
Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Morphological and Physiological Differences under the Two N Conditions
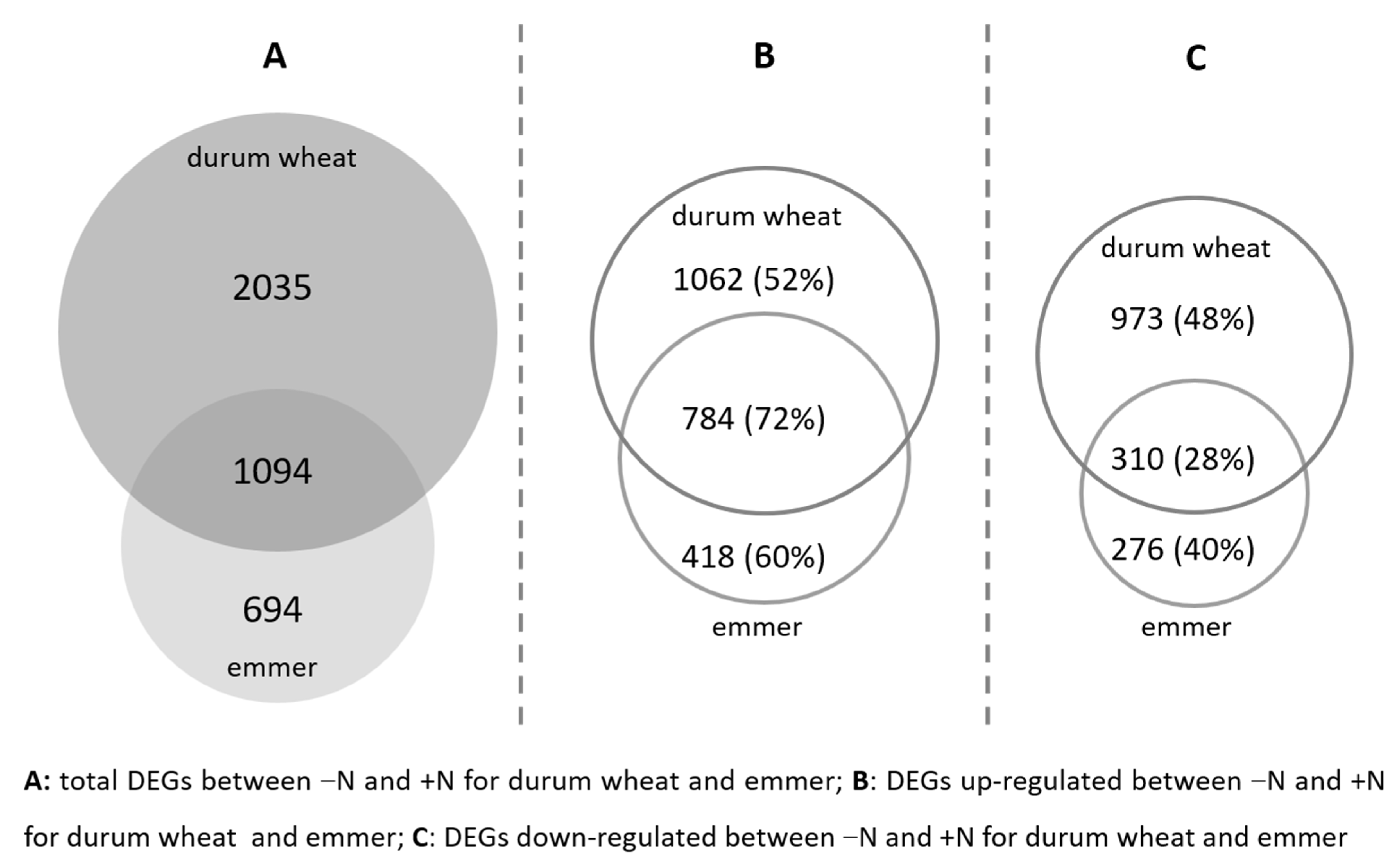
2.2. Transcriptomic Differences between the Two N Conditions
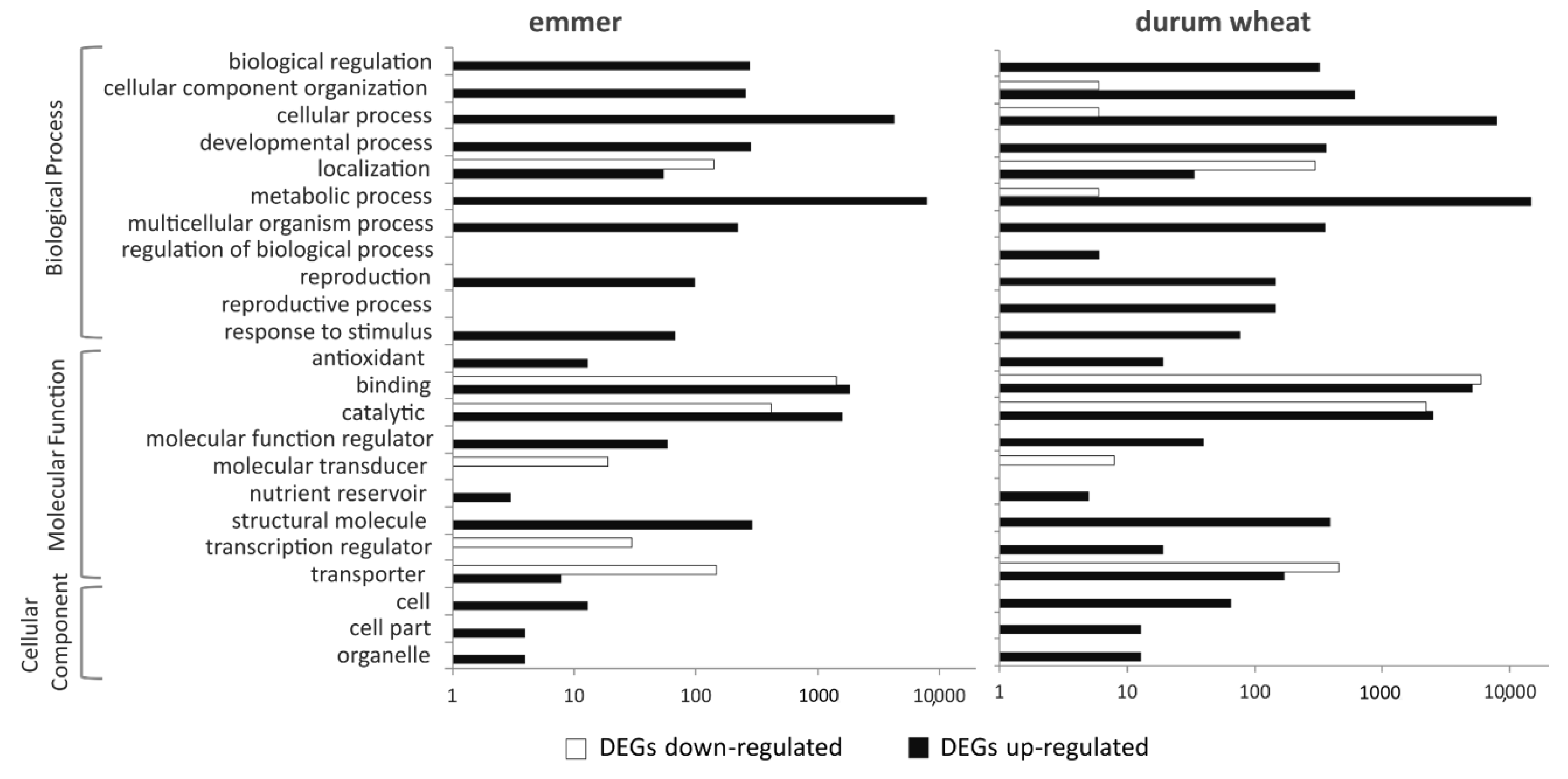
2.3. Functions of DEGs in Emmer and Durum Wheat between the Two N Conditions
2.4. GO Enrichment Analysis of DEGs
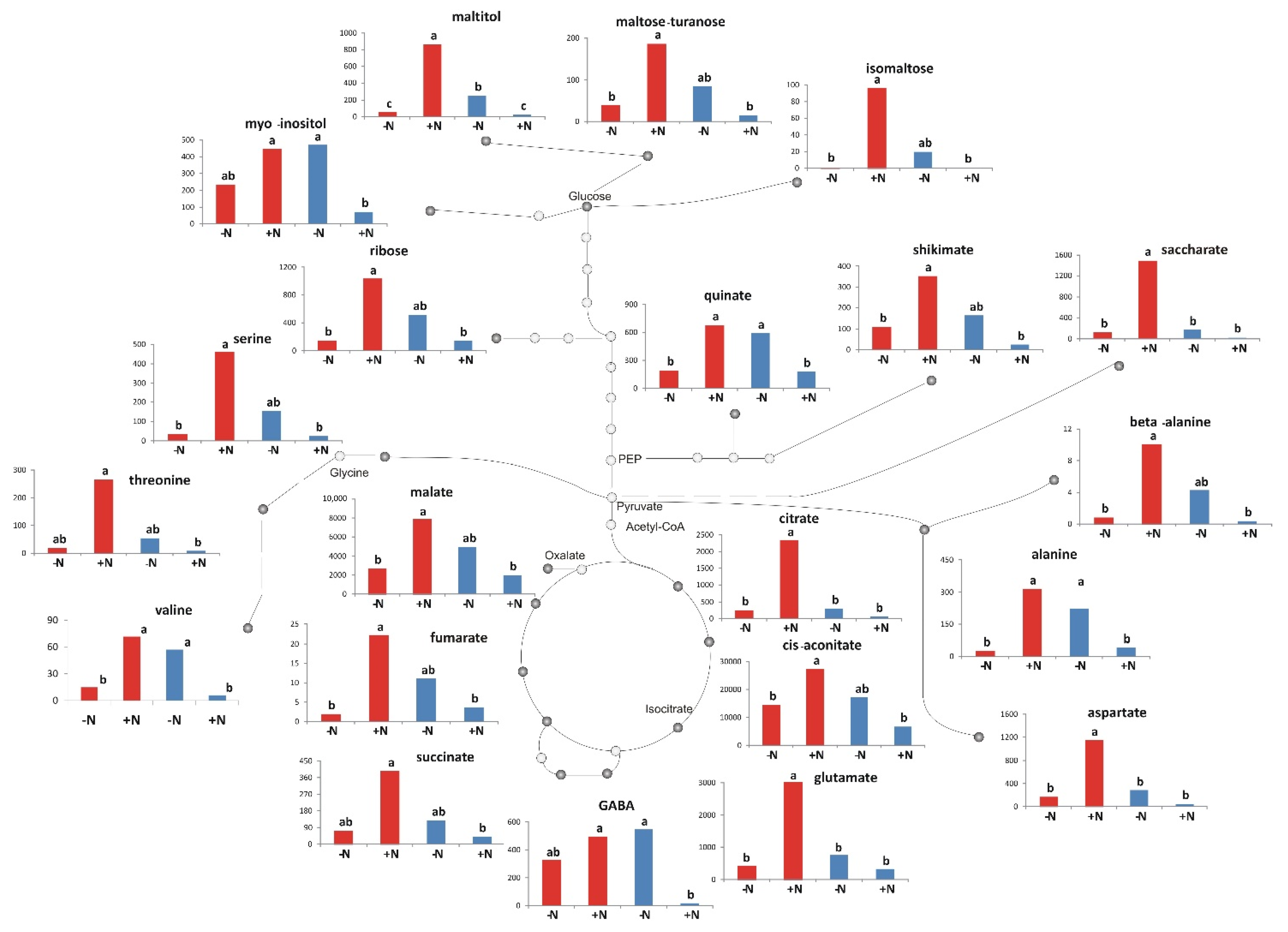
2.5. Metabolic Differences between the Two N Conditions
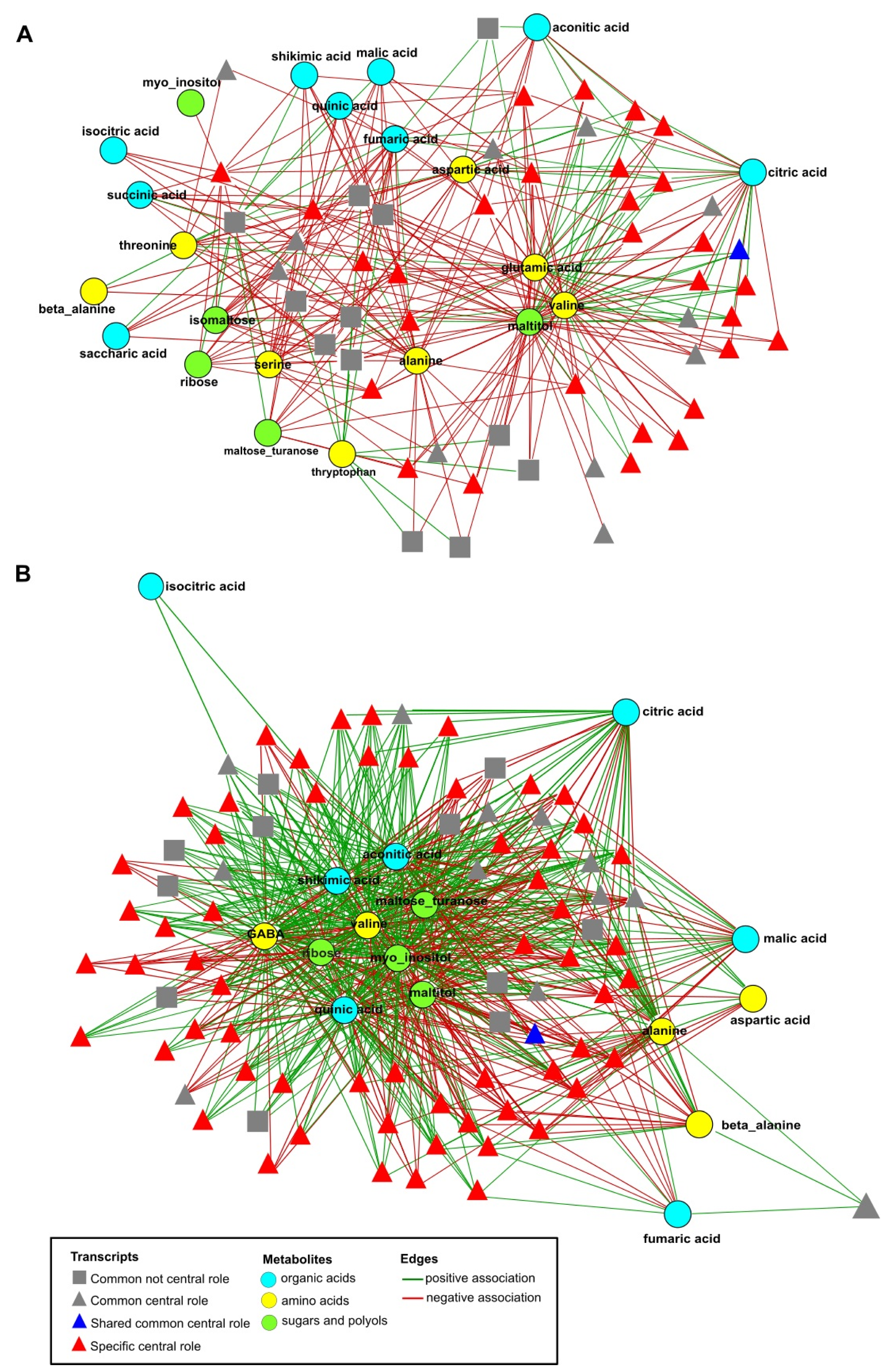
2.6. Network Analysis of Combined Data Sets
2.7. Function of DEGs Having a Central Role in the Networks
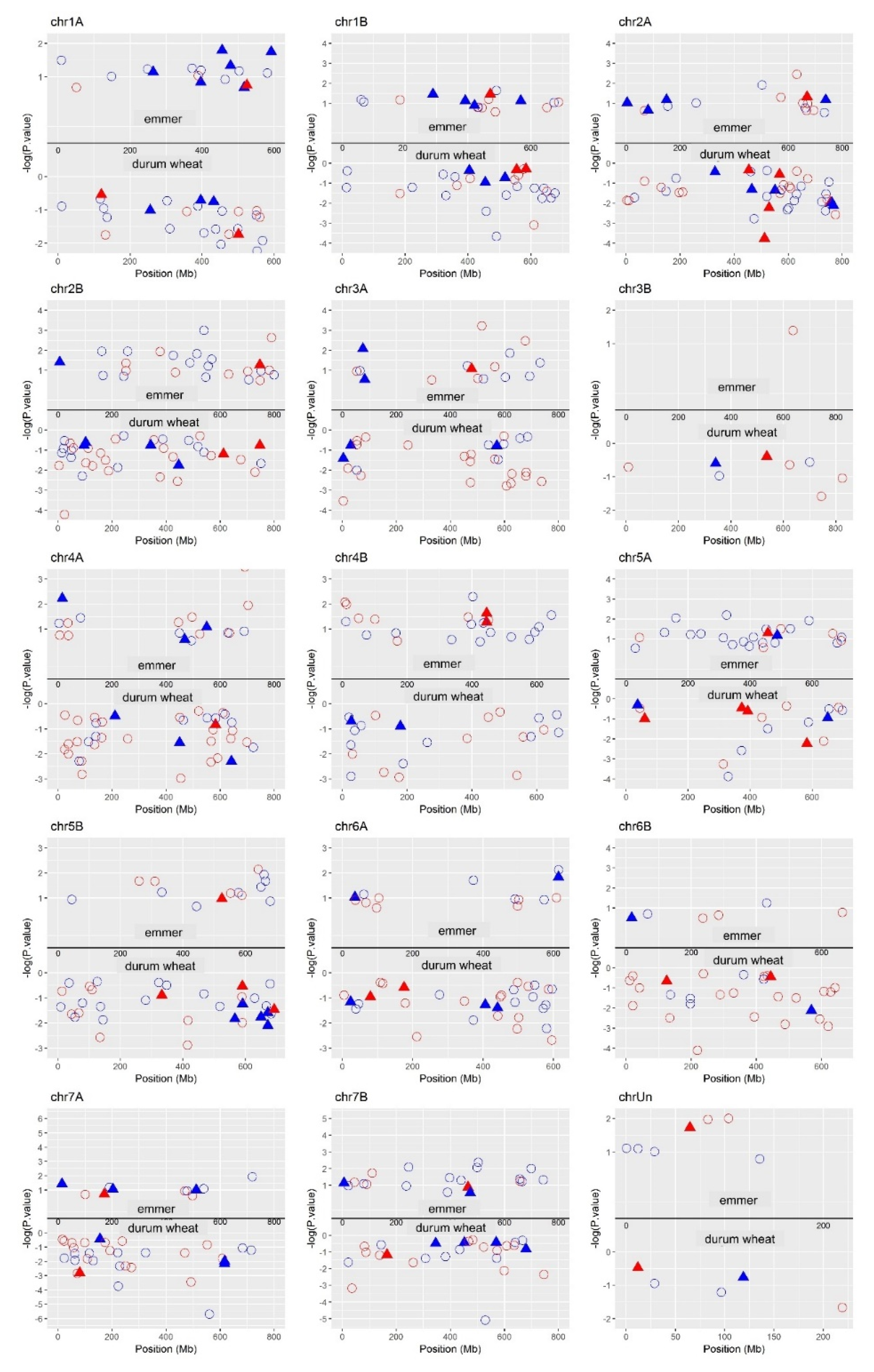
2.8. DEGs Position on the Genome
2.9. Environmental Effect
3. Discussion
4. Materials and Methods
4.1. Plant Material and Experimental Design
4.2. Phenotypic Traits
4.3. Transcriptomic Analysis
4.4. Metabolite Profiling
4.5. Statistical Analysis
4.6. Bioinformatics Analysis and Network Construction
4.6.1. Data Preprocessing
4.6.2. Analysis of Differential Expression
4.6.3. GO Enrichment Analysis
4.6.4. Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lea, P.J.; Azevedo, R.A. Nitrogen use efficency. 1. Uptake of nitrogen from the soil. Ann. Appl. Biol. 2006, 149, 243–247. [Google Scholar] [CrossRef]
- Miller, A.J.; Fan, X.; Orsel, M.; Smith, S.J.; Wells, D.M. Nitrate transport and signalling. J. Exp. Bot. 2007, 58, 2297–2306. [Google Scholar] [CrossRef]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef]
- Raun, W.R.; Johnson, G.V. Improving Nitrogen Use Efficiency for Cereal Production. Agron. J. 1999, 91, 357–363. [Google Scholar] [CrossRef]
- Cassman, K.G.; Dobermann, A.; Walters, D.T.; Yang, H. Meeting cereal demand while protecting natural resources and improving environmental quality. Annu. Rev. Environ. Resour. 2003, 28, 315–358. [Google Scholar] [CrossRef]
- Ayadi, S.; Karmous, C.; Hammami, Z.; Trifa, Y.; Rezgui, S. Variation of Durum Wheat Yield and Nitrogen Use Efficiency under Mediterranean Rainfed Environment. Intl. J. Agric. Crop Sci. 2014, 7, 693–699. [Google Scholar] [CrossRef]
- Ruisi, P.; Frangipane, B.; Amato, G.; Frenda, A.S.; Plaia, A.; Giambalvo, D.; Saia, S. Nitrogen uptake and nitrogen fertilizer recovery in old and modern wheat genotypes grown in the presence or absence of interspecific competition. Front. Plant Sci. 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, M.; Samuel, D. Wheat domestication: Archaeobotanical evidence. Science 1998, 279, 1431. [Google Scholar] [CrossRef]
- Tanno, K.I.; Willcox, G. How fast was wild wheat domesticated? Science 2006, 311, 1886. [Google Scholar] [CrossRef]
- Luo, M.-C.; Yang, Z.-L.; You, F.M.; Kawahara, T.; Waines, J.G.; Dvorak, J. The structure of wild and domesticated emmer wheat populations, gene flow between them, and the site of emmer domestication. Theor. Appl. Genet. 2007, 114, 947–959. [Google Scholar] [CrossRef]
- Nevo, E. Evolution of wild emmer wheat and crop improvement. J. Syst. Evol. 2014, 52, 673–696. [Google Scholar] [CrossRef]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food Security: The Challenge of Feeding 9 Billion People. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.-M.; Rothstein, S.J. Understanding plant response to nitrogen limitation for the improvement of crop nitrogen use efficiency. J. Exp. Bot. 2010, 62, 1499–1509. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant Nitrogen Assimilation and Use Efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef]
- Hawkesford, M.J. Genetic variation in traits for nitrogen use efficiency in wheat. J. Exp. Bot. 2017, 68, 2627–2632. [Google Scholar] [CrossRef]
- Gioia, T.; Nagel, K.A.; Beleggia, R.; Fragasso, M.; Ficco, D.B.M.; Pieruschka, R.; De Vita, P.; Fiorani, F.; Papa, R. Impact of domestication on the phenotypic architecture of durum wheat under contrasting nitrogen fertilization. J. Exp. Bot. 2015, 66, 5519–5530. [Google Scholar] [CrossRef]
- Krapp, A.; Berthomé, R.; Orsel, M.; Mercey-Boutet, S.; Yu, A.; Castaings, L.; Elftieh, S.; Major, H.; Renou, J.-P.; Daniel-Vedele, F. Arabidopsis roots show distinct temporal adaptation patterns towards nitrogen starvation. Plant Physiol. 2011, 157, 1255–1282. [Google Scholar] [CrossRef]
- Humbert, S.; Subedi, S.; Cohn, J.; Zeng, B.; Bi, Y.-M.; Chen, X.; Zhu, T.; McNicholas, P.D.; Rothstein, S.J. Genome-wide expression profiling of maize in response to individual and combined water and nitrogen stresses. BMC Genom. 2013, 14, 3. [Google Scholar] [CrossRef]
- Simons, M.; Saha, R.; Guillard, L.; Clément, G.; Armengaud, P.; Cañas, R.; Maranas, C.D.; Lea, P.J.; Hirel, B. Nitrogen-use efficiency in maize (Zea mays L.): From ’omics’ studies to metabolic modelling. J. Exp. Bot. 2014, 65, 5657–5671. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; Cigliano, R.A.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; De Paola, D.; Janni, M.; Curci, P.L.; Sonnante, G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS ONE 2017, 12, e0183253. [Google Scholar] [CrossRef]
- Yang, J.; Chen, X.; Zhu, C.; Peng, X.; He, X.; Fu, J.; Ouyang, L.; Bian, J.; Hu, L.; Sun, X.; et al. RNA-seq reveals differentially expressed genes of rice (Oryza sativa) spikelet in response to temperature interacting with nitrogen at meiosis stage. BMC Genom. 2015, 16, 959. [Google Scholar] [CrossRef]
- Gelli, M.; Duo, Y.; Konda, A.R.; Zhang, C.; Holding, D.; Dweikat, I. Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genom. 2014, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Xuan, H.; Yang, Y.; Wang, L.; Wei, L.; Wang, Y.; Kang, G. Transcription Analysis of Genes Encoding the Wheat Root Transporter NRT1 and NRT2 Families During Nitrogen Starvation. J. Plant Growth Regul. 2014, 33, 837–848. [Google Scholar] [CrossRef]
- Chen, R.; Tian, M.; Wu, X.; Huang, Y. Differential global gene expression changes in response to low nitrogen stress in two maize inbred lines with contrasting low nitrogen tolerance. Genes Genom. 2011, 33, 491–497. [Google Scholar] [CrossRef]
- Hao, Q.N.; Zhou, X.A.; Sha, A.H.; Wang, C.; Zhou, R.; Chen, S.L. Identification of genes associated with nitrogen-use efficiency by genome-wide transcriptional analysis of two soybean genotypes. BMC Genom. 2011, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Scheible, W.-R.; Morcuende, R.; Czechowski, T.; Fritz, C.; Osuna, D.; Palacios-Rojas, N.; Schindelasch, D.; Thimm, O.; Udvardi, M.K.; Stitt, M. Genome-Wide Reprogramming of Primary and Secondary Metabolism, Protein Synthesis, Cellular Growth Processes, and the Regulatory Infrastructure of Arabidopsis in Response to Nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar] [CrossRef]
- Amiour, N.; Imbaud, S.; Clément, G.; Agier, N.; Zivy, M.; Balot, B.; Balliau, T.; Armengaud, P.; Quilleré, I.; Cañas, R.; et al. The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J. Exp. Bot. 2012, 63, 5017–5033. [Google Scholar] [CrossRef]
- Bielecka, M.; Watanabe, M.; Morcuende, R.; Scheible, W.-R.; Hawkesford, M.J.; Hesse, H.; Hoefgen, R. Transcriptome and metabolome analysis of plant sulfate starvation and resupply provides novel information on transcriptional regulation of metabolism associated with sulfur, nitrogen and phosphorus nutritional responses in Arabidopsis. Front. Plant Sci. 2015, 5, 805. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Pérez, P.; Martínez-Carrasco, R.; Feil, R.; Lunn, J.E.; Watanabe, M.; Arrivault, S.; Stitt, M.; Hoefgen, R.; Morcuende, R. Metabolic and Transcriptional Analysis of Durum Wheat Responses to Elevated CO2 at Low and High Nitrate Supply. Plant Cell Physiol. 2016, 57, 2133–2146. [Google Scholar] [CrossRef]
- Yu, C.; Zhao, X.; Qi, G.; Bai, Z.; Wang, Y.; Wang, S.; Ma, Y.; Liu, Q.; Shumin, W.; Zhou, G. Integrated analysis of transcriptome and metabolites reveals an essential role of metabolic flux in starch accumulation under nitrogen starvation in duckweed. Biotechnol. Biofuels 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H.; et al. Elucidation of Gene-to-Gene and Metabolite-to-Gene Networks inArabidopsis by Integration of Metabolomics andTranscriptomics*. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar] [CrossRef]
- Caldana, C.; Degenkolbe, T.; Cuadros‒Inostroza, A.; Klie, S.; Sulpice, R.; Leisse, A.; Steinhauser, D.; Fernie, A.R.; Willmitzer, L.; Hannah, M.A. High‒density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 2011, 67, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, D.; Fernie, A.R.; Nikoloski, Z.; Fait, A. Network analysis: Tackling complex data to study plant metabolism. Trends Biotechnol. 2013, 31, 29–36. [Google Scholar] [CrossRef]
- Toubiana, D.; Xue, W.; Zhang, N.; Kremling, K.; Gur, A.; Pilosof, S.; Gibon, Y.; Stitt, M.; Buckler, E.S.; Fernie, A.R.; et al. Correlation-Based Network Analysis of Metabolite and Enzyme Profiles Reveals a Role of Citrate Biosynthesis in Modulating N and C Metabolism in Zea mays. Front. Plant Sci. 2016, 7, 1022. [Google Scholar] [CrossRef]
- Beleggia, R.; Rau, D.; Laidò, G.; Platani, C.; Nigro, F.; Fragasso, M.; De Vita, P.; Scossa, F.; Fernie, A.R.; Nikoloski, Z.; et al. Evolutionary Metabolomics Reveals Domestication-Associated Changes in Tetraploid Wheat Kernels. Mol. Biol. Evol. 2016, 33, 1740–1753. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.A.; Ficco, D.B.M.; Laidò, G.; Marone, D.; Papa, R.; Blanco, A.; Gadaleta, A.; De Vita, P.; Mastrangelo, A.M. A dense durum wheat T. dicoccum linkage map based on SNP markers for the study of seed morphology. Mol. Breed. 2014, 34, 1579–1597. [Google Scholar] [CrossRef]
- Huang, A.; Sang, Y.; Sun, W.; Fu, Y.; Yang, Z. Transcriptomic Analysis of Responses to Imbalanced Carbon: Nitrogen Availabilities in Rice Seedlings. PLoS ONE 2016, 11, e0165732. [Google Scholar] [CrossRef]
- Shaar-Moshe, L.; Hayouka, R.; Roessner, U.; Peleg, Z. Phenotypic and metabolic plasticity shapes life-history strategies under combinations of abiotic stresses. Plant Direct 2019, 3, e00113. [Google Scholar] [CrossRef] [PubMed]
- Tschoep, H.; Gibon, Y.; Carillo, P.; Armengaud, P.; Szecowka, M.; Nunes-Nesi, A.; Fernie, A.R.; Koehl, K.; Stitt, M. Adjustment of growth and central metabolism to a mild but sustained nitrogen-limitation inArabidopsis. Plant Cell Environ. 2009, 32, 300–318. [Google Scholar] [CrossRef]
- Bouché, N.; Fromm, H. GABA in plants: Just a metabolite? Trends Plant Sci. 2004, 9, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Fait, A.; Fromm, H.; Walter, D.; Galili, G.; Fernie, A.R. Highway or byway: The metabolic role of the GABA shunt in plants. Trends Plant Sci. 2008, 13, 14–19. [Google Scholar] [CrossRef]
- Beuve, N.; Rispail, N.; Laine, P.; Cliquet, J.B.; Ourry, A.; Le Deunff, E. Putative role of g-aminobutyric acid (GABA) as a long-distance signal in up-regulation of nitrate uptake in Brassica napus L. Plant Cell Environ. 2004, 27, 1035–1046. [Google Scholar] [CrossRef]
- Barbosa, J.M.; Singh, N.K.; Cherry, J.H.; Locy, R.D. Nitrate uptake and utilization is modulated by exogenous g-aminobutyric acid in Arabidopsis thaliana seedlings. Plant Physiol. Biochem. 2010, 48, 443–450. [Google Scholar] [CrossRef]
- Sulieman, S. Does GABA increase the efficiency of symbiotic N2 fixation in legumes? Plant Sign. Behav. 2011, 6, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Michaeli, S.; Fromm, H. Closing the loop on the GABA shunt in plants: Are GABA metabolism and signaling entwined? Front. Plant Sci. 2015, 6, 419. [Google Scholar] [CrossRef]
- Carillo, P. GABA Shunt in Durum Wheat. Front. Plant Sci. 2018, 9, 100. [Google Scholar] [CrossRef]
- Forde, B.G.; Lea, P.J. Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 2007, 58, 2339–2358. [Google Scholar] [CrossRef]
- Umate, P. Genome-wide analysis of the family of light-harvesting chlorophyll a/b-binding proteins in Arabidopsis and rice. Plant Signal. Behav. 2010, 5, 1537–1542. [Google Scholar] [CrossRef]
- Heddad, M.; Adamska, I. Light stress-regulated two-helix proteins in Arabidopsis thaliana related to the chlorophyll a/b-binding gene family. PNAS 2000, 97, 3741–3746. [Google Scholar] [CrossRef]
- Vashisht, A.A.; Tuteja, N. Stress responsive DEAD-box helicases: A new pathway to engineer plant stress tolerance. J. Photochem. Photobiol. B: Biol. 2006, 84, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, H.; Zhang, H.; Wang, X.; Song, F. OsBIRH1, a DEAD-box RNA helicase with functions in modulating defence responses against pathogen infection and oxidative stress. J. Exp. Bot. 2008, 59, 2133–2146. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, G.; Dong, T.; Wang, L.; Zhang, J.; Zhao, Z.; Hu, Z. SlDEAD31, a Putative DEAD-Box RNA Helicase Gene, Regulates Salt and Drought Tolerance and Stress-Related Genes in Tomato. PLoS ONE 2015, 10, e0133849. [Google Scholar] [CrossRef]
- Gu, L.; Xu, T.; Lee, K.; Lee, K.H.; Kang, H. A chloroplast-localized DEAD-box RNA helicase AtRH3 is essential for intron splicing and plays an important role in the growth and stress response in Arabidopsis thaliana. Plant Physiol. Biochem. 2014, 82, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, Y.; Luo, W.; Yang, C.; Ding, P.; Liu, Y.; Qiao, L.; Chang, Z.; Geng, H.; Wang, P.; et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PLoS ONE 2017, 12, e0181443. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Zhao, M.; Zhang, W.; Li, B.; An, D.; Li, J.; Zhang, A.; Liu, R.; Liu, X. A Genome-wide View of Transcriptome Dynamics during Early Spike Development in Bread Wheat. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Caverzan, A.; Casassola, A.; Brammer, S.P. Antioxidant responses of wheat plants under stress. Genet. Mol. Biol. 2016, 39, 1–6. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, S.K.; Goswami, S.; Singh, K.; Gadpayle, K.A.; Singh, G.; Pathak, H.; Rai, R. Transcript profiling and biochemical characterization of mitochondrial superoxide dismutase (mtSOD) in wheat (Triticum aestivum) under different exogenous stresses. Aust. J. Crop Sci. 2013, 7, 414–424. [Google Scholar]
- Liu, Y.; Bassham, D.C. Autophagy: Pathways for Self-Eating in Plant Cells. Annu. Rev. Plant Biol. 2012, 63, 215–237. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.E.; Lemaire, S.D.; Crespo, J.L. Reactive Oxygen Species and Autophagy in Plants and Algae. Plant Physiol. 2012, 160, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiong, Y.; Bassham, D.C. Autophagy is required for tolerance of drought and salt stress in plants. Autophagy 2009, 5, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Ishida, H.; Izumi, M.; Yoshimoto, K.; Ohsumi, Y.; Mae, T.; Makino, A. Autophagy Plays a Role in Chloroplast Degradation during Senescence in Individually Darkened Leaves. Plant Physiol. 2009, 149, 885–893. [Google Scholar] [CrossRef] [PubMed]
- De Vita, P.; Riefolo, C.; Codianni, P.; Cattivelli, L.; Fares, C. Agronomic and qualitative traits of T. turgidum ssp. dicoccum genotypes cultivated in Italy. Euphytica 2006, 150, 195–205. [Google Scholar] [CrossRef]
- De Vita, P.; Mastrangelo, A.M.; Codianni, P.; Fornara, M. Bio-agronomic Evaluation of Old and Modern Wheat, Spelt and Emmer Genotypes for Low-input Farming in Mediterranean Environment. Ital. J. Agron. 2007, 2, 291–302. [Google Scholar] [CrossRef]
- Iannucci, A.; Marone, D.; Russo, M.A.; De Vita, P.; Miullo, V.; Ferragonio, P.; Blanco, A.; Gadaleta, A.; Mastrangelo, A.M. Mapping QTL for Root and Shoot Morphological Traits in a Durum Wheat × T. dicoccum Segregating Population at Seedling Stage. Int. J. Genom. 2017, 2017, 1–17. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The Water-Culture Method for Growing Plants without Soil. Circular 347; California Agricultural Experiment Station: Berkeley, CA, USA, 1950. [Google Scholar]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- David, J.; Holtz, Y.; Ranwez, V.; Santoni, S.; Sarah, G.; Ardisson, M.; Poux, G.; Choulet, F.; Genthon, C.; Roumet, P.; et al. Genotyping by sequencing transcriptomes in an evolutionary pre-breeding durum wheat population. Mol. Breed. 2014, 34, 1531–1548. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 2014, 30, 1837–1843. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 1720. [Google Scholar] [CrossRef]
- Beleggia, R.; Platani, C.; Nigro, F.; DeVita, P.; Cattivelli, L.; Papa, R. Effect of genotype, environment and genotype-by-environment interaction on metabolite profiling in durum wheat (Triticum durum Desf.) grain. J. Cereal Sci. 2013, 57, 183–192. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor; Gentleman, R., Carey, V., Dudoit, S., Irizarry, R., Huber, W., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef]
- Kachitvichyanukul, V.; Schmeiser, B. Computer generation of hypergeometric random variates†. J. Stat. Comput. Simul. 1985, 22, 127–145. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Inter. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype Effect (G) | |||||
| Emmer | Durum Wheat | p Value | |||
| TLN | 3.88 ± 0.54 | 4.81 ± 0.48 | n.s. | ||
| TLA (cm2) | 24.85 ± 5.66 b | 41.92 ± 8.39 a | 0.0304 | ||
| SFW (g) | 0.63 ± 0.19 b | 1.17 ± 0.30 a | 0.0453 | ||
| PRL (cm) | 155.58 ± 26.79 | 172.72 ± 25.42 | n.s. | ||
| LRL (cm) | 20.49 ± 7.00 | 10.89 ± 4.04 | n.s. | ||
| TRL (cm) | 176.07 ± 32.17 | 183.62 ± 27.96 | n.s. | ||
| RSD (cm) | 62.84 ± 5.38 | 65.34 ± 3.66 | n.s. | ||
| RSW (cm) | 23.31 ± 3.26 | 22.82 ± 3.22 | n.s. | ||
| RDW (g) | 0.02 ± 0.01 | 0.03 ± 0.00 | n.s. | ||
| SRL (m g−1) | 107.31 ± 15.77 a | 67.69 ± 5.99 b | 0.0094 | ||
| TRL/TLA (cm cm−2) | 7.92 ± 1.65 | 5.98 ± 1.62 | n.s. | ||
| LRL/PRL | 0.11 ± 0.04 | 0.06 ± 0.02 | n.s. | ||
| SPAD | 26.55 ± 1.22 b | 34.91 ± 2.24 a | <0.0001 | ||
| Nitrogen Effect (N) | |||||
| −N | +N | p Value | |||
| TLN | 3.50 ± 0.19 b | 5.19 ± 0.59 a | 0.0173 | ||
| TLA (cm2) | 19.98 ± 1.43 b | 46.79 ± 8.35 a | 0.0023 | ||
| SFW (g) | 0.42 ± 0.05 b | 1.39 ± 0.28 a | 0.0017 | ||
| PRL (cm) | 191.72 ± 21.60 | 136.59 ± 26.47 | n.s. | ||
| LRL (cm) | 15.28 ± 5.19 | 16.10 ± 6.70 | n.s. | ||
| TRL (cm) | 206.99 ± 24.99 | 152.69 ± 31.39 | n.s. | ||
| RSD (cm) | 70.28 ± 2.54 | 57.90 ± 5.04 | n.s. | ||
| RSW (cm) | 24.79 ± 3.08 | 21.34 ± 3.27 | n.s. | ||
| RDW (g) | 0.03 ± 0.00 a | 0.02 ± 0.00 b | 0.0228 | ||
| SRL (m g−1) | 70.31 ± 5.93 b | 104.69 ± 16.65 a | 0.0201 | ||
| TRL/TLA (cm cm−2) | 10.57 ± 1.28 a | 3.34 ± 0.45 b | 0.0002 | ||
| LRL/PRL | 0.07 ± 0.02 | 0.10 ± 0.04 | n.s. | ||
| SPAD | 26.91 ± 1.24 b | 34.55 ± 2.41 a | 0.0001 | ||
| Genotype × Nitrogen Interaction Effect (G × N) | |||||
| Emmer × (−N) | Emmer × (+N) | Durum Wheat× (−N) | Durum Wheat× (+N) | p Value | |
| TLN | 3.00 ± 0.00 | 4.75 ± 0.92 | 4.00 ± 0.00 | 5.63 ± 0.80 | n.s. |
| TLA (cm2) | 17.22 ± 1.21 | 32.48 ± 10.45 | 22.74 ± 1.75 | 61.10 ± 8.94 | n.s. |
| SFW (g) | 0.34 ± 0.04 | 0.93 ± 0.32 | 0.50 ± 0.07 | 1.85 ± 0.35 | n.s. |
| PRL (cm) | 189.27 ± 31.72 | 121.89 ± 39.82 | 194.16 ± 34.17 | 151.28 ± 39.25 | n.s. |
| LRL (cm) | 18.60 ± 8.34 | 22.38 ± 12.53 | 11.96 ± 6.99 | 9.83 ± 5.13 | n.s. |
| TRL (cm) | 207.87 ± 39.64 | 144.27 ± 50.83 | 206.12 ± 36.65 | 161.11 ± 44.36 | n.s. |
| RSD (cm) | 68.69 ± 4.09 | 56.98 ± 9.76 | 71.86 ± 3.41 | 58.82 ± 4.74 | n.s. |
| RSW (cm) | 28.72 ± 3.47 | 17.89 ± 4.25 | 20.87 ± 4.68 | 24.78 ± 4.89 | n.s. |
| RDW (g) | 0.03 ± 0.01 | 0.01 ± 0.00 | 0.03 ± 0.01 | 0.02 ± 0.00 | n.s. |
| SRL (m g−1) | 75.75 ± 7.06 b | 138.87 ± 21.15 a | 64.87 ± 9.72 b | 70.52 ± 8.21 b | 0.0449 |
| TRL/TLA (cm cm−2) | 11.68 ± 1.71 | 4.17 ± 0.54 | 9.46 ± 1.98 | 2.50 ± 0.43 | n.s. |
| LRL/PRL | 0.09 ± 0.04 | 0.14 ± 0.07 | 0.06 ± 0.03 | 0.05 ± 0.02 | n.s. |
| SPAD | 24.35 ± 0.91 b | 28.75 ± 1.72 b | 29.48 ± 1.40 b | 40.35 ± 1.31 a | 0.0353 |
| Emmer | Durum Wheat | (Durum Wheat/Emmer) | Intersection | Common (Accepting the Fisher Ztest NULL Hypothesis) | |
|---|---|---|---|---|---|
| Number of edges in total | 1,249,637 | 3,500,971 | 2.8 | 396,571 | 393,779 |
| Number of edges DEG-DEG | 1,237,748 | 3,473,768 | 2.8 | 394,015 | 393,719 |
| Number of edges metabolite-metabolite | 185 | 157 | 0.85 | 65 | 60 |
| Number of edges DEG-metabolites | 11,704 | 27,046 | 2.3 | 2491 | 0 |
| Number of nodes | 1829 | 3167 | 1.7 | 1129 | 1127 |
| Number of central nodes | 260 | 479 | 1.8 | 367 | 398 |
| Number of edges to the central nodes: DEGs—significantly behaved metabolites * | 1898 | 4590 | 2.4 | 1217 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beleggia, R.; Omranian, N.; Holtz, Y.; Gioia, T.; Fiorani, F.; Nigro, F.M.; Pecchioni, N.; De Vita, P.; Schurr, U.; David, J.L.; et al. Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation. Int. J. Mol. Sci. 2021, 22, 4790. https://doi.org/10.3390/ijms22094790
Beleggia R, Omranian N, Holtz Y, Gioia T, Fiorani F, Nigro FM, Pecchioni N, De Vita P, Schurr U, David JL, et al. Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation. International Journal of Molecular Sciences. 2021; 22(9):4790. https://doi.org/10.3390/ijms22094790
Chicago/Turabian StyleBeleggia, Romina, Nooshin Omranian, Yan Holtz, Tania Gioia, Fabio Fiorani, Franca M. Nigro, Nicola Pecchioni, Pasquale De Vita, Ulrich Schurr, Jacques L. David, and et al. 2021. "Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation" International Journal of Molecular Sciences 22, no. 9: 4790. https://doi.org/10.3390/ijms22094790
APA StyleBeleggia, R., Omranian, N., Holtz, Y., Gioia, T., Fiorani, F., Nigro, F. M., Pecchioni, N., De Vita, P., Schurr, U., David, J. L., Nikoloski, Z., & Papa, R. (2021). Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation. International Journal of Molecular Sciences, 22(9), 4790. https://doi.org/10.3390/ijms22094790