
A Proteomic Approach for Systematic Mapping of Substrates of Human Deubiquitinating Enzymes

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Results and Discussion
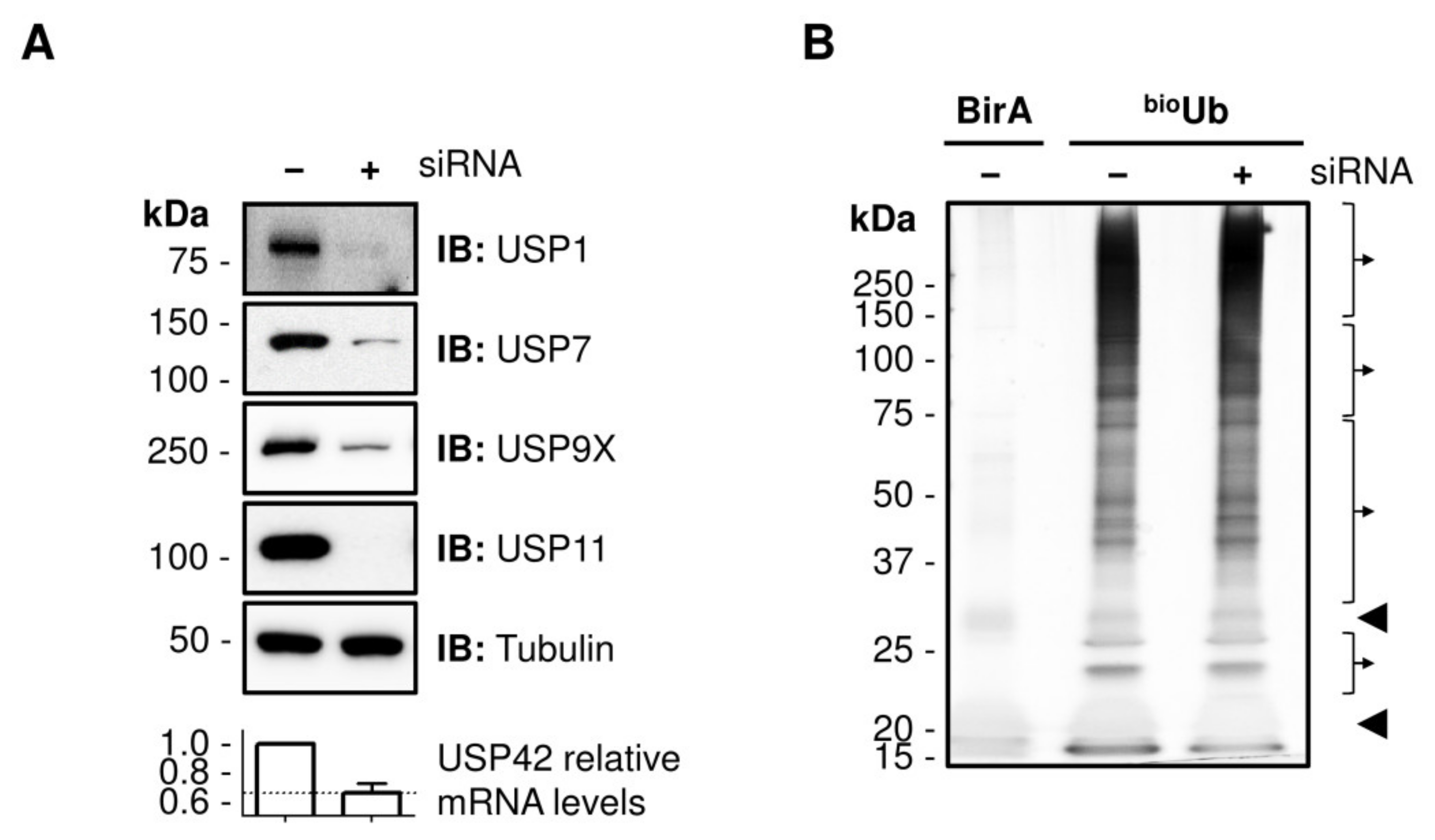
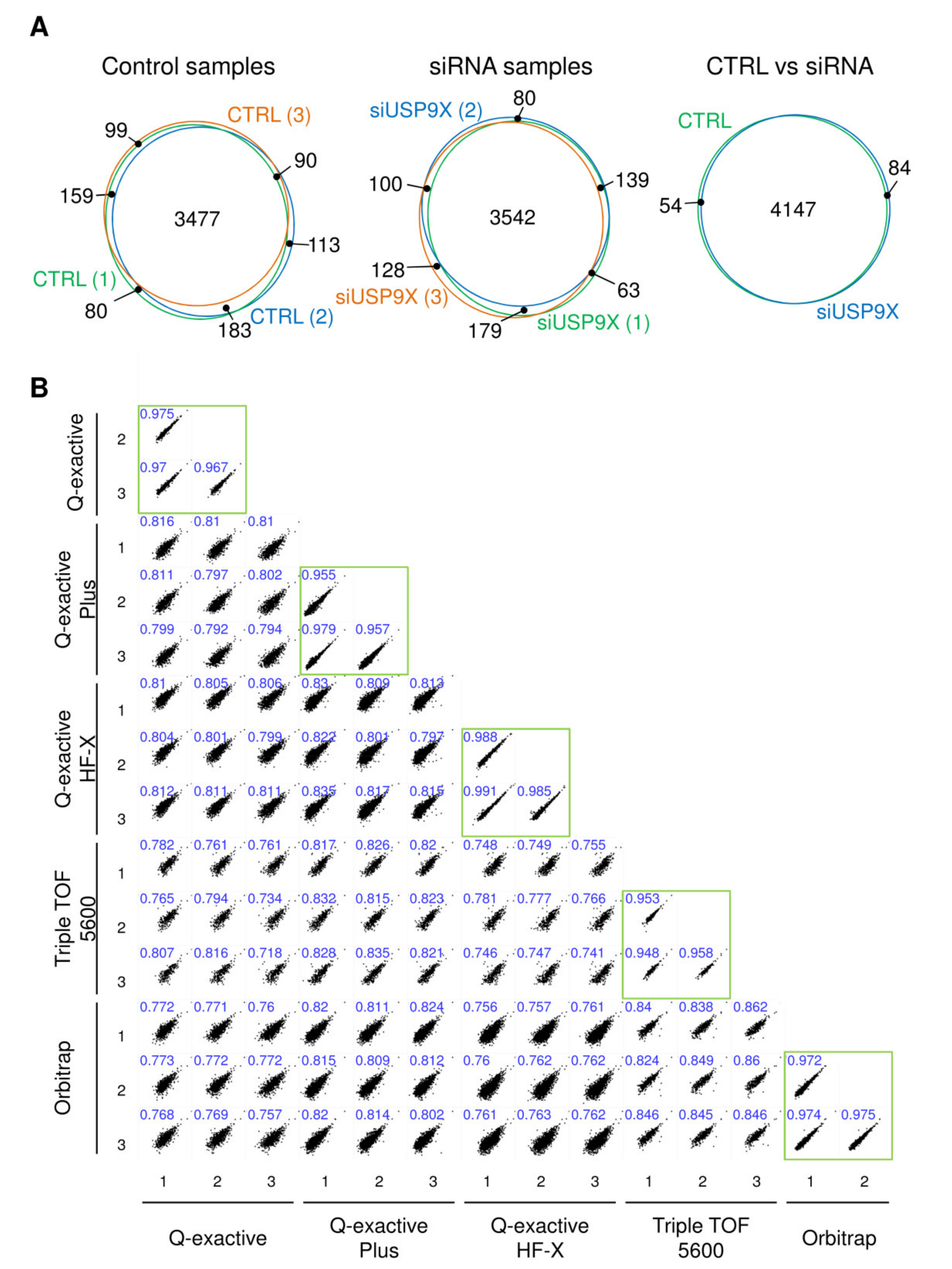
2.1. Identification of DUB Substrates in Cell Culture
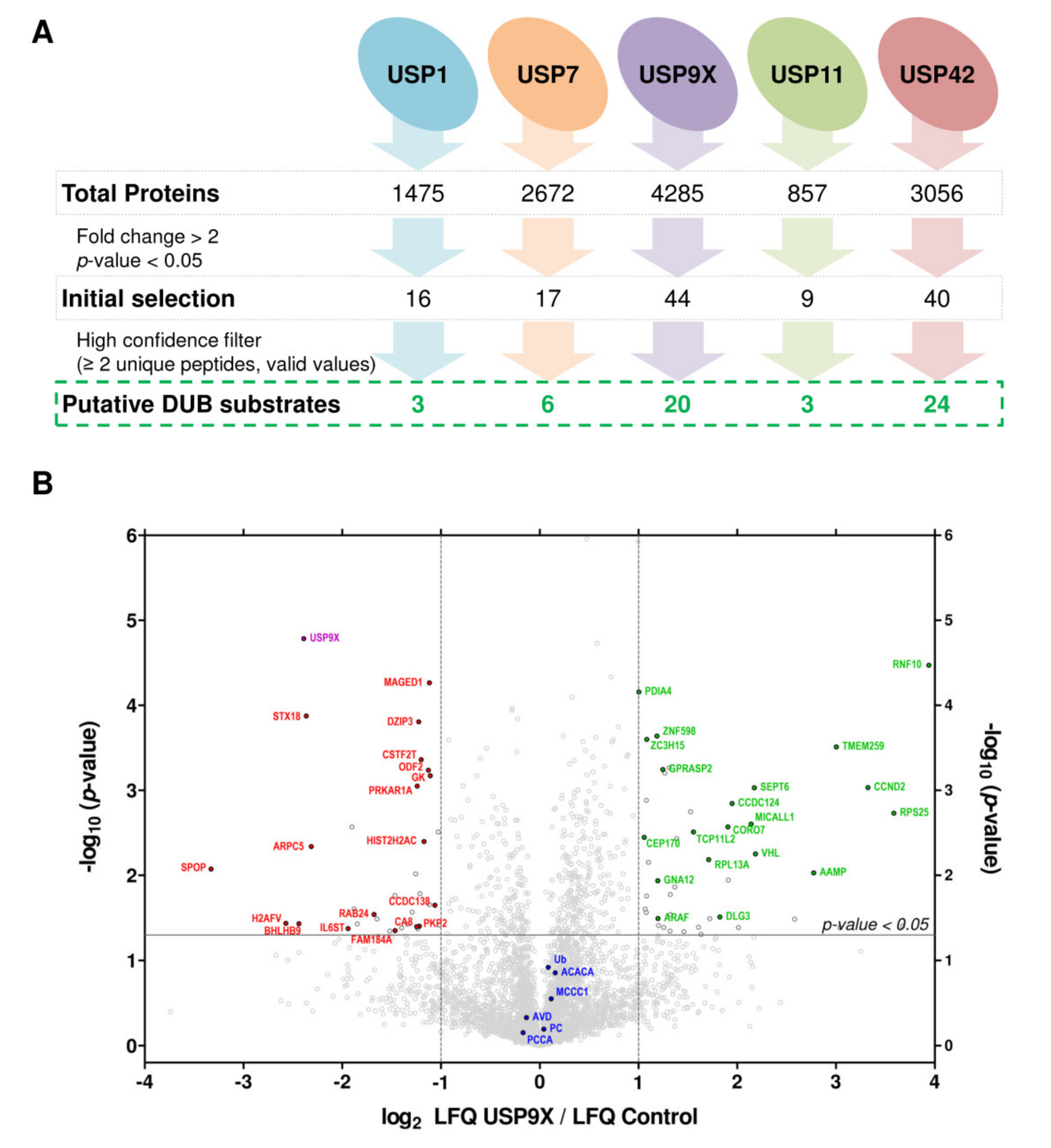
2.2. Large Scale Detection of In Vivo Substrates for USP1, USP7, USP9X, USP11 and USP42
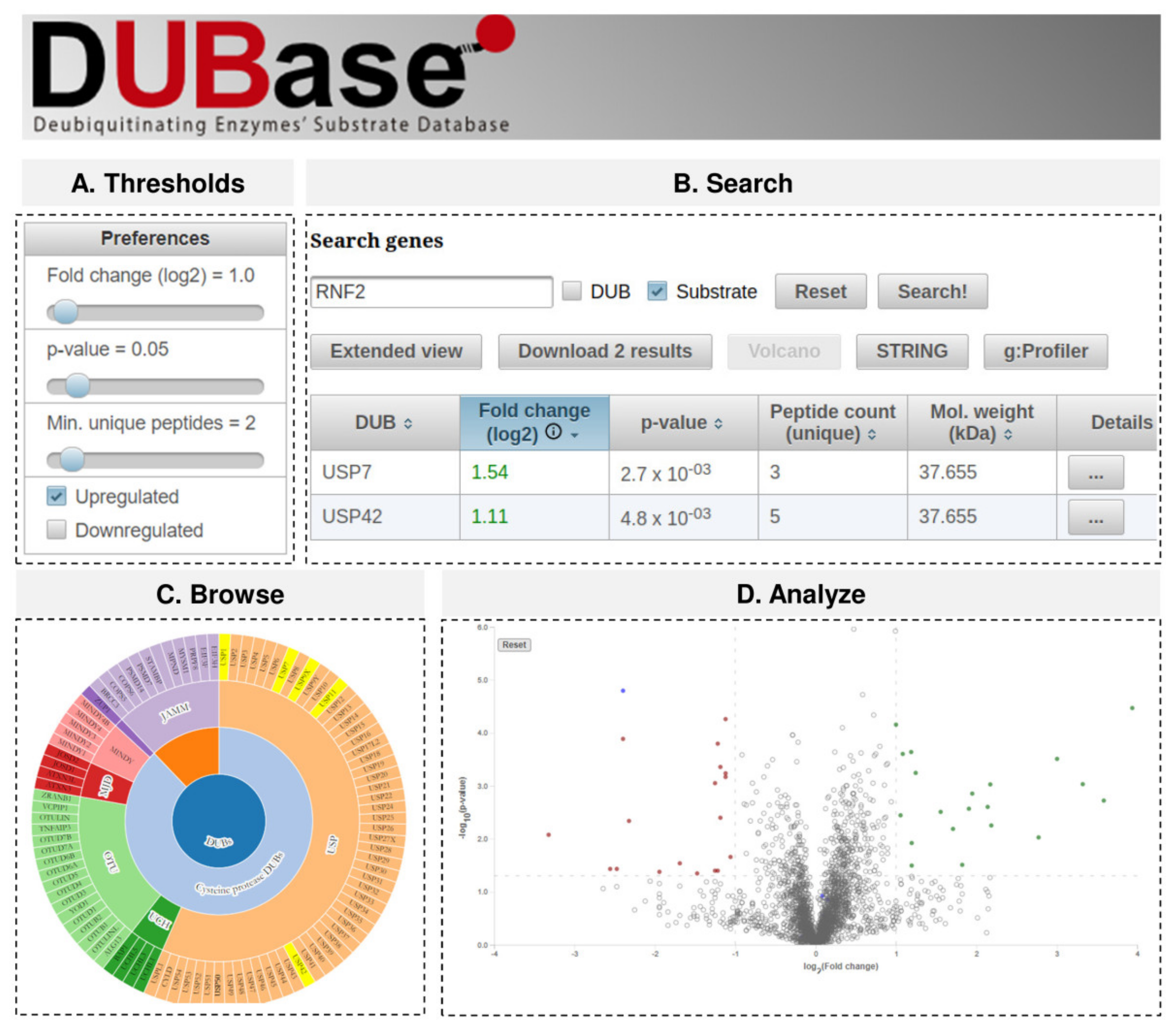
2.3. DUBase: A Novel Database for DUB Substrates
3. Conclusions
4. Materials and Methods
4.1. Cell Culture and RNA Silencing
4.2. Real-Time Quantitative PCR
- Forward: 5-TGTGGGCATCAATGGATTTGG-3′
- Reverse: 5-ACACCATGTATTCCGGGTCAAT-3′
- Forward: 5-AATCTTCAGACCCATCAGCCT-3′
- Reverse: 5-AGAACCTGCATCCATGTCTCC-3′
4.3. Western Blot
4.4. Biotin Pull-Down
4.5. Silver Staining
4.6. In-Gel Trypsin Digestion and Peptide Extraction
4.7. LC-MS/MS Analysis
4.8. Data Processing and Bioinformatics Analysis
4.9. Data Analysis
4.10. Web Application
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the Polyubiquitin Proteolytic Signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Terrell, J.; Shih, S.; Dunn, R.; Hicke, L. A Function for Monoubiquitination in the Internalization of a G Protein-Coupled Receptor. Mol. Cell 1998, 1, 193–202. [Google Scholar] [CrossRef]
- Flick, K.; Raasi, S.; Zhang, H.; Yen, J.L.; Kaiser, P. A Ubiquitin-Interacting Motif Protects Polyubiquitinated Met4 from Degradation by the 26S Proteasome. Nat. Cell Biol. 2006, 8, 509–515. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the Chains: Deubiquitylating Enzyme Specificity Begets Function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- Kimura, Y.; Tanaka, K. Regulatory Mechanisms Involved in the Control of Ubiquitin Homeostasis. J. Biochem. 2010, 147, 793–798. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. P53 Ubiquitination: Mdm2 and Beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Worden, E.J.; Dong, K.C.; Martin, A. An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol. Cell 2017, 67, 799–811. [Google Scholar] [CrossRef]
- Chen, H.; Polo, S.; Di Fiore, P.P.; De Camilli, P.V. Rapid Ca2+-Dependent Decrease of Protein Ubiquitination at Synapses. Proc. Natl. Acad. Sci. USA 2003, 100, 14908–14913. [Google Scholar] [CrossRef]
- Rinetti, G.V.; Schweizer, F.E. Ubiquitination Acutely Regulates Presynaptic Neurotransmitter Release in Mammalian Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3157–3166. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING Finger Families of E3 Ubiquitin Ligases at a Glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson Disease Gene Product, Parkin, Is a Ubiquitin-Protein Ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in Disease Pathogenesis and Treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP Mutations Cause Angelman Syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef]
- Osinalde, N.; Duarri, A.; Ramirez, J.; Barrio, R.; Perez de Nanclares, G.; Mayor, U. Impaired Proteostasis in Rare Neurological Diseases. Semin. Cell Dev. Biol. 2019, 93, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The Mitochondrial Deubiquitinase USP30 Opposes Parkin-Mediated Mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A Proteomics Approach to Understanding Protein Ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global Analysis of Lysine Ubiquitination by Ubiquitin Remnant Immunoaffinity Profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using Ubiquitin Binders to Decipher the Ubiquitin Code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef]
- Franco, M.; Seyfried, N.T.; Brand, A.H.; Peng, J.; Mayor, U. A Novel Strategy to Isolate Ubiquitin Conjugates Reveals Wide Role for Ubiquitination during Neural Development. Mol. Cell. Proteom. 2011, 10, M110.002188. [Google Scholar] [CrossRef]
- Mayor, U.; Peng, J. Deciphering Tissue-Specific Ubiquitylation by Mass Spectrometry. Methods Mol. Biol. Clifton 2012, 832, 65–80. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ramirez, J.; Franco, M.; Lectez, B.; Gonzalez, M.; Barrio, R.; Mayor, U. Ube3a, the E3 Ubiquitin Ligase Causing Angelman Syndrome and Linked to Autism, Regulates Protein Homeostasis through the Proteasomal Shuttle Rpn10. Cell. Mol. Life Sci. 2014, 71, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Min, M.; Mayor, U.; Dittmar, G.; Lindon, C. Using in Vivo Biotinylated Ubiquitin to Describe a Mitotic Exit Ubiquitome from Human Cells. Mol. Cell. Proteom. 2014, 13, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin Profiling in Liver Using a Transgenic Mouse with Biotinylated Ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef]
- Ramirez, J.; Martinez, A.; Lectez, B.; Lee, S.Y.; Franco, M.; Barrio, R.; Dittmar, G.; Mayor, U. Proteomic Analysis of the Ubiquitin Landscape in the Drosophila Embryonic Nervous System and the Adult Photoreceptor Cells. PLoS ONE 2015, 10, e0139083. [Google Scholar] [CrossRef]
- Ramirez, J.; Min, M.; Barrio, R.; Lindon, C.; Mayor, U. Isolation of Ubiquitinated Proteins to High Purity from In Vivo Samples. Methods Mol. Biol. Clifton 2016, 1449, 193–202. [Google Scholar] [CrossRef]
- Martinez, A.; Lectez, B.; Ramirez, J.; Popp, O.; Sutherland, J.D.; Urbé, S.; Dittmar, G.; Clague, M.J.; Mayor, U. Quantitative Proteomic Analysis of Parkin Substrates in Drosophila Neurons. Mol. Neurodegener. 2017, 12, 29. [Google Scholar] [CrossRef]
- Ramirez, J.; Lectez, B.; Osinalde, N.; Sivá, M.; Elu, N.; Aloria, K.; Procházková, M.; Perez, C.; Martínez-Hernández, J.; Barrio, R.; et al. Quantitative Proteomics Reveals Neuronal Ubiquitination of Rngo/Ddi1 and Several Proteasomal Subunits by Ube3a, Accounting for the Complexity of Angelman Syndrome. Hum. Mol. Genet. 2018, 27, 1955–1971. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, J.; Morales, M.; Osinalde, N.; Martínez-Padrón, I.; Mayor, U.; Ferrús, A. The Ubiquitin Ligase Ariadne-1 Regulates Neurotransmitter Release via Ubiquitination of NSF. J. Biol. Chem. 2021, 296, 100408. [Google Scholar] [CrossRef] [PubMed]
- Elu, N.; Osinalde, N.; Beaskoetxea, J.; Ramirez, J.; Lectez, B.; Aloria, K.; Rodriguez, J.A.; Arizmendi, J.M.; Mayor, U. Detailed Dissection of UBE3A-Mediated DDI1 Ubiquitination. Front. Physiol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A Minimal Peptide Substrate in Biotin Holoenzyme Synthetase-Catalyzed Biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Nijman, S.M.B.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of Monoubiquitinated PCNA by DUB Autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- García-Santisteban, I.; Peters, G.J.; Giovannetti, E.; Rodríguez, J.A. USP1 Deubiquitinase: Cellular Functions, Regulatory Mechanisms and Emerging Potential as Target in Cancer Therapy. Mol. Cancer 2013, 12, 91. [Google Scholar] [CrossRef]
- Jelluma, N.; Brenkman, A.B.; van den Broek, N.J.F.; Cruijsen, C.W.A.; van Osch, M.H.J.; Lens, S.M.A.; Medema, R.H.; Kops, G.J.P.L. Mps1 Phosphorylates Borealin to Control Aurora B Activity and Chromosome Alignment. Cell 2008, 132, 233–246. [Google Scholar] [CrossRef]
- Valles, G.J.; Bezsonova, I.; Woodgate, R.; Ashton, N.W. USP7 Is a Master Regulator of Genome Stability. Front. Cell Dev. Biol. 2020, 8, 717. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, S.; Ogata, T.; Fujiwara, S.; Nakamura, K.; Tanaka, S. A Novel Function of RING Finger Protein 10 in Transcriptional Regulation of the Myelin-Associated Glycoprotein Gene and Myelin Formation in Schwann Cells. PLoS ONE 2008, 3, e3464. [Google Scholar] [CrossRef]
- Murtaza, M.; Jolly, L.A.; Gecz, J.; Wood, S.A. La FAM Fatale: USP9X in Development and Disease. Cell. Mol. Life Sci. 2015, 72, 2075–2089. [Google Scholar] [CrossRef]
- Whitcomb, E.A.; Dudek, E.J.; Liu, Q.; Taylor, A. Novel Control of S Phase of the Cell Cycle by Ubiquitin-Conjugating Enzyme H7. Mol. Biol. Cell 2009, 20, 1–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y.; et al. Deubiquitylation and Stabilization of P21 by USP11 Is Critical for Cell-Cycle Progression and DNA Damage Responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef]
- Wakasugi, K.; Slike, B.M.; Hood, J.; Otani, A.; Ewalt, K.L.; Friedlander, M.; Cheresh, D.A.; Schimmel, P. A Human Aminoacyl-TRNA Synthetase as a Regulator of Angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 173–177. [Google Scholar] [CrossRef]
- Liu, S.; Wang, T.; Shi, Y.; Bai, L.; Wang, S.; Guo, D.; Zhang, Y.; Qi, Y.; Chen, C.; Zhang, J.; et al. USP42 Drives Nuclear Speckle MRNA Splicing via Directing Dynamic Phase Separation to Promote Tumorigenesis. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Reimand, J.; Arak, T.; Vilo, J. G:Profiler—A Web Server for Functional Interpretation of Gene Lists (2011 Update). Nucleic Acids Res. 2011, 39, W307–W315. [Google Scholar] [CrossRef] [PubMed]
- Osinalde, N.; Sánchez-Quiles, V.; Akimov, V.; Blagoev, B.; Kratchmarova, I. SILAC-Based Quantification of Changes in Protein Tyrosine Phosphorylation Induced by Interleukin-2 (IL-2) and IL-15 in T-Lymphocytes. Data Brief 2015, 5, 53–58. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Silenced DUB | Gene Name | Description | FC 1 |
|---|---|---|---|
| USP1 | BCOR | BCL-6 corepressor | 2.43 |
| PCNA | Proliferating cell nuclear antigen | 1.49 | |
| GLUL | Glutamine synthetase | 1.12 | |
| USP7 | TTK | Dual specificity protein kinase TTK | 2.33 |
| GABPA | GA-binding protein alpha chain | 1.66 | |
| RPL29 | 60S ribosomal protein L29 | 1.61 | |
| RNF2 | E3 ubiquitin-protein ligase RING2 | 1.54 | |
| CREB1 | Cyclic AMP-responsive element-binding protein 1 | 1.44 | |
| MARCKS | Myristoylated alanine-rich C-kinase substrate | 1.21 | |
| USP9X | RNF10 | RING finger protein 10 | 3.94 |
| RPS25 | 40S ribosomal protein S25 | 3.59 | |
| CCND2 | G1/S-specific cyclin-D2 | 3.32 | |
| TMEM259 | Membralin | 3.00 | |
| AAMP | Angio-associated migratory cell protein | 2.77 | |
| VHL | Von Hippel-Lindau disease tumor suppressor | 2.18 | |
| SEPT6 | Septin-6 | 2.17 | |
| MICALL1 | MICAL-like protein 1 | 2.14 | |
| CCDC124 | Coiled-coil domain-containing protein 124 | 1.95 | |
| CORO7 | Coronin-7 | 1.91 | |
| DLG3 | Disks large homolog 3 | 1.82 | |
| RPL13A | 60S ribosomal protein L13a | 1.71 | |
| TCP11L2 | T-complex protein 11-like protein 2 | 1.56 | |
| GPRASP2 | G-protein coupled receptor-associated sorting protein 2 | 1.25 | |
| ARAF | Serine/threonine-protein kinase A-Raf | 1.20 | |
| GNA12 | Guanine nucleotide-binding protein subunit alpha-12 | 1.19 | |
| ZNF598 | Zinc finger protein 598 | 1.19 | |
| ZC3H15 | Zinc finger CCCH domain-containing protein 15 | 1.08 | |
| CEP170 | Centrosomal protein of 170 kDa | 1.06 | |
| PDIA4 | Protein disulfide-isomerase A4 | 1.00 | |
| USP11 | RPS2 | 40S ribosomal protein S2 | 4.17 |
| UBE2L3 | Ubiquitin-conjugating enzyme E2 L3 | 2.15 | |
| RPS7 | 40S ribosomal protein S7 | 1.33 | |
| USP42 | SLC20A1 | Sodium-dependent phosphate transporter 1 | 4.09 |
| WARS | Tryptophan--tRNA ligase, cytoplasmic | 3.86 | |
| CUL2 | Cullin-2 | 2.94 | |
| UBR7 | Putative E3 ubiquitin-protein ligase UBR7 | 2.86 | |
| CPD | Carboxypeptidase D | 2.31 | |
| ARHGAP17 | Rho GTPase-activating protein 17 | 2.30 | |
| SETD3 | Histone-lysine N-methyltransferase setd3 | 2.10 | |
| GTF2H1 | General transcription factor IIH subunit 1 | 2.04 | |
| CORO1C | Coronin-1C | 1.95 | |
| HMOX1 | Heme oxygenase 1 | 1.69 | |
| PRKAR2A | cAMP-dependent protein kinase type II-alpha regulatory subunit | 1.65 | |
| DNAJC21 | DnaJ homolog subfamily C member 21 | 1.52 | |
| FUBP3 | Far upstream element-binding protein 3 | 1.40 | |
| PES1 | Pescadillo homolog | 1.38 | |
| SMARCD2 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member 2 | 1.36 | |
| LSR | Lipolysis-stimulated lipoprotein receptor | 1.26 | |
| IFT81 | Intraflagellar transport protein 81 homolog | 1.25 | |
| RAD50 | DNA repair protein RAD50 | 1.23 | |
| RNF2 | E3 ubiquitin-protein ligase RING2 | 1.11 | |
| PJA2 | E3 ubiquitin-protein ligase Praja-2 | 1.10 | |
| PSMC6 | 26S protease regulatory subunit 10B | 1.07 | |
| BUB3 | Mitotic checkpoint protein BUB3 | 1.06 | |
| BTBD2 | BTB/POZ domain-containing protein 2 | 1.05 | |
| FABP5 | Fatty acid-binding protein, epidermal | 1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, J.; Prieto, G.; Olazabal-Herrero, A.; Borràs, E.; Fernandez-Vigo, E.; Alduntzin, U.; Osinalde, N.; Beaskoetxea, J.; Lectez, B.; Aloria, K.; et al. A Proteomic Approach for Systematic Mapping of Substrates of Human Deubiquitinating Enzymes. Int. J. Mol. Sci. 2021, 22, 4851. https://doi.org/10.3390/ijms22094851
Ramirez J, Prieto G, Olazabal-Herrero A, Borràs E, Fernandez-Vigo E, Alduntzin U, Osinalde N, Beaskoetxea J, Lectez B, Aloria K, et al. A Proteomic Approach for Systematic Mapping of Substrates of Human Deubiquitinating Enzymes. International Journal of Molecular Sciences. 2021; 22(9):4851. https://doi.org/10.3390/ijms22094851
Chicago/Turabian StyleRamirez, Juanma, Gorka Prieto, Anne Olazabal-Herrero, Eva Borràs, Elvira Fernandez-Vigo, Unai Alduntzin, Nerea Osinalde, Javier Beaskoetxea, Benoit Lectez, Kerman Aloria, and et al. 2021. "A Proteomic Approach for Systematic Mapping of Substrates of Human Deubiquitinating Enzymes" International Journal of Molecular Sciences 22, no. 9: 4851. https://doi.org/10.3390/ijms22094851
APA StyleRamirez, J., Prieto, G., Olazabal-Herrero, A., Borràs, E., Fernandez-Vigo, E., Alduntzin, U., Osinalde, N., Beaskoetxea, J., Lectez, B., Aloria, K., Rodriguez, J. A., Paradela, A., Sabidó, E., Muñoz, J., Corrales, F., Arizmendi, J. M., & Mayor, U. (2021). A Proteomic Approach for Systematic Mapping of Substrates of Human Deubiquitinating Enzymes. International Journal of Molecular Sciences, 22(9), 4851. https://doi.org/10.3390/ijms22094851