCL2-dependent chemotactic activity for basophils. Clin. Transl. Allergy 2020, 10. [Google Scholar] [CrossRef]
- Kim, M.; Kwon, Y.; Jung, H.S.; Kim, Y.; Jeoung, D. FcεRI-HDAC3-MCP1 Signaling Axis Promotes Passive Anaphylaxis Mediated by Cellular Interactions. Int. J. Mol. Sci. 2019, 20, 4964. [Google Scholar] [CrossRef]
- Ma, P.; Mali, R.S.; Munugalavadla, V.; Krishnan, S.; Ramdas, B.; Sims, E.; Martin, H.; Ghosh, J.; Li, S.; Chan, R.J.; et al. The PI3K pathway drives the maturation of mast cells via microphthalmia transcription factor. Blood 2011, 118, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Tshori, S.; Nechushtan, H. Mast cell transcription factors—Regulators of cell fate and phenotype. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 42–48. [Google Scholar] [CrossRef]
- Sasaki, H.; Kurotaki, D.; Tamura, T. Regulation of basophil and mast cell development by transcription factors. Allergol. Int. 2016, 65, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef]
- Romagnani, S. Immunologic influences on allergy and the TH1/TH2 balance. J. Allergy Clin. Immunol. 2004, 113, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Phan, V.; Ito, T.; Inaba, M.; Azuma, Y.; Kibata, K.; Inagaki-Katashiba, N.; Tanaka, A.; Satake, A.; Nomura, S. Immunomodulatory drugs suppress Th1-inducing ability of dendritic cells but enhance Th2-mediated allergic responses. Blood Adv. 2020, 4, 3572–3585. [Google Scholar] [CrossRef]
- Ferreira, R.; Ohneda, K.; Yamamoto, M.; Philipsen, S. GATA1 Function, a Paradigm for Transcription Factors in Hematopoiesis. Mol. Cell. Biol. 2005, 25, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Orkin, S.H. GATA transcription factors: Key regulators of hematopoiesis. Exp. Hematol. 1995, 23, 99–107. [Google Scholar]
- Molkentin, J.D. The zinc finger-containing transcription factors GATA-4, -5, and -6: Ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000, 275, 38949–38952. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, S.; Takai, J.; Ishijima, Y.; Suzuki, M.; Moriguchi, T.; Philipsen, S.; Yamamoto, M.; Ohneda, K. Regulation of GATA Factor Expression Is Distinct between Erythroid and Mast Cell Lineages. Mol. Cell. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, L.; Tsukamoto, S.; Suzuki, M.; Yamamoto-Mukai, H.; Yamamoto, M.; Philipsen, S.; Ohneda, K. Ablation of Gatal in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood 2008, 111, 4375–4385. [Google Scholar] [CrossRef]
- Migliaccio, A.R.; Rana, R.A.; Sanchez, M.; Lorenzini, R.; Centurione, L.; Bianchi, L.; Vannucchi, A.M.; Migliaccio, G.; Orkin, S.H. GATA-1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA-1low mouse mutant. J. Exp. Med. 2003, 197, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Ghinassi, B.; Sanchez, M.; Martelli, F.; Amabile, G.; Vannucchi, A.M.; Migliaccio, G.; Orkin, S.H.; Migliaccio, A.R. The hypomorphic Gata1low mutation alters the proliferation/differentiation potential of the common megakaryocytic-erythroid progenitor. Blood 2007, 109, 1460–1471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nishiyama, C.; Ito, T.; Nishiyama, M.; Masaki, S.; Maeda, K.; Nakano, N.; Ng, W.; Fukuyama, K.; Yamamoto, M.; Okumura, K.; et al. GATA-1 is required for expression of FcεRI on mast cells: Analysis of mast cells derived from GATA-1 knockdown mouse bone marrow. Int. Immunol. 2005. [Google Scholar] [CrossRef]
- Masuda, A.; Hashimoto, K.; Yokoi, T.; Doi, T.; Kodama, T.; Kume, H.; Ohno, K.; Matsuguchi, T. Essential Role of GATA Transcriptional Factors in the Activation of Mast Cells. J. Immunol. 2007, 178, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Ohneda, K.; Moriguchi, T.; Ohmori, S.; Ishijima, Y.; Satoh, H.; Philipsen, S.; Yamamoto, M. Transcription Factor GATA1 Is Dispensable for Mast Cell Differentiation in Adult Mice. Mol. Cell. Biol. 2014, 34, 1812–1826. [Google Scholar] [CrossRef]
- Nei, Y.; Obata-Ninomiya, K.; Tsutsui, H.; Ishiwata, K.; Miyasaka, M.; Matsumoto, K.; Nakae, S.; Kanuka, H.; Inase, N.; Karasuyama, H. GATA-1 regulates the generation and function of basophils. Proc. Natl. Acad. Sci. USA 2013, 110, 18620–18625. [Google Scholar] [CrossRef]
- Ohmori, S.; Moriguchi, T.; Noguchi, Y.; Ikeda, M.; Kobayashi, K.; Tomaru, N.; Ishijima, Y.; Ohneda, O.; Yamamoto, M.; Ohneda, K. GATA2 is critical for the maintenance of cellular identity in differentiated mast cells derived from mouse bone marrow. Blood 2015, 125, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Inage, E.; Kasakura, K.; Yashiro, T.; Suzuki, R.; Baba, Y.; Nakano, N.; Hara, M.; Tanabe, A.; Oboki, K.; Matsumoto, K.; et al. Critical Roles for PU.1, GATA1, and GATA2 in the Expression of Human FcεRI on Mast Cells: PU.1 and GATA1 Transactivate FCER1A, and GATA2 Transactivates FCER1A and MS4A2. J. Immunol. 2014, 192, 3936–3946. [Google Scholar] [CrossRef]
- Desai, A.; Sowerwine, K.; Liu, Y.; Lawrence, M.G.; Chovanec, J.; Hsu, A.P.; O’Connell, M.P.; Kim, J.; Boris, L.; Jones, N.; et al. GATA-2–deficient mast cells limit IgE-mediated immediate hypersensitivity reactions in human subjects. J. Allergy Clin. Immunol. 2019, 144, 613–617.e14. [Google Scholar] [CrossRef]
- Kasakura, K.; Nagata, K.; Miura, R.; Iida, M.; Nakaya, H.; Okada, H.; Arai, T.; Arai, T.; Kawakami, Y.; Kawakami, T.; et al. Cooperative Regulation of the Mucosal Mast Cell–Specific Protease Genes Mcpt1 and Mcpt2 by GATA and Smad Transcription Factors. J. Immunol. 2020, 204, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.N.; Olson, M.C.; Barton, K.P.; Leiden, J.M. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 1996, 384, 474–475. [Google Scholar] [CrossRef]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; Van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Cote-Sierra, J.; Guo, L.; Paul, W.E. GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006, 16, 3–10. [Google Scholar] [CrossRef]
- Taghon, T.; Yui, M.A.; Rothenberg, E.V. Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat. Immunol. 2007, 8, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, U.; Chen, J.S.; Zhang, B.; Flynn, W.F.; Lu, Y.; Song, W.; Joseph, J.; Gertie, J.A.; Xu, L.; Collet, M.A.; et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.M.; Heller, N.M. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [PubMed]
- LaPorte, S.L.; Juo, Z.S.; Vaclavikova, J.; Colf, L.A.; Qi, X.; Heller, N.M.; Keegan, A.D.; Garcia, K.C. Molecular and Structural Basis of Cytokine Receptor Pleiotropy in the Interleukin-4/13 System. Cell 2008, 132, 259–272. [Google Scholar] [CrossRef]
- Ashley, S.E.; Tan, H.T.T.; Peters, R.; Allen, K.J.; Vuillermin, P.; Dharmage, S.C.; Tang, M.L.K.; Koplin, J.; Lowe, A.; Ponsonby, A.L.; et al. Genetic variation at the Th2 immune gene IL13 is associated with IgE-mediated paediatric food allergy. Clin. Exp. Allergy 2017, 47, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Pykäläinen, M.; Kinos, R.; Valkonen, S.; Rydman, P.; Kilpeläinen, M.; Laitinen, L.A.; Karjalainen, J.; Nieminen, M.; Hurme, M.; Kere, J.; et al. Association analysis of common variants of STAT6, GATA3, and STAT4 to asthma and high serum IgE phenotypes. J. Allergy Clin. Immunol. 2005, 115, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Huebner, M.; Kim, D.Y.; Ewart, S.; Karmaus, W.; Sadeghnejad, A.; Arshad, S.H. Patterns of GATA3 and IL13 gene polymorphisms associated with childhood rhinitis and atopy in a birth cohort. J. Allergy Clin. Immunol. 2008, 121, 408–414. [Google Scholar] [CrossRef][Green Version]
- McKenzie, G.J.; Fallon, P.G.; Emson, C.L.; Grencis, R.K.; McKenzie, A.N.J. Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J. Exp. Med. 1999, 189, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Grassmann, J.D.S.; Gowthaman, U.; Olyha, S.J.; Simoneau, T.; Berin, M.C.; Eisenbarth, S.C.; Williams, A. Flow cytometric identification of T 13 cells in mouse and human. J. Allergy Clin. Immunol. 2021, 147, 470–483. [Google Scholar] [CrossRef]
- Krug, N.; Hohlfeld, J.M.; Buhl, R.; Renz, J.; Garn, H.; Renz, H. Blood eosinophils predict therapeutic effects of a GATA3-specific DNAzyme in asthma patients. J. Allergy Clin. Immunol. 2017, 140, 625–628.e5. [Google Scholar] [CrossRef]
- Jang, H.Y.; Gu, S.; Lee, S.M.; Park, B.H. Overexpression of sirtuin 6 suppresses allergic airway inflammation through deacetylation of GATA3. J. Allergy Clin. Immunol. 2016, 138, 1452–1455.e13. [Google Scholar] [CrossRef]
- Malaviya, R.; Zhu, D.M.; Dibirdik, I.; Uckun, F.M. Targeting Janus kinase 3 in mast cells prevents immediate hypersensitivity reactions and anaphylaxis. J. Biol. Chem. 1999, 274, 27028–27033. [Google Scholar] [CrossRef]
- Yamaki, K.; Yoshino, S. Remission of food allergy by the Janus kinase inhibitor ruxolitinib in mice. Int. Immunopharmacol. 2014, 18, 217–224. [Google Scholar] [CrossRef]
- Hermans, M.A.W.; Schrijver, B.; van Holten-Neelen, C.C.P.A.; Gerth van Wijk, R.; van Hagen, P.M.; van Daele, P.L.A.; Dik, W.A. The JAK1/JAK2- inhibitor ruxolitinib inhibits mast cell degranulation and cytokine release. Clin. Exp. Allergy 2018, 48, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; Prochaska, L. Ruxolitinib improves symptoms and quality of life in a patient with systemic mastocytosis. Biomark. Res. 2016, 4, 2. [Google Scholar] [CrossRef][Green Version]
- Akbar, M.; Garcia-Melchor, E.; Chilaka, S.; Little, K.J.; Sood, S.; Reilly, J.H.; Liew, F.Y.; McInnes, I.B.; Millar, N.L. Attenuation of Dupuytren s fibrosis via targeting of the STAT1 modulated IL-13R-1 response. Sci. Adv. 2020, 6, eaaz8272. [Google Scholar] [CrossRef]
- Erlich, T.H.; Yagil, Z.; Kay, G.; Peretz, A.; Migalovich-Sheikhet, H.; Tshori, S.; Nechushtan, H.; Levi-Schaffer, F.; Saada, A.; Razin, E. Mitochondrial STAT3 plays a major role in IgE-antigen-mediated mast cell exocytosis. J. Allergy Clin. Immunol. 2014, 134, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Erlich, T.H.; Sharkia, I.; Landolina, N.; Assayag, M.; Goldberger, O.; Berkman, N.; Levi-Schaffer, F.; Razin, E. Modulation of allergic responses by mitochondrial STAT3 inhibitors. Allergy 2018, 73, 2160–2171. [Google Scholar] [CrossRef]
- Hox, V.; O’Connell, M.P.; Lyons, J.J.; Sackstein, P.; Dimaggio, T.; Jones, N.; Nelson, C.; Boehm, M.; Holland, S.M.; Freeman, A.F.; et al. Diminution of signal transducer and activator of transcription 3 signaling inhibits vascular permeability and anaphylaxis. J. Allergy Clin. Immunol. 2016, 138, 187–199. [Google Scholar] [CrossRef]
- Siegel, A.M.; Stone, K.D.; Cruse, G.; Lawrence, M.G.; Olivera, A.; Jung, M.; Barber, J.S.; Freeman, A.F.; Holland, S.M.; O’Brien, M.; et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J. Allergy Clin. Immunol. 2013, 132, 1388–1396.e3. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Casassus, P.; Caillat-Vigneron, N.; Martin, A.; Simon, J.; Gallais, V.; Beaudry, P.; Eclache, V.; Laroche, L.; Lortholary, P.; Raphaël, M.; et al. Treatment of adult systemic mastocytosis with interferon-α: Results of a multicentre phase II trial on 20 patients. Br. J. Haematol. 2002, 119, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Pardanani, A.; Butterfield, J.H.; Li, C.-Y.; Tefferi, A. Cytoreductive therapy in 108 adults with systemic mastocytosis: Outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am. J. Hematol. 2009, 84, 790–794. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shimabukuro-Demoto, S.; Tsutsui, H.; Toyama-Sorimachi, N. Type i interferon limits mast cell-mediated anaphylaxis by controlling secretory granule homeostasis. PLoS Biol. 2019, 17, e3000530. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tsutsui, H.; Shimabukuro-Demoto, S.; Yoshida-Sugitani, R.; Karyu, H.; Furuyama-Tanaka, K.; Ohshima, D.; Kato, N.; Okamura, T.; Toyama-Sorimachi, N. Lysosome biogenesis regulated by the amino-acid transporter SLC15A4 is critical for functional integrity of mast cells. Int. Immunol. 2017, 29, 551–566. [Google Scholar] [CrossRef]
- Li, L.; Sun, B.; Gao, Y.; Niu, H.; Yuan, H.; Lou, H. STAT3 contributes to lysosomal-mediated cell death in a novel derivative of riccardin D-treated breast cancer cells in association with TFEB. Biochem. Pharmacol. 2018, 150, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.R.; Komazawa, N.; Morii, E.; Oboki, K.; Nakano, T. Involvement of connective tissue-type mast cells in Th1 immune responses via Stat4 expression. Blood 2005, 105, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.R.; Nishizawa, Y. Stat4 suppresses the proliferation of connective tissue-type mast cells. Lab. Investig. 2008, 88, 856–864. [Google Scholar] [CrossRef]
- Li, Y.; Qi, X.; Liu, B.; Huang, H. The STAT5–GATA2 Pathway Is Critical in Basophil and Mast Cell Differentiation and Maintenance. J. Immunol. 2015, 194, 4328–4338. [Google Scholar] [CrossRef]
- Shelburne, C.P.; McCoy, M.E.; Piekorz, R.; Sexl, V.; Roh, K.H.; Jacobs-Helber, S.M.; Gillespie, S.R.; Bailey, D.P.; Mirmonsef, P.; Mann, M.N.; et al. Stat5 expression is critical for mast cell development and survival. Blood 2003, 102, 1290–1297. [Google Scholar] [CrossRef]
- Witte, O.N. Steel locus defines new multipotent growth factor. Cell 1990, 63, 5–6. [Google Scholar] [CrossRef]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.; Connell, M.P.O.; Komarow, H.D.; Carter, M.C.; Smrz, D. Oncogenic D816V-KIT signaling in mast cells causes persistent IL-6 production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef]
- Ando, T.; Xiao, W.; Gao, P.; Namiranian, S.; Matsumoto, K.; Tomimori, Y.; Hong, H.; Yamashita, H.; Kimura, M.; Kashiwakura, J.; et al. Critical Role for Mast Cell Stat5 Activity in Skin Inflammation. Cell Rep. 2014, 6, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Barnstein, B.O.; Li, G.; Wang, Z.; Kennedy, S.; Chalfant, C.; Nakajima, H.; Bunting, K.D.; Ryan, J.J. Stat5 Expression Is Required for IgE-Mediated Mast Cell Function. J. Immunol. 2006, 177, 3421–3426. [Google Scholar] [CrossRef]
- Pullen, N.A.; Barnstein, B.O.; Falanga, Y.T.; Wang, Z.; Suzuki, R.; Tamang, T.D.L.; Khurana, M.C.; Harry, E.A.; Draber, P.; Bunting, K.D.; et al. Novel mechanism for FcεRI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J. Biol. Chem. 2012, 287, 2045–2054. [Google Scholar] [CrossRef]
- Babina, M.; Wang, Z.; Franke, K.; Zuberbier, T. Thymic Stromal Lymphopoietin Promotes MRGPRX2-Triggered Degranulation of Skin Mast Cells in a STAT5-Dependent Manner with Further Support from JNK. Cells 2021, 10, 102. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Malaviya, R.; Uckun, F.M. Role of STAT6 in IgE Receptor/FcεRI-Mediated Late Phase Allergic Responses of Mast Cells. J. Immunol. 2002, 168, 421–426. [Google Scholar] [CrossRef]
- Tamura, K.; Suzuki, M.; Arakawa, H.; Tokuyama, K.; Morikawa, A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int. Arch. Allergy Immunol. 2003, 131, 33–38. [Google Scholar] [CrossRef]
- Hussein, Y.M.; Alzahrani, S.S.; Alharthi, A.A.; Alhazmi, A.S.; Ghonaim, M.M.; Alghamdy, A.A.N.; El Askary, A. Gene polymorphism of interleukin-4, interleukin-4 receptor and STAT6 in children with atopic dermatitis in Taif, Saudi Arabia. Immunol. Investig. 2016, 45, 223–234. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed]
- Rehli, M.; Lichanska, A.; Cassady, A.I.; Ostrowski, M.C.; Hume, D.A. TFEC is a macrophage-restricted member of the microphthalmia-TFE subfamily of basic helix-loop-helix leucine zipper transcription factors. J. Immunol. 1999, 162, 1559–1565. [Google Scholar] [PubMed]
- Beckmann, H.; Su, L.K.; Kadesch, T. TFE3: A helix-loop-helix protein that activates transcription through the immunoglobulin enhancer μE3 motif. Genes Dev. 1990, 4, 167–179. [Google Scholar] [CrossRef]
- Steingrimsson, E.; Tessarollo, L.; Pathak, B.; Hou, L.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc. Natl. Acad. Sci. USA 2002, 99, 4477–4482. [Google Scholar] [CrossRef]
- Oppezzo, A.; Rosselli, F. The underestimated role of the microphthalmia-associated transcription factor (MiTF) in normal and pathological haematopoiesis. Cell Biosci. 2021, 11, 18. [Google Scholar] [CrossRef]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.; Shao, H.; Liu, J.; Jin, M.; Chen, J.; Huang, Y. MiRNA-340 inhibits osteoclast differentiation via repression of MITF. Biosci. Rep. 2017, 37, 1–8. [Google Scholar] [CrossRef]
- Wen, B.; Li, S.; Li, H.; Chen, Y.; Ma, X.; Wang, J.; Lu, F.; Qu, J.; Hou, L. Microphthalmia-associated transcription factor regulates the visual cycle genes Rlbp1 and Rdh5 in the retinal pigment epithelium. Sci. Rep. 2016, 6, 21208. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Morii, E.; Oboki, K.; Ishihara, K.; Jippo, T.; Hirano, T.; Kitamura, Y. Roles of MITF for development of mast cells in mice: Effects on both precursors and tissue environments. Blood 2004, 104, 1656–1661. [Google Scholar] [CrossRef]
- Qi, X.; Hong, J.; Chaves, L.; Zhuang, Y.; Chen, Y.; Wang, D.; Chabon, J.; Graham, B.; Ohmori, K.; Li, Y.; et al. Antagonistic Regulation by the Transcription Factors C/EBPα and MITF Specifies Basophil and Mast Cell Fates. Immunity 2013, 39, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Shahlaee, A.H.; Brandal, S.; Lee, Y.-N.; Jie, C.; Takemoto, C.M. Distinct and Shared Transcriptomes Are Regulated by Microphthalmia-Associated Transcription Factor Isoforms in Mast Cells. J. Immunol. 2007, 178, 378–388. [Google Scholar] [CrossRef]
- Lee, S.-H.H.; Lee, J.-H.H.; Lee, Y.-M.M.; Kim, D.-K.K. Involvement of MITF-A, an alternative isoform of mi transcription factor, on the expression of tryptase gene in human mast cells. Exp. Mol. Med. 2010, 42, 366. [Google Scholar] [CrossRef]
- Moore, K.J. Insight into the microphthalmia gene. Trends Genet. 1995, 11, 442–448. [Google Scholar] [CrossRef]
- Lee, Y.-N.; Noel, P.; Shahlaee, A.; Carter, M.; Kapur, R.; Wayne, A.; Metcalfe, D.D.; Takemoto, C. Kit Signaling Regulates Mitf Expression in Mastocytosis. Blood 2006, 108, 3601. [Google Scholar] [CrossRef]
- Lee, Y.-N.; Brandal, S.; Noel, P.; Wentzel, E.; Mendell, J.T.T.; McDevitt, M.A.; Kapur, R.; Carter, M.; Metcalfe, D.D.D.; Takemoto, C.M.M. KIT signaling regulates MITF expression through miRNAs in normal and malignant mast cell proliferation. Blood 2011, 117, 3629–3640. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.; Harmacek, L.; Long, Z.; Liang, J.; Lukin, K.; Leach, S.M.; O’Connor, B.; Gerber, A.N.; Hagman, J.; et al. The transcription factors GATA2 and microphthalmia-associated transcription factor regulate Hdc gene expression in mast cells and are required for IgE/mast cell–mediated anaphylaxis. J. Allergy Clin. Immunol. 2018, 142, 1173–1184. [Google Scholar] [CrossRef]
- Morii, E.; Oboki, K. MITF Is Necessary for Generation of Prostaglandin D 2 in Mouse Mast Cells. J. Biol. Chem. 2004, 279, 48923–48929. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-N.N.; Nechushtan, H.; Figov, N.; Razin, E. The Function of Lysyl-tRNA Synthetase and Ap4A as Signaling Regulators of MITF Activity in FcϵRI-Activated Mast Cells. Immunity 2004, 20, 145–151. [Google Scholar] [CrossRef]
- Yu, J.; Liu, Z.; Liang, Y.; Luo, F.; Zhang, J.; Tian, C.; Motzik, A.; Zheng, M.; Kang, J.; Zhong, G.; et al. Second messenger Ap4A polymerizes target protein HINT1 to transduce signals in FcεRI-activated mast cells. Nat. Commun. 2019, 10, 4664. [Google Scholar] [CrossRef] [PubMed]
- Yagil, Z.; Hadad Erlich, T.; Ofir-Birin, Y.; Tshori, S.; Kay, G.; Yekhtin, Z.; Fisher, D.E.; Cheng, C.; Wong, W.S.F.; Hartmann, K.; et al. Transcription factor E3, a major regulator of mast cell-mediated allergic response. J. Allergy Clin. Immunol. 2012, 129, 1357–1366.e5. [Google Scholar] [CrossRef] [PubMed]
- Kasakura, K.; Takahashi, K.; Itoh, T.; Hosono, A.; Nunomura, S.; Ra, C.; Momose, Y.; Itoh, K.; Nishiyama, C.; Kaminogawa, S. C/EBPα controls mast cell function. FEBS Lett. 2014, 588, 4645–4653. [Google Scholar] [CrossRef]
- Oda, Y.; Kasakura, K.; Fujigaki, I.; Kageyama, A.; Okumura, K.; Ogawa, H.; Yashiro, T.; Nishiyama, C. The effect of PU.1 knockdown on gene expression and function of mast cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Baba, Y.; Maeda, K.; Yashiro, T.; Inage, E.; Niyonsaba, F.; Hara, M.; Suzuki, R.; Ohtsuka, Y.; Shimizu, T.; Ogawa, H.; et al. Involvement of PU.1 in mast cell/ basophil-specific function of the human IL1RL1/ST2 promoter. Allergol. Int. 2012, 61, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Jayapal, M.; Tay, H.K.; Reghunathan, R.; Zhi, L.; Chow, K.K.; Rauff, M.; Melendez, A.J. Genome-wide gene expression profiling of human mast cells stimulated by IgE or FcεRI-aggregation reveals a complex network of genes involved in inflammatory responses. BMC Genom. 2006, 7, 210. [Google Scholar] [CrossRef]
- Lorentz, A.; Klopp, I.; Gebhardt, T.; Manns, M.P.; Bischoff, S.C. Role of activator protein 1, nuclear factor-κB, and nuclear factor of activated T cells in IgE receptor-mediated cytokine expression in mature human mast cells. J. Allergy Clin. Immunol. 2003, 111, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Klein-Hessling, S.; Palmetshofer, A.; Serfling, E.; Tertilt, C.; Bopp, T.; Heib, V.; Becker, M.; Taube, C.; Schild, H.; et al. Specific and Redundant Roles for NFAT Transcription Factors in the Expression of Mast Cell-Derived Cytokines. J. Immunol. 2006, 177, 6667–6674. [Google Scholar] [CrossRef]
- Lee, Y.-N.; Tuckerman, J.; Nechushtan, H.; Schutz, G.; Razin, E.; Angel, P. c-Fos as a Regulator of Degranulation and Cytokine Production in FcεRI-Activated Mast Cells. J. Immunol. 2004, 173, 2571–2577. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.L.; Walker, L.L. Dependence of mast cell IgE-mediated cytokine production on nuclear factor-κB activity. J. Allergy Clin. Immunol. 2000, 105, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Power, M.R.; Lin, T.-J.J. De novo synthesis of early growth response factor-1 is required for the full responsiveness of mast cells to produce TNF and IL-13 by IgE and antigen stimulation. Blood 2006, 107, 2814–2820. [Google Scholar] [CrossRef]
- Li, B.; Berman, J.; Tang, J.T.; Lin, T.J. The early growth response factor-1 is involved in stem cell factor (SCF)-induced interleukin 13 production by mast cells, but is dispensable for SCF-dependent mast cell growth. J. Biol. Chem. 2007, 282, 22573–22581. [Google Scholar] [CrossRef]
- Kalesnikoff, J.; Rios, E.J.; Chen, C.-C.; Nakae, S.; Zabel, B.A.; Butcher, E.C.; Tsai, M.; Tam, S.-Y.; Galli, S.J.; Herzenberg, L.A. RabGEF1 Regulates Stem Cell Factorc-Kit-Mediated Signaling Events and Biological Responses in Mast Cells. In Proceedings of the National Academy of Sciences, Stanford, CA, USA, 26 December 2005; Volume 103. [Google Scholar]
- Iwaki, S.; Tkaczyk, C.; Satterthwaite, A.B.; Halcomb, K.; Beaven, M.A.; Metcalfe, D.D.; Gilfillan, A.M. Btk plays a crucial role in the amplification of FcεRI-mediated mast cell activation by Kit. J. Biol. Chem. 2005, 280, 40261–40270. [Google Scholar] [CrossRef]
- Yannay-Cohen, N.; Carmi-Levy, I.; Kay, G.; Yang, C.M.; Han, J.M.; Kemeny, D.M.; Kim, S.; Nechushtan, H.; Razin, E. LysRS Serves as a Key Signaling Molecule in the Immune Response by Regulating Gene Expression. Mol. Cell 2009, 34, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Birin, Y.; Fang, P.; Bennett, S.P.; Zhang, H.-M.; Wang, J.; Rachmin, I.; Shapiro, R.; Song, J.; Dagan, A.; Pozo, J.; et al. Structural Switch of Lysyl-tRNA Synthetase between Translation and Transcription. Mol. Cell 2013, 49, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, L.; Fontaine, C.; Gougat, C.; Avinens, O.; Eliaou, J.-F.; Guglielmi, P.; Demoly, P. IL-10 promoter and IL4-Ralpha gene SNPs are associated with immediate beta-lactam allergy in atopic women. Allergy 2006, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Strait, R.T.; Morris, S.C.; Smiley, K.; Urban, J.F.; Finkelman, F.D. IL-4 Exacerbates Anaphylaxis. J. Immunol. 2003, 170, 3835–3842. [Google Scholar] [CrossRef]
- Sanders, N.L.; Mishra, A. Role of interleukin-18 in the pathophysiology of allergic diseases. Cytokine Growth Factor Rev. 2016, 32, 31–39. [Google Scholar] [CrossRef]
- Worm, M.; Francuzik, W.; Renaudin, J.-M.; Bilo, M.B.; Cardona, V.; Scherer Hofmeier, K.; Köhli, A.; Bauer, A.; Christoff, G.; Cichocka-Jarosz, E.; et al. Factors increasing the risk for a severe reaction in anaphylaxis An analysis of data from The European Anaphylaxis Registry. Allergy 2018, 73, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Gülen, T.; Oude Elberink, J.N.G.; Brockow, K. Anaphylaxis in Mastocytosis. In Mastocytosis; Springer International Publishing: Cham, Switzerland, 2020; pp. 141–155. [Google Scholar]
- Macleod, A.C.; Klug, L.R.; Patterson, J.; Griffith, D.J.; Beadling, C.; Town, A.; Heinrich, M.C. Combination therapy for KIT-mutant mast cells: Targeting constitutive NFAT and KIT activity. Mol. Cancer Ther. 2014, 13, 2840–2851. [Google Scholar] [CrossRef]
- Ribó, P.; Guo, Y.; Aranda, J.; Ainsua-Enrich, E.; Navinés-Ferrer, A.; Guerrero, M.; Pascal, M.; de la Cruz, C.; Orozco, M.; Muñoz-Cano, R.; et al. Mutation in KARS: A novel mechanism for severe anaphylaxis. J. Allergy Clin. Immunol. 2020, 147, 1855–1864.e9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TF | Target or Action Related to Anaphylaxis | Cells | References |
|---|---|---|---|
| GATA1 | FcɛRI αchain Tryptases IL-4 | MC Basophils | [27,47,48,49,50,52] |
| GATA2 | FcɛRI α/ β chain Tryptases Histamine Proteases | MC Basophils | [27,51,52,53,54] |
| GATA3 | Anaphylactic IgE synthesis IL-13, IL-4, IL-5 | Thf13 Th2 | [57,59,60,61,66] |
| STAT1 | IL-13Rα1 | MC | [73] |
| STAT3 | Increase degranulation | MC | [74,77] |
| STAT4 | IL-6 | MC | [84,85] |
| STAT5 | Increase degranulation Leukotriene synthesis IL-4, IL-13 and TNFα mRNA stabilization | MC | [92,93] |
| STAT6 | IL-6 TNFα | MC | [96,97,98] |
| MITF | Histamine Chymases Tryptases Granzyme B PGD2 | MC | [111,112,116,117] |
| TFE3 | FcɛRI KIT | MC | [120] |
| TFEB | Granule Biogenesis | MC | [81,82] |
| PU.1 | FcɛRI IL33 | MC Basophils | [52,122,123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Proaño-Pérez, E.; Muñoz-Cano, R.; Martin, M. Anaphylaxis: Focus on Transcription Factor Activity. Int. J. Mol. Sci. 2021, 22, 4935. https://doi.org/10.3390/ijms22094935
Guo Y, Proaño-Pérez E, Muñoz-Cano R, Martin M. Anaphylaxis: Focus on Transcription Factor Activity. International Journal of Molecular Sciences. 2021; 22(9):4935. https://doi.org/10.3390/ijms22094935
Chicago/Turabian StyleGuo, Yanru, Elizabeth Proaño-Pérez, Rosa Muñoz-Cano, and Margarita Martin. 2021. "Anaphylaxis: Focus on Transcription Factor Activity" International Journal of Molecular Sciences 22, no. 9: 4935. https://doi.org/10.3390/ijms22094935
APA StyleGuo, Y., Proaño-Pérez, E., Muñoz-Cano, R., & Martin, M. (2021). Anaphylaxis: Focus on Transcription Factor Activity. International Journal of Molecular Sciences, 22(9), 4935. https://doi.org/10.3390/ijms22094935