
Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line

 ,
, 
 , , ,
, , , 

Abstract
1. Introduction
2. Results
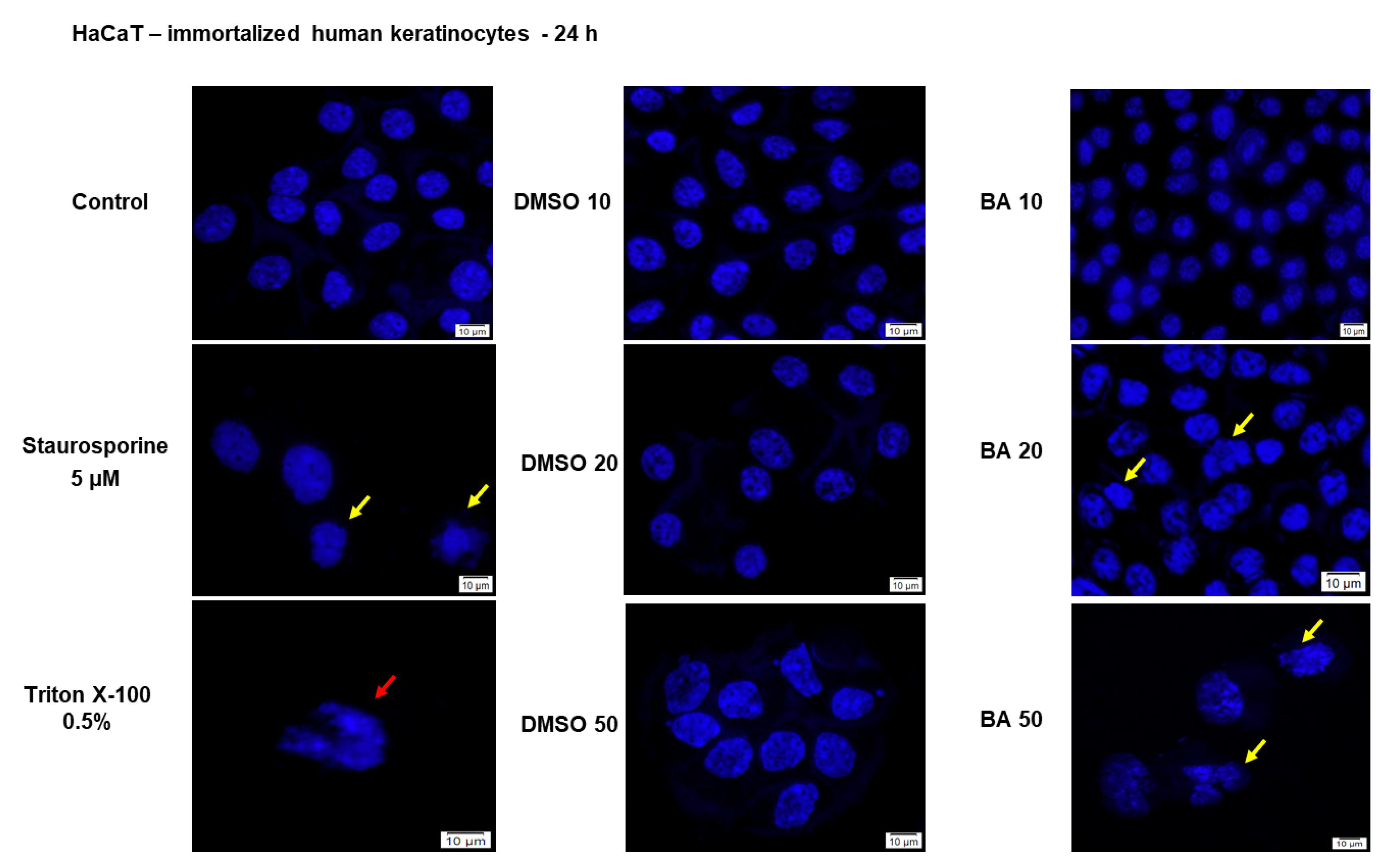
2.1. High Concentrations of BA Promote Cytotoxicity and Morphological Changes in HaCaT Cells
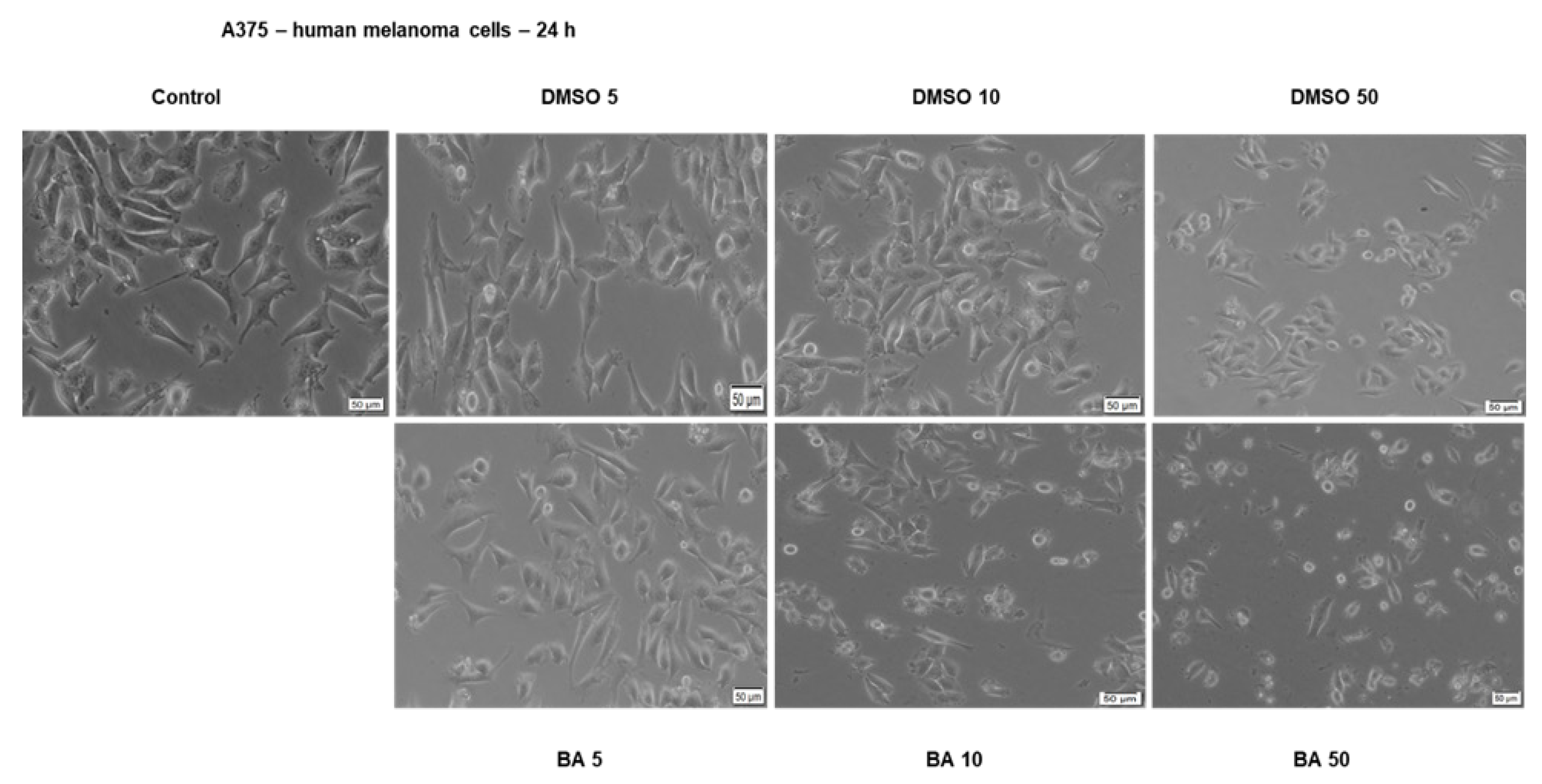
2.2. BA Exerts a Dose-Dependent Cytotoxic Effect in A375 Human Melanoma Cells
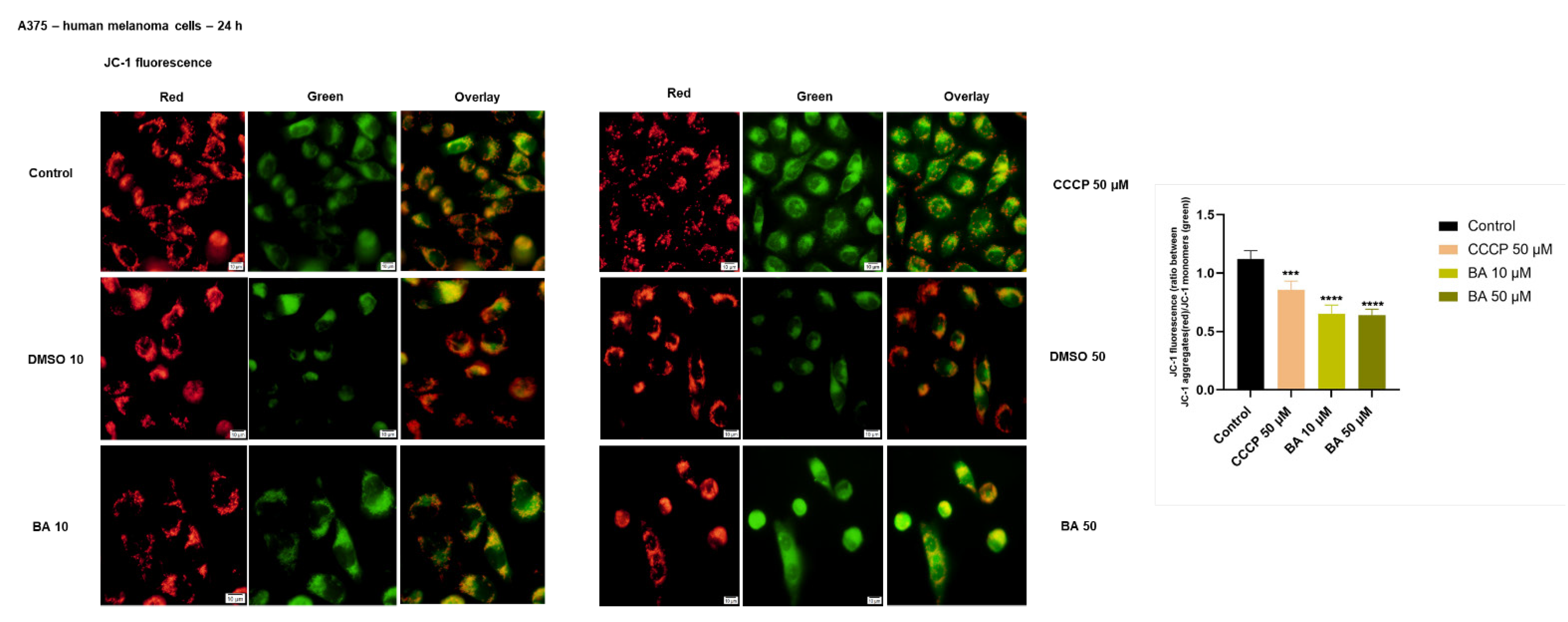
2.3. BA Treatment Impacts on Mitochondrial Membrane Potential in a Dose-Dependent Manner
2.4. BA Stimulation Induces Changes in Mitochondrial Markers mRNA Expression
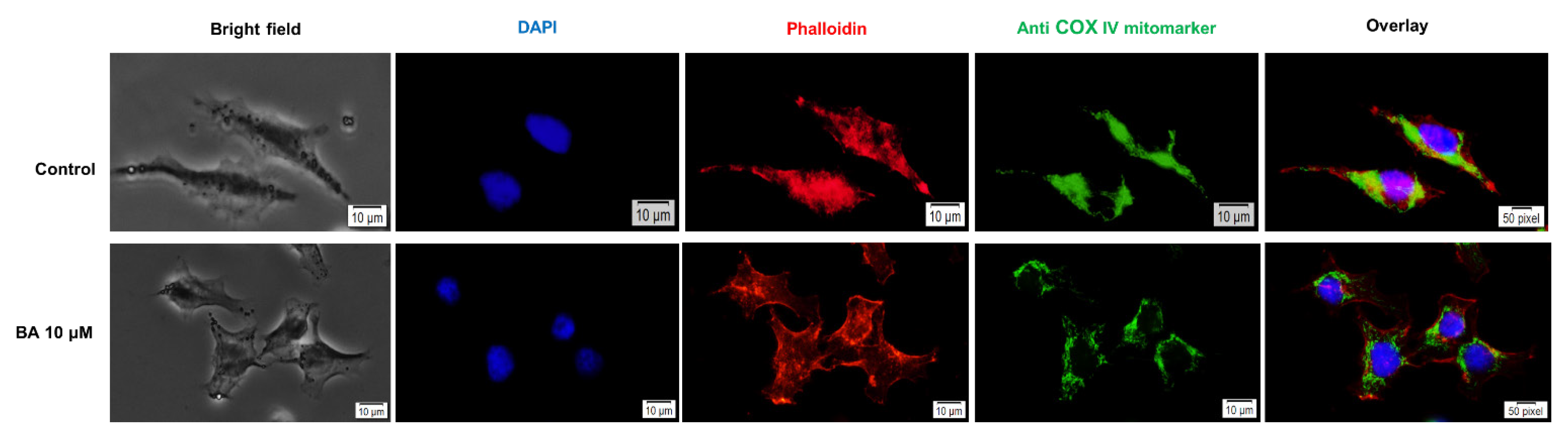
2.5. Impact of BA on Human Melanoma Cells Morphology and Cellular Organelles
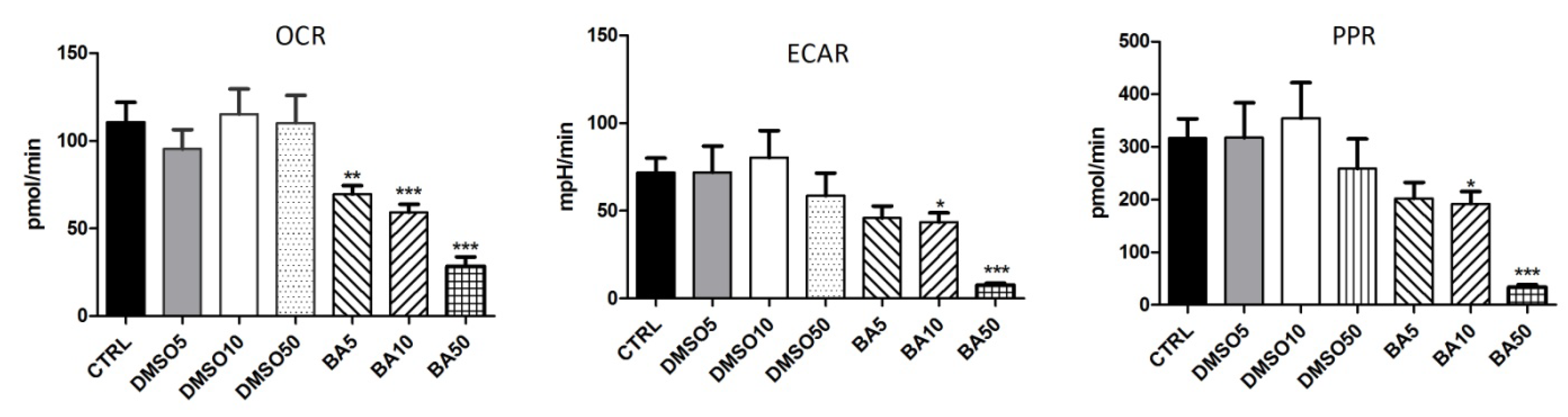
2.6. BA Treatment Modulates the Bioenergetic Profile of A375 Cells
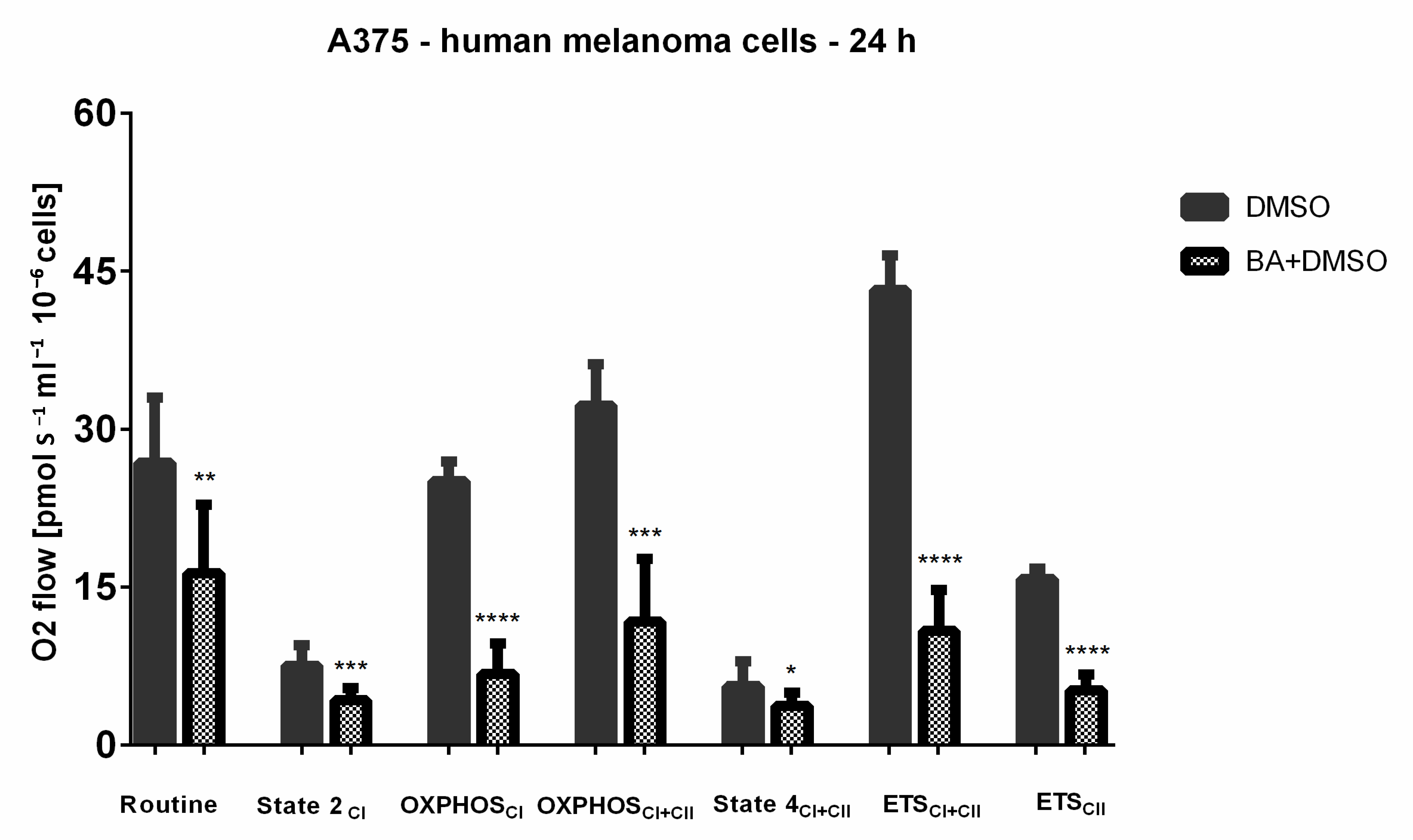
2.7. BA-Mediated Changes on Mitochondrial Respiratory Parameters in A375 Human Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture Conditions
4.3. Cytotoxicity Assessment
4.4. Mitochondrial Membrane Potential (ΔΨ) Assay (JC-1)
4.5. Quantification of Apoptotic Markers by Real-Time PCR
4.6. Mitochondria Localization Using Immunofluorescence
4.7. Bioenergetic Profile of A375 Human Melanoma Cells
4.8. High-Resolution Respirometry Studies in Permeabilized Cells
- Routine respiration (basal respiration)—R—represents the physiological respiratory activity in intact cells; is dependent of cellular energy need, energy turnover and the degree of coupling to phosphorylation to ADP [65]
- State 2CI—Leak state—L—basal respiration measured in the presence of malate (M) and glutamate (G)—complex I dependent—substrates (NADH-generating substrates).
- OXPHOSCI (active respiration)—P—mitochondrial respiratory capacity in the ADP-activated state of oxidative phosphorylation, measured after addition of ADP and in the presence of reducing complex I-dependent substrates: M and G.
- State 4CI+CII—Leak state—obtained after addition of oligomycin—an inhibitor of ATP synthase, this residual respiration rate is assigned to proton leak, yields a non-phosphorylating state and a return to basal respiration dependent on both CI and CII.
- ETSCI+CII—the maximal respiratory capacity of the electron transport system mimics a physiological energy need inducing an increase of oxygen consumption. It is acquired after successive titrations of an uncoupler as FCCP (carbonyl cyanide p-(trifluoro-methoxy) phenyl-hydrazone [67].
- Residual oxygen consumption—ROX—indicates the remaining mitochondrial respiration after addition of rotenone—an inhibitor of complex I and antimycin—an inhibitor of complex III [67].
- R/E (Routine/ ETSCI+II)—routine respiration normalized to ETS capacity; indicates how tight functions Routine respiration to the maximum capacity of the system.
- L/E (State 4CI+II/ ETSCI+II)—Leak respiration normalized to ETS capacity; represents the part of maximum respiratory capacity linked to proton leak. Values as 0.0 indicate fully coupled mitochondria, whereas 1.0 is attributed to fully uncoupled mitochondria [44].
- RCR—(OXPHOSCI+II/State 4CI+II)—respiratory control ratio is defined as the ratio between active respiration and leak state obtained after oligomycin addition; is strongly affected by any change in oxidative phosphorylation and is considered a useful indicator of mitochondrial dysfunction [35].
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prado, G.; Svoboda, R.M.; Rigel, D.S. What’s New in Melanoma. Dermatol. Clin. 2019, 37, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Gajos-Michniewicz, A.; Czyz, M. Role of miRNAs in Melanoma Metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef]
- Coricovac, D.; Dehelean, C.; Moaca, E.A.; Pinzaru, I.; Bratu, T.; Navolan, D.; Boruga, O. Cutaneous Melanoma-A Long Road from Experimental Models to Clinical Outcome: A Review. Int. J. Mol. Sci. 2018, 19, 1566. [Google Scholar] [CrossRef] [PubMed]
- European Cancer Information System. ECIS. Available online: https://ecis.jrc.ec.europa.eu/ (accessed on 25 February 2021).
- Wróbel, S.; Przybyło, M.; Stępień, E. The Clinical Trial Landscape for Melanoma Therapies. J. Clin. Med. 2019, 8, 368. [Google Scholar] [CrossRef]
- Hosseini, M.; Kasraian, Z.; Rezvani, H.R. Energy metabolism in skin cancers: A therapeutic perspective. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 712–722. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine uti-lization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh-Gohari, S.; Weber, D.D.; Catalano, L.; Feichtinger, R.G.; Kofler, B.; Lang, R. Targeting Mitochondria in Melanoma. Biomolecules 2020, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef]
- Theodosakis, N.; Micevic, G.; Kelly, D.P.; Bosenberg, M. Mitochondrial function in melanoma. Arch. Biochem. Biophys. 2014, 563, 56–59. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096. [Google Scholar] [PubMed]
- Ríos, J.L.; Máñez, S. New Pharmacological Opportunities for Betulinic Acid. Planta Med. 2018, 84, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Pascale, A.V.; Finelli, R.; Carillo, A.L.; Annunziata, R.; Trimarco, B.; Iaccarino, G. Targeting mitochondria as therapeutic strategy for metabolic disorders. Sci. World J. 2014, 2014, 604685. [Google Scholar] [CrossRef] [PubMed]
- Gheorgheosu, D.; Duicu, O.; Dehelean, C.; Soica, C.; Muntean, D. Betulinic acid as a potent and complex antitumor phytochemical: A minireview. Anticancer Agents Med. Chem. 2014, 14, 936. [Google Scholar]
- Heiss, E.H.; Kramer, M.P.; Atanasov, A.G.; Beres, H.; Schachner, D.; Dirsch, V.M. Glycolytic switch in response to betulinic acid in non-cancer cells. PLoS ONE 2014, 9, e115683. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, R.; Pezzuto, J.M. Betulinic acid-induced programmed cell death in human melanoma cells involves mitogen-activated protein kinase activation. Clin. Cancer Res. 2003, 9, 2866. [Google Scholar]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Gomide, A.B.; Costa, É.T.; Junqueira, H.C.; Stolf, B.S.; Itri, R.; Baptista, M.S. Membrane damage by betulinic acid provides insights into cellular aging. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3129–3143. [Google Scholar] [CrossRef] [PubMed]
- Surowiak, P.; Drag, M.; Materna, V.; Dietel, M.; Lage, H. Betulinic acid exhibits stronger cytotoxic activity on the normal melanocyte NHEM-neo cell line than on drug-resistant and drug-sensitive MeWo melanoma cell lines. Mol. Med. Rep. 2009, 2, 543–548. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Pinto, D.C.G.A. Plant Secondary Metabolites as Anticancer Agents: Successes in Clinical Trials and Therapeutic Application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef]
- Clinical trial—Evaluation of 20% Betulinic Acid Ointment for Treatment of Dysplastic Nevi (Moderate to Severe Dysplasia). Available online: https://www.clinicaltrials.gov/ct2/show/NCT00346502?term=betulinic+acid&draw=2&rank=1 (accessed on 20 January 2021).
- Dehelean, C.A.; Marcovici, I.; Soica, C.; Mioc, M.; Coricovac, D.; Iurciuc, S.; Cretu, O.M.; Pinzaru, I. Plant-Derived Anticancer Compounds as New Perspectives in Drug Discovery and Alternative Therapy. Molecules 2021, 26, 1109. [Google Scholar] [CrossRef]
- Ramsay, E.E.; Hogg, P.J.; Dilda, P.J. Mitochondrial metabolism inhibitors for cancer therapy. Pharm. Res. 2011, 28, 2731–2744. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Muntean, D.M.; Sturza, A.; Pavel, I.Z.; Duicu, O.M. Modulation of Cancer Metabolism by Phytochemicals—A Brief Overview. Anticancer Agents Med. Chem. 2018, 18, 684–692. [Google Scholar] [CrossRef]
- Sturza, A.; Pavel, I.; Ancușa, S.; Danciu, C.; Dehelean, C.; Duicu, O.; Muntean, D. Quercetin exerts an inhibitory effect on cellular bioenergetics of the B164A5 murine melanoma cell line. Mol. Cell. Biochem. 2018, 447, 103–109. [Google Scholar] [CrossRef]
- Wang, G.; Yu, Y.; Wang, Y.Z.; Zhu, Z.M.; Yin, P.H.; Xu, K. Effects and mechanisms of fatty acid metabolism-mediated glycolysis regulated by betulinic acid-loaded nanoliposomes in colorectal cancer. Oncol. Rep. 2020, 44, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; García-García, J.D.; Belmont-Díaz, J.; López-Marure, R.; Hernández-Esquivel, L.; et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Sundstrøm, T.; Prestegarden, L.; Azuaje, F.; Aasen, S.N.; Røsland, G.V.; Varughese, J.K.; Bahador, M.; Bernatz, S.; Braun, Y.; Harter, P.N.; et al. Inhibition of mitochondrial respiration prevents BRAF-mutant melanoma brain metastasis. Acta Neuropathol. Commun. 2019, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities and opportunities. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 655–664. [Google Scholar] [CrossRef]
- Brennan, J.P.; Southworth, R.; Medina, R.A.; Davidson, S.M.; Duchen, M.R.; Shattock, M.J. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc. Res. 2006, 72, 313–321. [Google Scholar] [CrossRef]
- Lim, H.W.; Lim, H.Y.; Wong, K.P. Uncoupling of oxidative phosphorylation by curcumin: Implication of its cellular mechanism of action. Biochem. Biophys. Res. Commun. 2009, 389, 187. [Google Scholar] [CrossRef]
- Terada, H. Uncouplers of oxidative phosphorylation. Environ. Health Perspect. 1990, 87, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25. [Google Scholar] [PubMed]
- Liebscher, G.; Vanchangiri, K.; Mueller, T.; Feige, K.; Cavalleri, J.M.; Paschke, R. In vitro anticancer activity of Betulinic acid and derivatives thereof on equine melanoma cell lines from grey horses and in vivo safety assessment of the compound NVX-207 in two horses. Chem. Biol. Interact. 2016, 246, 20–29. [Google Scholar] [CrossRef]
- Gheorgheosu, D.; Jung, M.; Ören, B.; Schmid, T.; Dehelean, C.; Muntean, D.; Brüne, B. Betulinic acid suppresses NGAL-induced epithelial-to-mesenchymal transition in melanoma. Biol. Chem. 2013, 394, 773–781. [Google Scholar] [CrossRef]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef]
- Fulda, S.; Scaffidi, G.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [PubMed]
- Anvekar, R.A.; Asciolla, J.J.; Missert, D.J.; Chipuk, J.E. Born to be alive: A role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front. Oncol. 2011, 1, 34. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Fragomeni, R.A.S.; Schalck, A.; Ferreiro-Neira, I.; Hoff, T.; Cooper, Z.A.; Haq, R.; Panka, D.J.; Kwong, L.N.; Davies, M.A.; et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS ONE 2014, 9, e101286. [Google Scholar] [CrossRef]
- Kumar, P.; Bhadauria, A.S.; Singh, A.K.; Saha, S. Betulinic acid as apoptosis activator: Molecular mechanisms, mathematical modeling and chemical modifications. Life Sci. 2018, 209, 24–33. [Google Scholar] [CrossRef]
- Pandey, M.K.; Sung, B.; Aggarwal, B.B. Betulinic acid suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase SHP-1 in human multiple myeloma cells. Int. J. Cancer 2010, 127, 282–292. [Google Scholar] [CrossRef]
- Xu, T.; Pang, Q.; Wang, Y.; Yan, X. Betulinic acid induces apoptosis by regulating PI3K/Akt signaling and mitochondrial pathways in human cervical cancer cells. Int. J. Mol. Med. 2017, 40, 1669–1678. [Google Scholar] [CrossRef]
- Wick, W.; Grimmel, C.; Wagenknecht, B.; Dichgans, J.; Weller, M. Betulinic acid-induced apoptosis in glioma cells: A sequential requirement for new protein synthesis, for-mation of reactive oxygen species, and caspase processing. J. Pharmacol. Exp. Ther. 1999, 289, 1306–1312. [Google Scholar]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nuñez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Thurnher, D.; Turhani, D.; Pelzmann, M.; Wannemacher, B.; Knerer, B.; Formanek, M.; Wacheck, V.; Selzer, E. Betulinic acid: A new cytotoxic com-pound against malignant head and neck cancer cells. Head Neck 2003, 25, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Selzer, E.; Thallinger, C.; Hoeller, C.; Oberkleiner, P.; Wacheck, V.; Pehamberger, H.; Jansen, B. Betulinic acid-induced Mcl-1 expression in human melanoma—mode of action and functional significance. Mol. Med. 2002, 8, 877–884. [Google Scholar] [CrossRef]
- Farcas, C.G.; Macasoi, I.; Pinzaru, I.; Chirita, M.; Mihaila, M.C.C.; Dehelean, C.; Avram, S.; Loghin, F.; Mocanu, L.; Rotaru, V.; et al. Controlled Synthesis and Characterization of Micrometric Single Crystalline Magnetite With Superparamagnetic Behavior and Cytocompatibility/Cytotoxicity Assessments. Front. Pharmacol. 2020, 11, 410. [Google Scholar] [CrossRef] [PubMed]
- Maghiari, A.L.; Coricovac, D.; Pinzaru, I.A.; Macașoi, I.G.; Marcovici, I.; Simu, S.; Navolan, D.; Dehelean, C. High Concentrations of Aspartame Induce Pro-Angiogenic Effects in Ovo and Cytotoxic Effects in HT-29 Human Colorectal Carcinoma Cells. Nutrients 2020, 12, 3600. [Google Scholar] [CrossRef]
- Farcas, C.G.; Dehelean, C.; Pinzaru, I.A.; Mioc, M.; Socoliuc, V.; Moaca, E.A.; Avram, S.; Ghiulai, R.; Coricovac, D.; Pavel, I.; et al. Thermosensitive Betulinic Acid-Loaded Magnetoliposomes: A Promising Antitumor Potential for Highly Aggressive Human Breast Adenocarcinoma Cells Under Hyperthermic Conditions. Int. J. Nanomed. 2020, 15, 8175–8200. [Google Scholar] [CrossRef] [PubMed]
- Pinzaru, I.; Tanase, A.; Enatescu, V.; Coricovac, D.; Bociort, F.; Marcovici, I.; Watz, C.; Vlaia, L.; Soica, C.; Dehelean, C. Proniosomal Gel for Topical Delivery of Rutin: Preparation, Physicochemical Characterization and In Vitro Toxicological Profile Using 3D Reconstructed Human Epidermis Tissue and 2D Cells. Antioxidants 2021, 10, 85. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio. Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Duicu, O.M.; Scurtu, I.; Popescu, R.; Sturza, A.; Coricovac, D.; Danila, D.M.; Privistirescu, A.; Muntean, D.M. Assessment of the effects of methylene blue on cellular bioenergetics in H9c2 cells. Rev. Chim. 2015, 66, 519–522. [Google Scholar]
- Makrecka-Kuka, M.; Krumschnabel, G.; Gnaiger, E. High-Resolution Respirometry for Simultaneous Measurement of Oxygen and Hydrogen Peroxide Fluxes in Permeabilized Cells, Tissue Homogenate and Isolated Mitochondria. Biomolecules 2015, 5, 1319–1338. [Google Scholar] [CrossRef] [PubMed]
- Hals, I.K.; Bruerberg, S.G.; Ma, Z.; Scholz, H.; Björklund, A.; Grill, V. Mitochondrial Respiration in Insulin-Producing β-Cells: General Characteristics and Adaptive Effects of Hypoxia. PLoS ONE 2015, 10, e0138558. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.H.; Greenwood, J.C.; Spyres, M.B.; Eckmann, D.M. Measurement of Mitochondrial Respiration and Motility in Acute Care: Sepsis, Trauma, and Poisoning. J. Intensive Care Med. 2017, 32, 86. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flux Control Ratios | 24 h Stimulation | |
|---|---|---|
| DMSO | BA | |
| R/E | 0.604 ± 0.145 | 0.975 ± 0.022 ** |
| L/E | 0.154 ± 0.040 | 0.299 ± 0.098 * |
| RCR | 4.530 ± 1.398 | 2.785 ± 0.953 * |
| Primer’s Name | Forward | Reverse |
|---|---|---|
| 18 S * | 5′ GTAACCCGTTGAACCCCATT 3′ | 5′ CCA-TCC-AAT-CGG-TAGTAG-CG 3′ |
| GAPDH * | 5′AAG-GTG-AAG-GTC-GGA-GTC-AAC 3′ | 5′GGG-GTC-ATT-GAT-GGC-AAC-AAT-A 3′ |
| Bax | 5′ GCCGGGTTGTCGCCCTTTT 3′ | 5′CCGCTCCCGGAGGAAGTCCA 3′ |
| Bid | 5′CCT-TGC-TCC-GTG-ATG-TCT-TTC 3′ | 5′GTA-GGT-GCC-TAG-GTT-CTG-GT 3′ |
| Bad | 5′CCC-AGA-GTT-TGA-GCC-GAG-TG 3′ | 5′CCC-ATC-CCT-TCG-TCC-T 3′ |
| Bak | 5′ATGGTCACCTTACCTCTGCAA 3′ | 5′TCATAGCGTCGGTTGATGTCG 3′ |
| Bcl-2 | 5′CGGGAGATGTCGCCCCTGGT 3′ | 5′GCATGCTGGGGCCGTACAGT 3′ |
| Bcl-XL | 5′GATCCCCATGGCAGCAGTAAAGCAAG 3′ | 5′ CCCCATCCCGGAAGAGTTCATTCACT 3′ |
| Caspase 3 | 5′GCGGTTGTAGAAGAGTTTCGTG 3′ | 5′CTCACGGCCTGGGATTTCAA 3′ |
| Caspase 8 | 5′AGAGTCTGTGCCCAAATCAAC 3′ | 5′GCTGCTTCTCTCTTTGCTGAA 3′ |
| Apaf-1 | 5′CACGTTCAAAGGTGGCTGAT 3′ | 5′TGGTCAACTGCAAGGACCAT 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coricovac, D.; Dehelean, C.A.; Pinzaru, I.; Mioc, A.; Aburel, O.-M.; Macasoi, I.; Draghici, G.A.; Petean, C.; Soica, C.; Boruga, M.; et al. Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line. Int. J. Mol. Sci. 2021, 22, 4870. https://doi.org/10.3390/ijms22094870
Coricovac D, Dehelean CA, Pinzaru I, Mioc A, Aburel O-M, Macasoi I, Draghici GA, Petean C, Soica C, Boruga M, et al. Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line. International Journal of Molecular Sciences. 2021; 22(9):4870. https://doi.org/10.3390/ijms22094870
Chicago/Turabian StyleCoricovac, Dorina, Cristina Adriana Dehelean, Iulia Pinzaru, Alexandra Mioc, Oana-Maria Aburel, Ioana Macasoi, George Andrei Draghici, Crina Petean, Codruta Soica, Madalina Boruga, and et al. 2021. "Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line" International Journal of Molecular Sciences 22, no. 9: 4870. https://doi.org/10.3390/ijms22094870
APA StyleCoricovac, D., Dehelean, C. A., Pinzaru, I., Mioc, A., Aburel, O.-M., Macasoi, I., Draghici, G. A., Petean, C., Soica, C., Boruga, M., Vlaicu, B., & Muntean, M. D. (2021). Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line. International Journal of Molecular Sciences, 22(9), 4870. https://doi.org/10.3390/ijms22094870