
Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
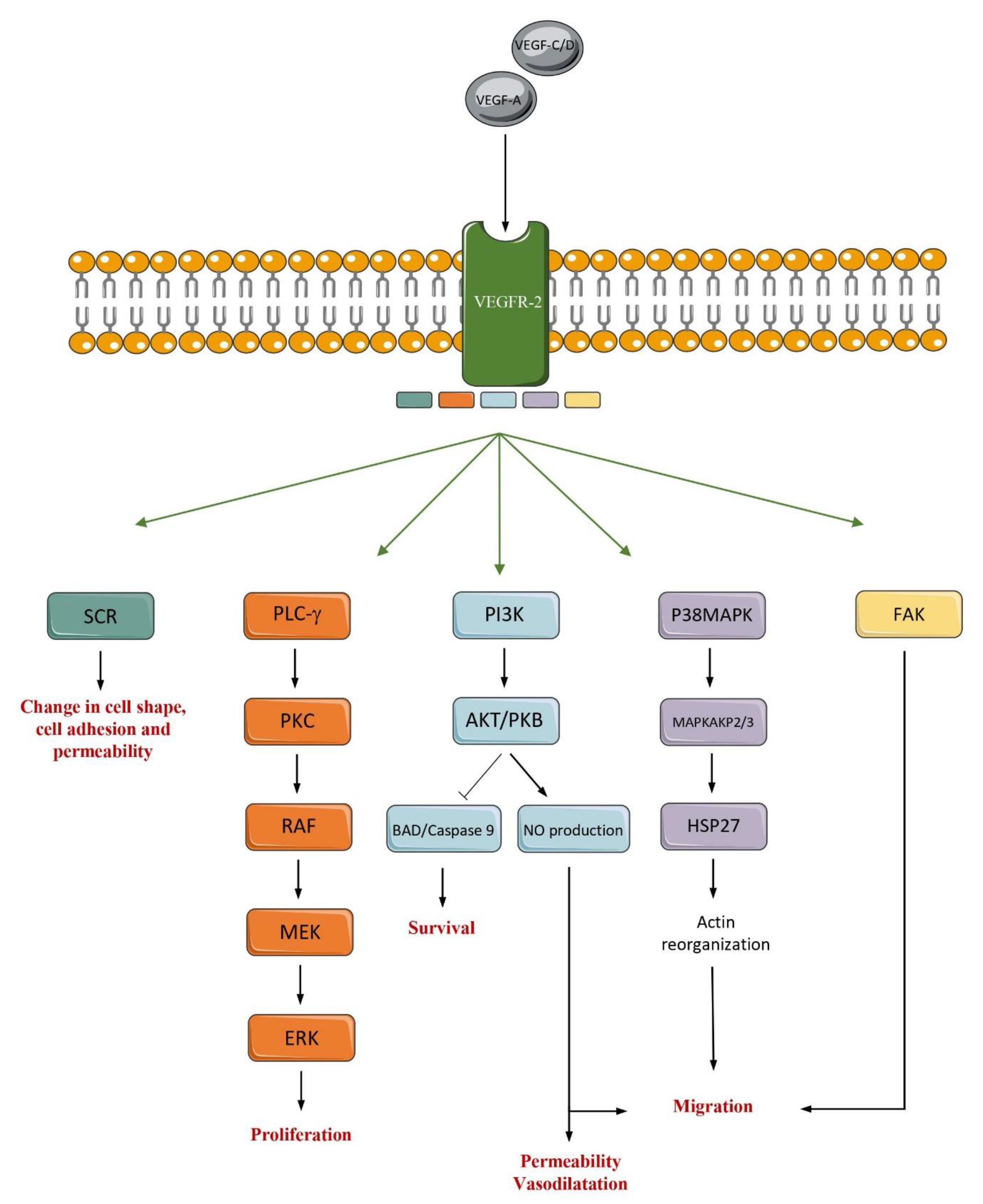
2. Overview of VEGF Signaling Pathways and Its Major Contributors
3. Insight into the VEGF Signaling Pathways Roles in Angiogenesis
3.1. The Angiogenic Switch
3.2. Tumor Blood Vessels Abnormalities
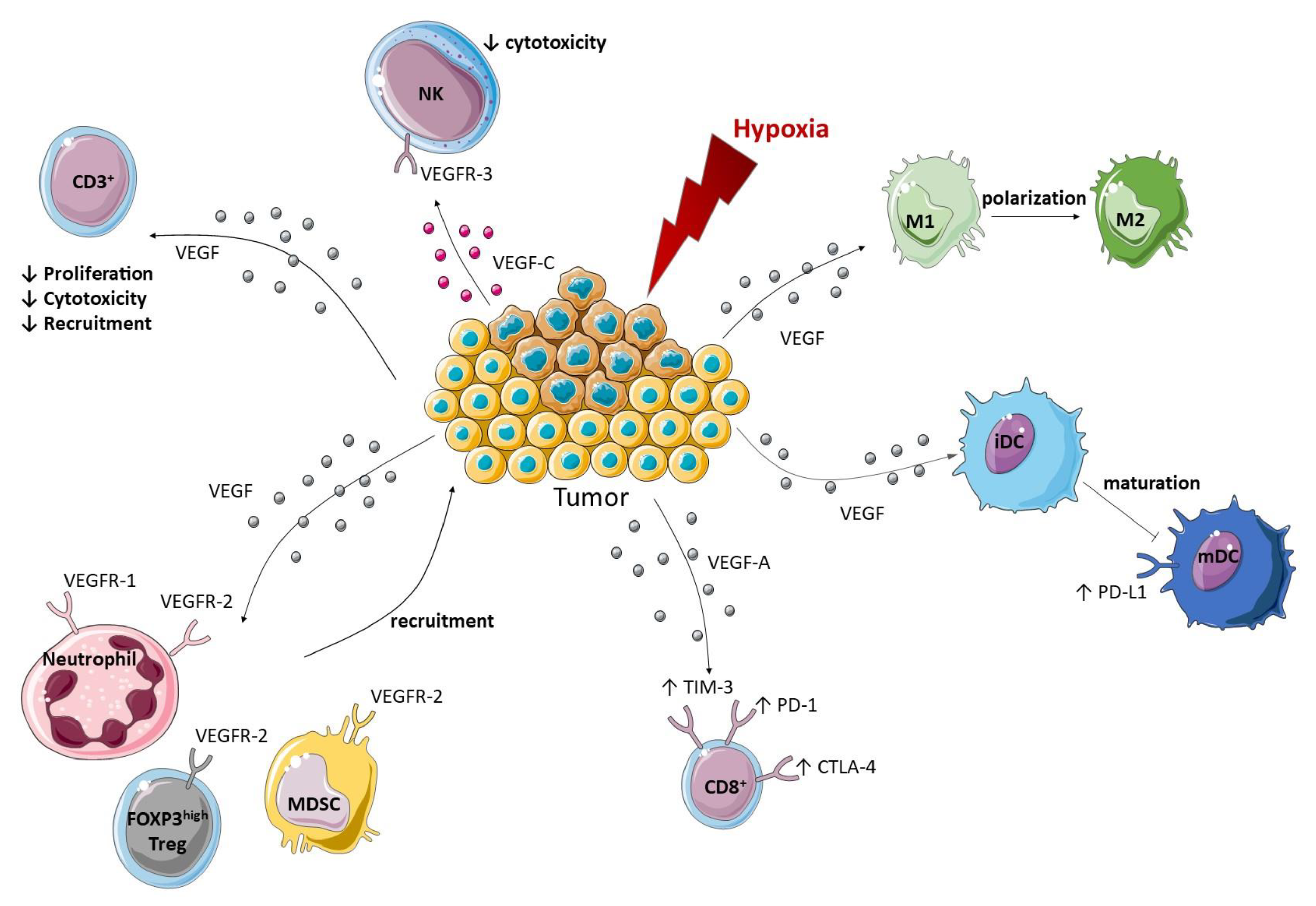
4. Cancer Immune Response and VEGF
4.1. Pro-Angiogenic VEGF Modulates Protein Expression on Endothelial Cells
4.2. Pro-Angiogenic VEGF Modulates the Innate Immune Response
4.2.1. Macrophages
4.2.2. Natural Killer
4.2.3. Neutrophils
4.2.4. Mast Cells
4.2.5. Myeloid Derived Suppressor Cells
4.2.6. Dendritic Cells
4.3. Pro-Angiogenic VEGF Modulates the Adaptive Immune Response
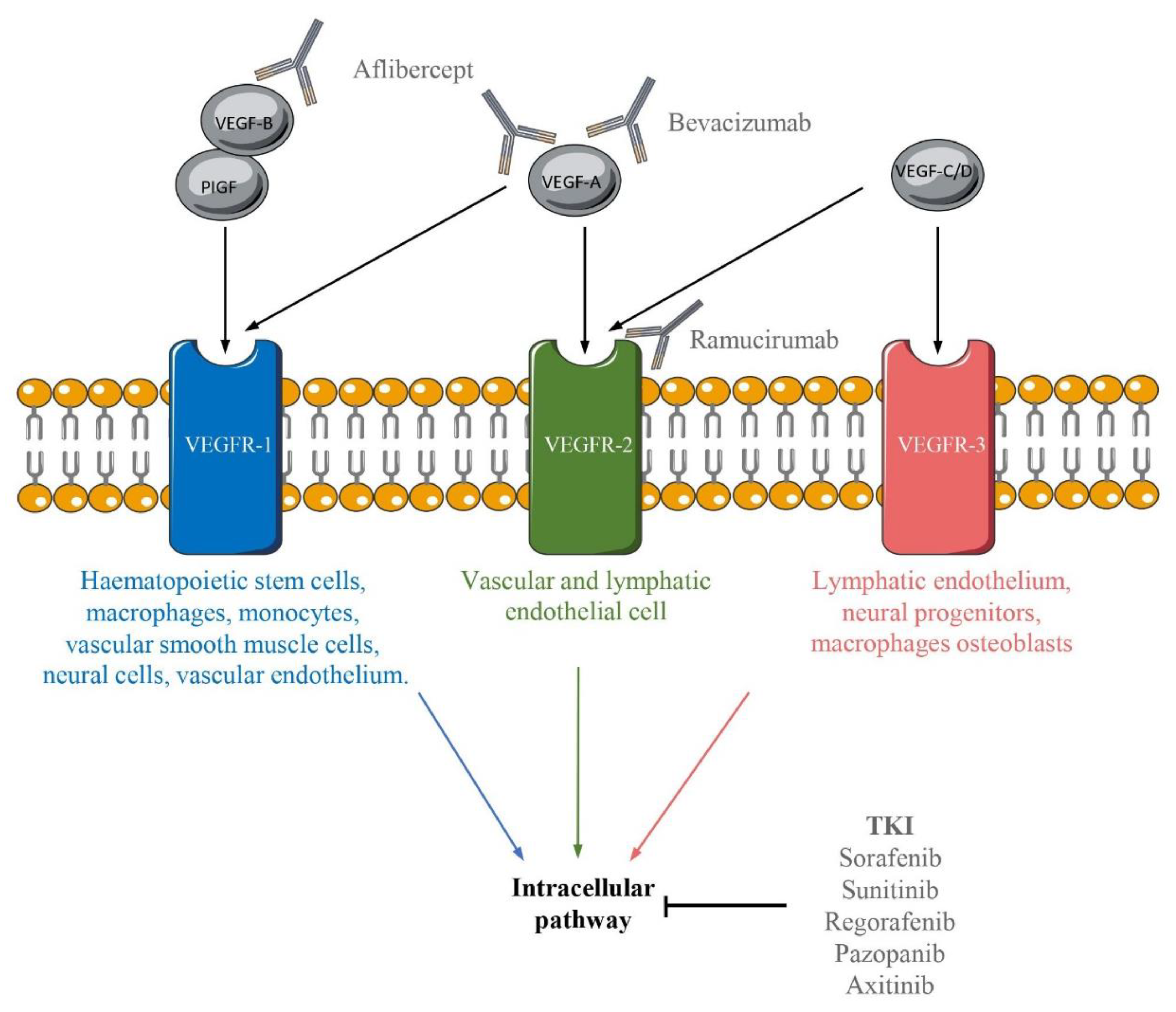
5. A Whole Range of Anti-VEGF/VEGFR Therapies
5.1. Antibodies Targeting VEGF Pathways
5.2. Tyrosine Kinase Inhibitor
6. The Limit of Anti-Angiogenic Therapies
7. Combined Therapies
7.1. Anti-VEGF and Other Anti-Angiogenic Mechanisms
7.2. Anti VEGF and Chemotherapy
7.3. VEGF and Immune Checkpoint Inhibition
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ANG-2 | Angiopoietin-2 |
| BC | Breast cancer |
| CRC | Colorectal cancer |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| CXCL10 | CXC-chemokine ligand 10 |
| CXCR3+ | C-X-C motif chemokine receptor 3 |
| DC | Dendritic cells |
| EMA | European Medial Agency |
| FGF | Fibroblast growth factor |
| FLT3 | Fms-like tyrosine kinase-3 receptor |
| HCC | Hepatocellular carcinoma |
| HIF-1 | Hypoxia-inducible factor 1 |
| HSPG | Heparan sulfate proteoglycans |
| ICAM1 | Intracellular adhesion molecule 1 |
| MAPK | Mitogen-activated protein kinase |
| MC | Mast cells |
| MDSC | Myeloid-derived suppressor cell |
| MMP-9 | Metallopeptidase 9 |
| NF-κB | Nuclear factor-kappa B |
| NK | Natural killer |
| NRP | Neuropilin |
| NSCLC | Non-small cell lung cancer |
| OC | Ovarian cancer |
| OS | Overall survival |
| PD-1 | Programmed cell death 1 |
| PDGF | Platelet-derived growth factor |
| PFS | Progression-free survival |
| PI3K | Phosphoinositide-3-kinase |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PLCγ | Phospholipase-Cγ |
| PlGF | Placental growth factor |
| RCC | Renal cell carcinoma |
| SCC | Squamous cell carcinoma |
| TAM | Tumor-associated macrophage |
| TGF-β | Transforming growth factor β |
| TIM-3 | T cell immunoglobulin and mucin-domain containing-3 |
| TKI | Tyrosine kinase inhibitors |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor-α |
| VCAM1 | Vascular cell adhesion protein 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptors |
References
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; McClung, J.A.; Aronow, W.S. Chapter 6—Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease; Aronow, W.S., McClung, J.A., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 49–65. ISBN 978-0-12-802385-3. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.-F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF Gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal Blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Catargi, B. VEGF in physiological process and thyroid disease. Ann. Endocrinol. 2007, 68, 438–448. [Google Scholar] [CrossRef]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular Endothelial Growth Factor (VEGF)—Key Factor in Normal and Pathological Angiogenesis. Romanian J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Y.; Zhang, J.; Xing, B.; Xuan, W.; Wang, H.; Huang, H.; Yang, J.; Tang, J. NRP-2 in Tumor Lymphangiogenesis and Lymphatic Metastasis. Cancer Lett. 2018, 418, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ch, B.; Khaled, Y.; Ammori, B.; Elkord, E. Neuropilin 1: Function and Therapeutic Potential in Cancer. Cancer Immunol. Immunother. 2013, 63. [Google Scholar] [CrossRef]
- Prud’homme, G.J.; Glinka, Y. Neuropilins Are Multifunctional Coreceptors Involved in Tumor Initiation, Growth, Metastasis and Immunity. Oncotarget 2012, 3, 921–939. [Google Scholar] [CrossRef]
- Van Wijk, X.M.R.; van Kuppevelt, T.H. Heparan Sulfate in Angiogenesis: A Target for Therapy. Angiogenesis 2014, 17, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Dixelius, J.; Matsumoto, T.; Claesson-Welsh, L. VEGF-Receptor Signal Transduction. Trends Biochem. Sci. 2003, 28, 488–494. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular Abnormalities of Blood Vessels as Targets in Cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and Mechanisms of Vessel Normalization for Cancer and Other Angiogenic Diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Darden, J.; Payne, L.B.; Zhao, H.; Chappell, J.C. Excess Vascular Endothelial Growth Factor-A Disrupts Pericyte Recruitment during Blood Vessel Formation. Angiogenesis 2019, 22, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R. Improving Immunotherapy Outcomes with Anti-Angiogenic Treatments and Vice Versa. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, S.M.; Damen, C.A.; van Adrichem, N.P.; Blijham, G.H.; Groenewegen, G.; Griffioen, A.W. Endothelial CD34 Is Suppressed in Human Malignancies: Role of Angiogenic Factors. Cancer Lett. 1997, 120, 203–211. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Oude Egbrink, M.G.A.; Kuijpers, M.J.E.; van der Niet, S.T.; Heijnen, V.V.T.; Bouma-ter Steege, J.C.A.; Wagstaff, J.; Griffioen, A.W. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in Vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of Vascular Endothelial Growth Factor on the Lymphocyte-Endothelium Interactions: Identification of Caveolin-1 and Nitric Oxide as Control Points of Endothelial Cell Anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-ΚB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Critical cells driving immune suppression in the tumor microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.K.; Pradhan, B.S.; Nayak, N.; Das, S.; Hsu, C.-D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13, e0191040. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The Proangiogenic Phenotype of Natural Killer Cells in Patients with Non-Small Cell Lung Cancer. Neoplasia US 2013, 15, 133–142. [Google Scholar] [CrossRef]
- Cerdeira, A.S.; Rajakumar, A.; Royle, C.M.; Lo, A.; Husain, Z.; Thadhani, R.I.; Sukhatme, V.P.; Karumanchi, S.A.; Kopcow, H.D. Conversion of Peripheral Blood NK Cells to a Decidual NK-like Phenotype by a Cocktail of Defined Factors. J. Immunol. 2013, 190, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Detoraki, A.; Staiano, R.I.; Granata, F.; Giannattasio, G.; Prevete, N.; de Paulis, A.; Ribatti, D.; Genovese, A.; Triggiani, M.; Marone, G. Vascular Endothelial Growth Factors Synthesized by Human Lung Mast Cells Exert Angiogenic Effects. J. Allergy Clin. Immunol. 2009, 123, 1142–1149. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-Angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages: Implications in Cancer Immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of Myeloid Immune Suppressor Gr+CD11b+ Cells in Tumor-Bearing Host Directly Promotes Tumor Angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Massena, S.; Christoffersson, G.; Vågesjö, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Herrera Hidalgo, C.; Giraud, A.; Lomei, J.; Weström, S.; et al. Identification and Characterization of VEGF-A–Responsive Neutrophils Expressing CD49d, VEGFR1, and CXCR4 in Mice and Humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef]
- Long, J.; Hu, Z.; Xue, H.; Wang, Y.; Chen, J.; Tang, F.; Zhou, J.; Liu, L.; Qiu, W.; Zhang, S.; et al. Vascular Endothelial Growth Factor (VEGF) Impairs the Motility and Immune Function of Human Mature Dendritic Cells through the VEGF Receptor 2-RhoA-Cofilin1 Pathway. Cancer Sci. 2019, 110, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, S.; Min, W.-S.; Kim, H.-J. Restoration of Natural Killer Cell Cytotoxicity by VEGFR-3 Inhibition in Myelogenous Leukemia. Cancer Lett. 2014, 354, 281–289. [Google Scholar] [CrossRef]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.M.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumor-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in Natural Killer Cells Inhibits Tumour Growth by Stimulating Non-Productive Angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D.; et al. Ly6Clo Monocytes Drive Immunosuppression and Confer Resistance to Anti-VEGFR2 Cancer Therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Tamma, R.; Guidolin, D.; Annese, T.; Tortorella, C.; Ruggieri, S.; Rega, S.; Zito, F.A.; Nico, B.; Ribatti, D. Spatial Distribution of Mast Cells and Macrophages around Tumor Glands in Human Breast Ductal Carcinoma. Exp. Cell Res. 2017, 359, 179–184. [Google Scholar] [CrossRef]
- De Souza Junior, D.A.; Santana, A.C.; da Silva, E.Z.M.; Oliver, C.; Jamur, M.C. The Role of Mast Cell Specific Chymases and Tryptases in Tumor Angiogenesis. Available online: https://www.hindawi.com/journals/bmri/2015/142359/ (accessed on 21 January 2021).
- Starkey, J.R.; Crowle, P.K.; Taubenberger, S. Mast-Cell-Deficient W/Wv Mice Exhibit A Decreased Rate of Tumor Angiogenesis. Int. J. Cancer 1988, 42, 48–52. [Google Scholar] [CrossRef]
- McHale, C.; Mohammed, Z.; Gomez, G. Human Skin-Derived Mast Cells Spontaneously Secrete Several Angiogenesis-Related Factors. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.; Mohammed, Z.; Deppen, J.; Gomez, G. Interleukin-6 Potentiates FcεRI-Induced PGD2 Biosynthesis and Induces VEGF from Human in Situ-Matured Skin Mast Cells. Biochim. Biophys. Acta 2018, 1862, 1069–1078. [Google Scholar] [CrossRef]
- Sismanopoulos, N.; Delivanis, D.; Alysandratos, K.; Angelidou, A.; Vasiadi, M.; Therianou, A.; Theoharides, T. IL-9 Induces VEGF Secretion from Human Mast Cells and IL-9/IL-9 Receptor Genes Are Overexpressed in Atopic Dermatitis. PLoS ONE 2012, 7, e33271. [Google Scholar] [CrossRef]
- Rm, A.-M.; Js, M. Prostaglandin E2 Induces Degranulation-Independent Production of Vascular Endothelial Growth Factor by Human Mast Cells. J. Immunol. Baltim. 2004, 172, 1227–1236. [Google Scholar] [CrossRef]
- Cao, J.; Papadopoulou, N.; Kempuraj, D.; Boucher, W.S.; Sugimoto, K.; Cetrulo, C.L.; Theoharides, T.C. Human Mast Cells Express Corticotropin-Releasing Hormone (CRH) Receptors and CRH Leads to Selective Secretion of Vascular Endothelial Growth Factor. J. Immunol. 2005, 174, 7665–7675. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Ryzhov, S.; Goldstein, A.E.; Biaggioni, I. Mast Cell-Mediated Stimulation of Angiogenesis: Cooperative Interaction between A2B and A3 Adenosine Receptors. Circ. Res. 2003, 92, 485–492. [Google Scholar] [CrossRef]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating Neutrophils Mediate the Initial Angiogenic Switch in a Mouse Model of Multistage Carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The Role of Vascular Endothelial Growth Factor in the Hypoxic and Immunosuppressive Tumor Microenvironment: Perspectives for Therapeutic Implications. Med. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of Myeloid Leukocytes and Soluble Mediators in Pancreatic Cancer: Importance of Myeloid-Derived Suppressor Cells. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.-P.; Ferrara, N. Tumor Refractoriness to Anti-VEGF Treatment Is Mediated by CD11b+Gr1+ Myeloid Cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 Improves Myeloid Dendritic Cell–Mediated Antitumor Immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical Significance of Defective Dendritic Cell Differentiation in Cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar] [PubMed]
- Alfaro, C.; Suarez, N.; Gonzalez, A.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervas-Stubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of Bevacizumab, Sunitinib and Sorafenib as Single Agents or in Combination on the Inhibitory Effects of VEGF on Human Dendritic Cell Differentiation from Monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular Endothelial Growth Factor Affects Dendritic Cell Maturation Through the Inhibition of Nuclear Factor-ΚB Activation in Hemopoietic Progenitor Cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar] [PubMed]
- Dikov, M.; Ohm, J.; Ray, N.; Tchekneva, E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D. Differential Roles of Vascular Endothelial Growth Factor Receptors 1 and 2 in Dendritic Cell Differentiation. J. Immunol. 2005, 174, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, T.; Olson, J.; Johnson, R.S.; Nizet, V. HIF-1α Influences Myeloid Cell Antigen Presentation and Response to Subcutaneous OVA Vaccination. J. Mol. Med. Berl. Ger. 2013, 91, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.W.; Gold, M.J.; Garcia-Batres, C.; Tai, K.; Elford, A.R.; Himmel, M.E.; Elia, A.J.; Ohashi, P.S. Hypoxia-Inducible Factor 1 Alpha Limits Dendritic Cell Stimulation of CD8 T Cell Immunity. PLoS ONE 2020, 15, e0244366. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF Inhibits T-Cell Development and May Contribute to Tumor-Induced Immune Suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF Directly Suppresses Activation of T Cells from Ascites Secondary to Ovarian Cancer via VEGF Receptor Type 2. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF Cooperatively Control the Immunotolerant Tumor Environment and the Efficacy of Cancer Immunotherapies. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 Deficiency on CD4+Foxp3+ Regulatory T Cells Impairs Mouse Melanoma Growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [PubMed]
- Assoun, S.; Brosseau, S.; Steinmetz, C.; Gounant, V.; Zalcman, G. Bevacizumab in Advanced Lung Cancer: State of the Art. Future Oncol. 2017, 13. [Google Scholar] [CrossRef]
- Aguiar, R.B.; de Moraes, J.Z. de Exploring the Immunological Mechanisms Underlying the Anti-Vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the Immune Response by Pro-Angiogenic Factors. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef]
- Wu, J.-B.; Tang, Y.-L.; Liang, X.-H. Targeting VEGF Pathway to Normalize the Vasculature: An Emerging Insight in Cancer Therapy. OncoTargets Ther. 2018, 11, 6901–6909. [Google Scholar] [CrossRef]
- Ricci, V.; Ronzoni, M.; Fabozzi, T. Aflibercept a New Target Therapy in Cancer Treatment: A Review. Crit. Rev. Oncol. Hematol. 2015, 96, 569–576. [Google Scholar] [CrossRef]
- Cyramza. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/cyramza (accessed on 26 January 2021).
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab Strongly Impacts Effector/ Activated Regulatory T Cells and CD8+ T Cells in the Tumor Microenvironment. J. Immunother. Cancer 2018, 6. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent Advances on Anti-Angiogenesis Receptor Tyrosine Kinase Inhibitors in Cancer Therapy. J. Hematol. Oncol. J Hematol Oncol 2019, 12, 27. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and Development of Sorafenib: A Multikinase Inhibitor for Treating Cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.-Y.; Chen, S.-H. The Novel Role of Tyrosine Kinase Inhibitor in the Reversal of Immune Suppression and Modulation of Tumor Microenvironment for Immune-Based Cancer Therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef]
- Chen, M.-L.; Yan, B.-S.; Lu, W.-C.; Chen, M.-H.; Yu, S.-L.; Yang, P.-C.; Cheng, A.-L. Sorafenib Relieves Cell-Intrinsic and Cell-Extrinsic Inhibitions of Effector T Cells in Tumor Microenvironment to Augment Antitumor Immunity. Int. J. Cancer 2014, 134, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V.; et al. A Decrease of Regulatory T Cells Correlates With Overall Survival After Sunitinib-Based Antiangiogenic Therapy in Metastatic Renal Cancer Patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic Targeting of the Angiopoietin-TIE Pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF Bispecific Antibody Reprograms Macrophages and Resident Microglia to Anti-Tumor Phenotype and Prolongs Glioblastoma Survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast Growth Factors (FGFs) in Cancer: FGF Traps as a New Therapeutic Approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the Fibroblast Growth Factor Receptor (FGFR) Pathway: The Current Landscape and Barriers to Clinical Application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef]
- Bockhorn, M.; Tsuzuki, Y.; Xu, L.; Frilling, A.; Broelsch, C.E.; Fukumura, D. Differential Vascular and Transcriptional Responses to Anti-Vascular Endothelial Growth Factor Antibody in Orthotopic Human Pancreatic Cancer Xenografts. Clin. Cancer Res. 2003, 9, 4221–4226. [Google Scholar]
- Meurer, S.K.; Weiskirchen, R. Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Ollauri-Ibáñez, C.; Núñez-Gómez, E.; Egido-Turrión, C.; Silva-Sousa, L.; Díaz-Rodríguez, E.; Rodríguez-Barbero, A.; López-Novoa, J.M.; Pericacho, M. Continuous Endoglin (CD105) Overexpression Disrupts Angiogenesis and Facilitates Tumor Cell Metastasis. Angiogenesis 2020, 23, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.; Jha, A.K.; Ameri, K.; Marcus, S.G.; Yeghiazarians, Y.; Healy, K.E. TGF-Β1/CD105 Signaling Controls Vascular Network Formation within Growth Factor Sequestering Hyaluronic Acid Hydrogels. PLoS ONE 2018, 13, e0194679. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.; Shi, Y.; et al. Vessel Co-Option Mediates Resistance to Anti-Angiogenic Therapy in Liver Metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef]
- Avastin. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/avastin (accessed on 23 April 2021).
- Zaltrap. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zaltrap (accessed on 26 January 2021).
- Carrato, A.; Swieboda-Sadlej, A.; Staszewska-Skurczynska, M.; Lim, R.; Roman, L.; Shparyk, Y.; Bondarenko, I.; Jonker, D.J.; Sun, Y.; De la Cruz, J.A.; et al. Fluorouracil, Leucovorin, and Irinotecan plus Either Sunitinib or Placebo in Metastatic Colorectal Cancer: A Randomized, Phase III Trial. J. Clin. Oncol. 2013, 31, 1341–1347. [Google Scholar] [CrossRef]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined Antiangiogenic and Anti-PD-L1 Therapy Stimulates Tumor Immunity through HEV Formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous Blockade of Programmed Death 1 and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) Induces Synergistic Anti-Tumour Effect in Vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, C. Anti-VEGF/VEGFR2 Monoclonal Antibodies and Their Combinations with PD-1/PD-L1 Inhibitors in Clinic. Curr. Cancer Drug Targets 2020, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geindreau, M.; Ghiringhelli, F.; Bruchard, M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. Int. J. Mol. Sci. 2021, 22, 4871. https://doi.org/10.3390/ijms22094871
Geindreau M, Ghiringhelli F, Bruchard M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. International Journal of Molecular Sciences. 2021; 22(9):4871. https://doi.org/10.3390/ijms22094871
Chicago/Turabian StyleGeindreau, Mannon, François Ghiringhelli, and Mélanie Bruchard. 2021. "Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response" International Journal of Molecular Sciences 22, no. 9: 4871. https://doi.org/10.3390/ijms22094871
APA StyleGeindreau, M., Ghiringhelli, F., & Bruchard, M. (2021). Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. International Journal of Molecular Sciences, 22(9), 4871. https://doi.org/10.3390/ijms22094871