
High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata


Abstract
1. Introduction
2. Results
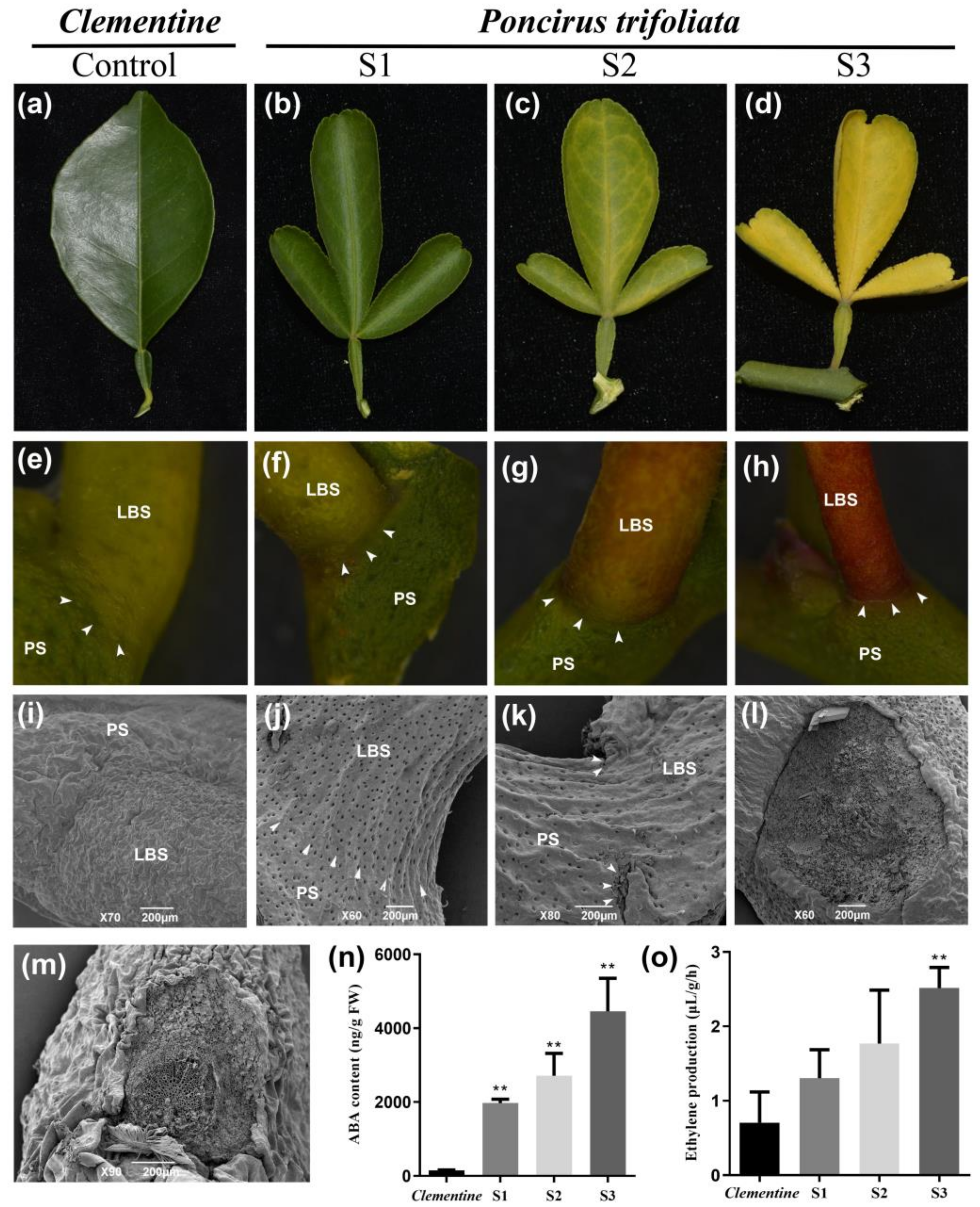
2.1. Morphological Characterization of Citrus Leaf Abscission and Hormone Quantification
2.2. Hybrid Population Construction
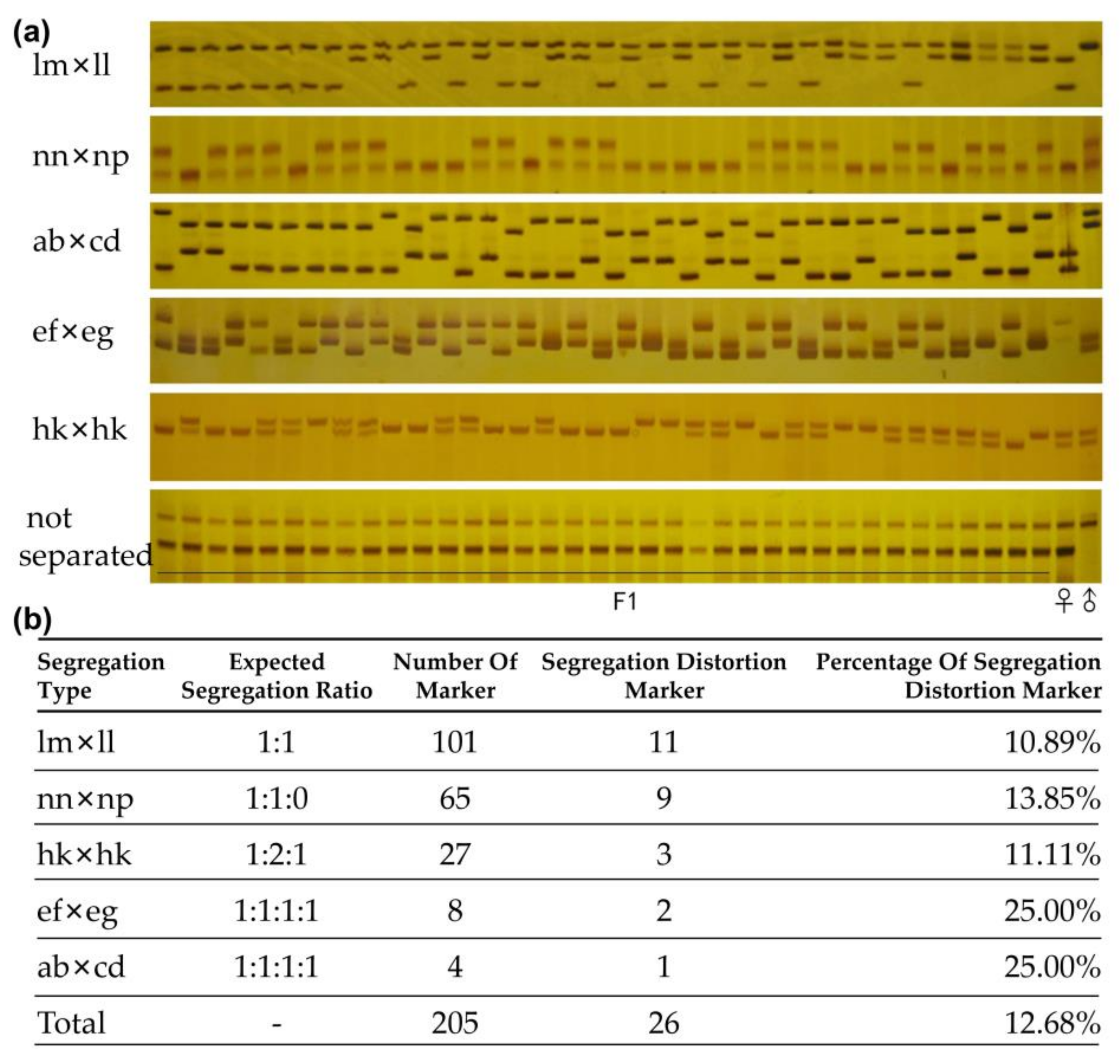
2.3. Screening and Verification of SSR and InDel Markers
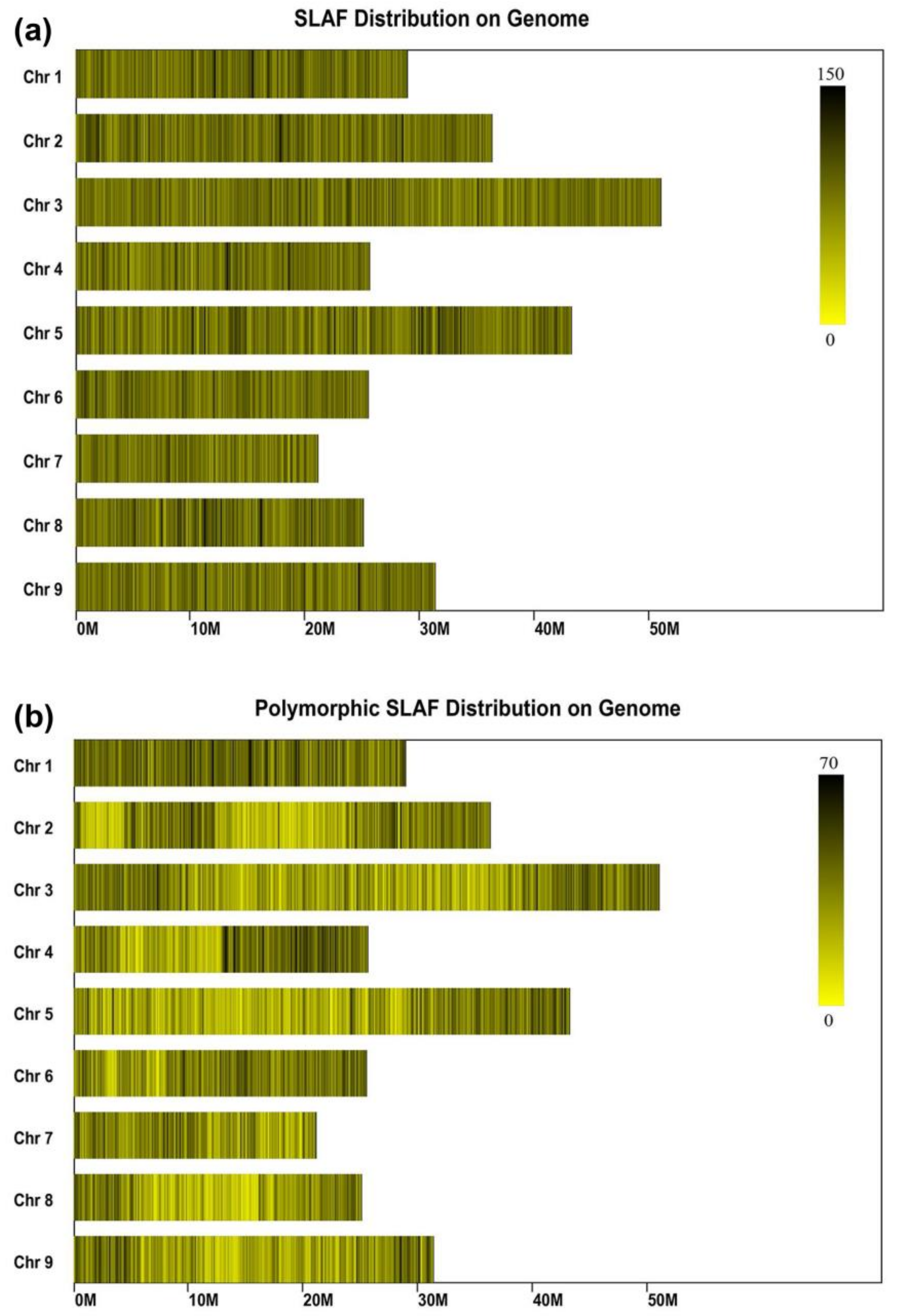
2.4. SLAF Sequencing and Genotyping
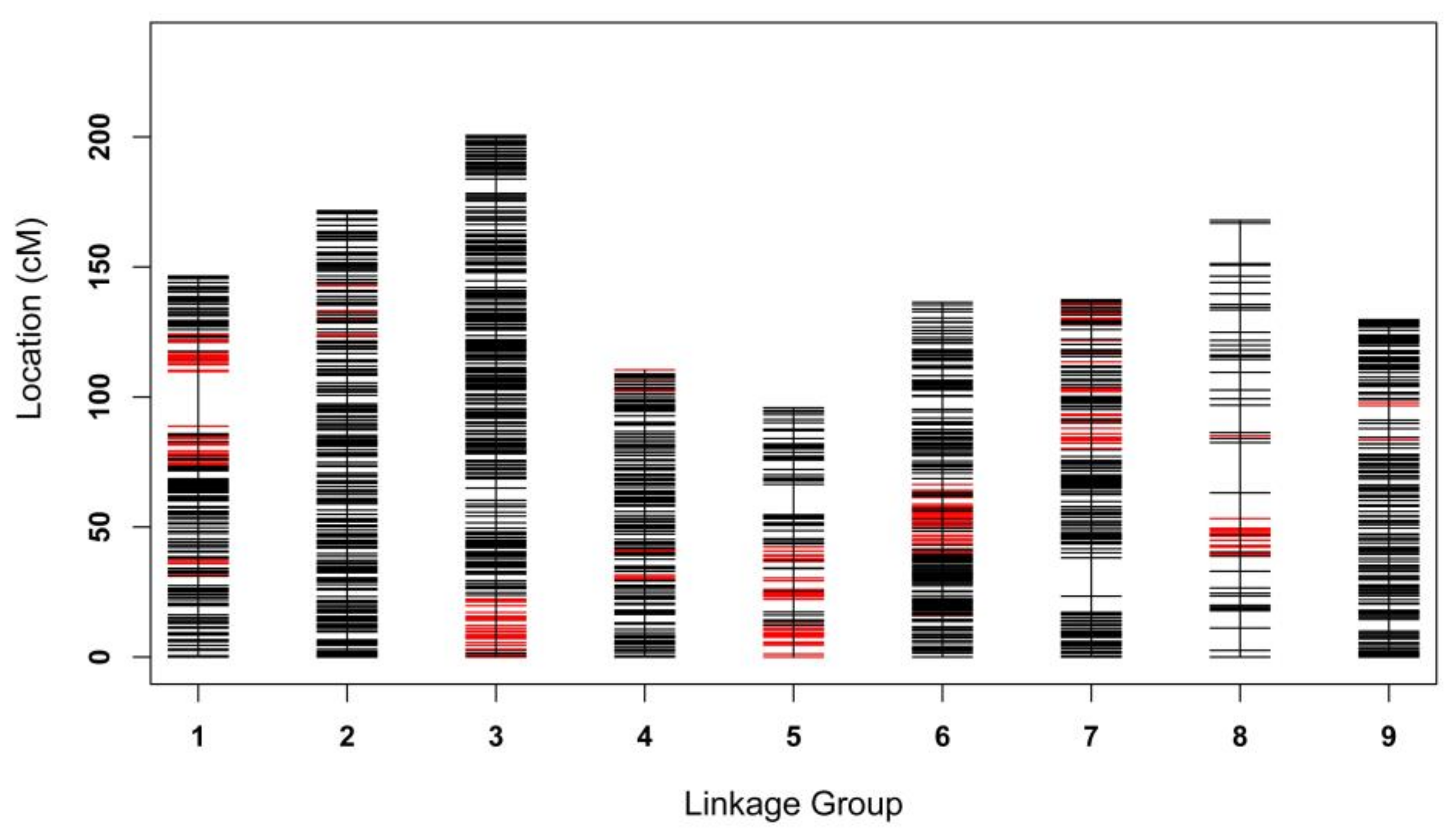
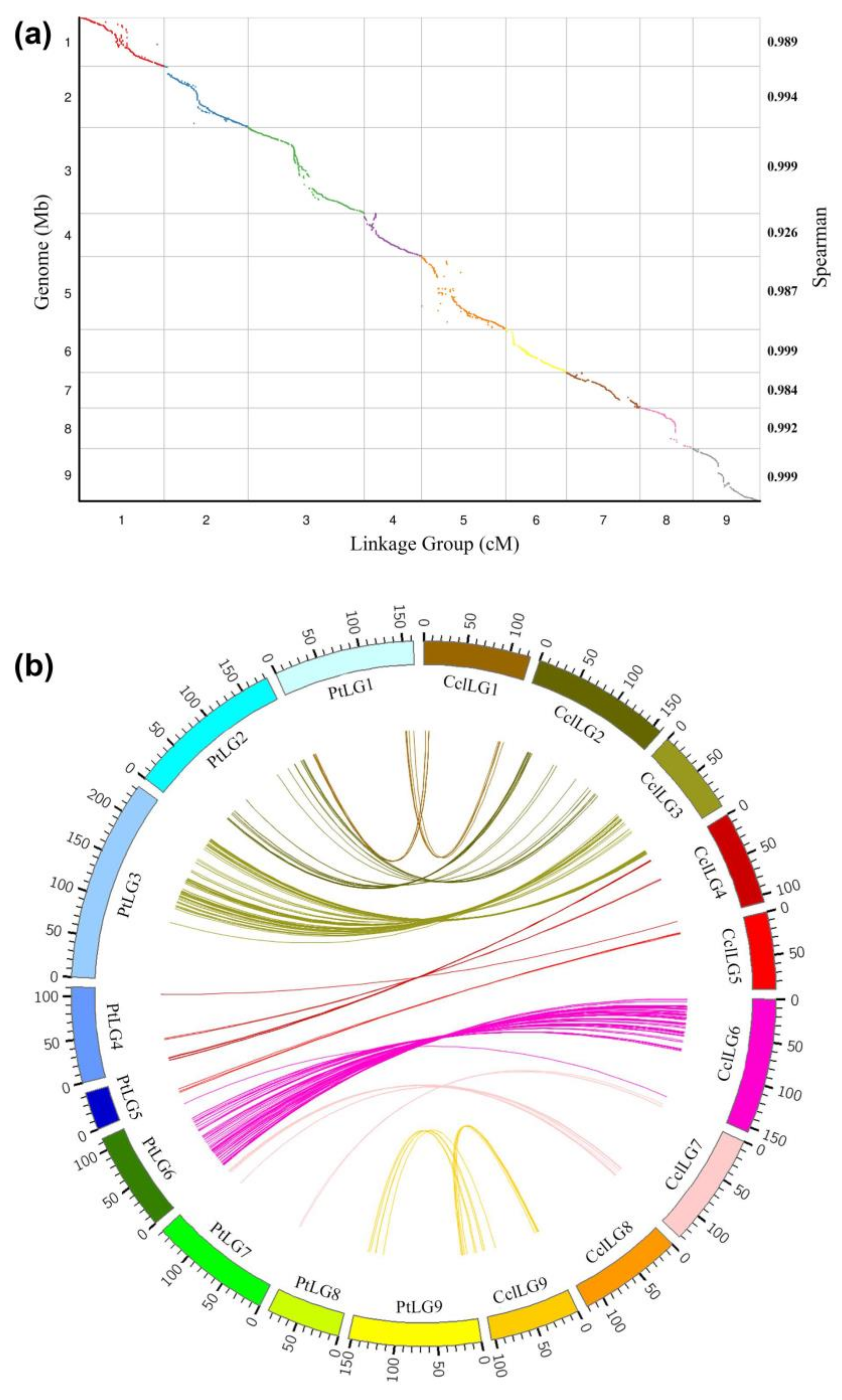
2.5. High-Density Linkage Map Construction Using SNP, SSR, and InDel Markers
2.6. Comparative Analysis of the High-Density Linkage Map
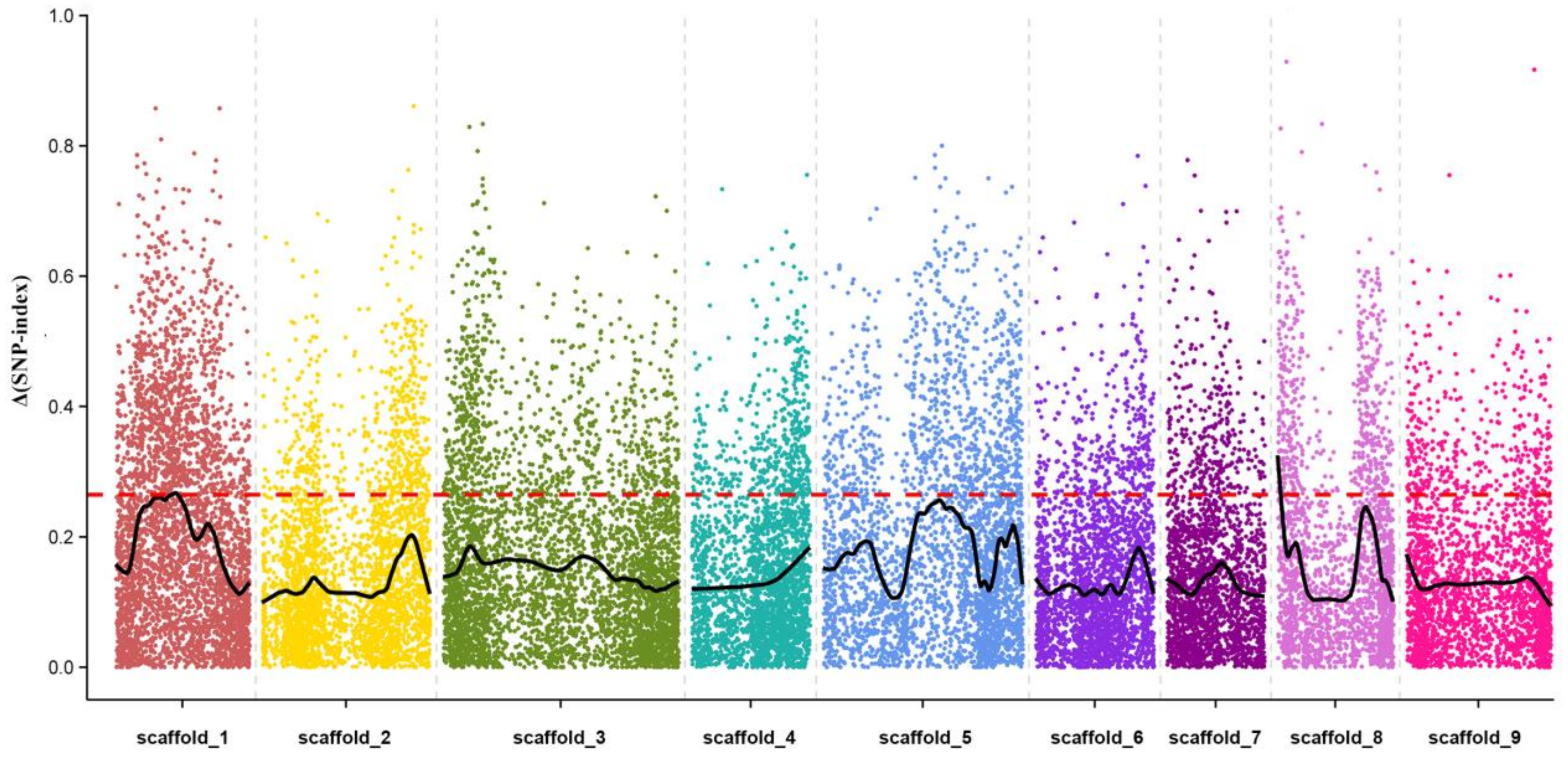
2.7. QTL Mapping of Leaf Abscission Trait in Citrus
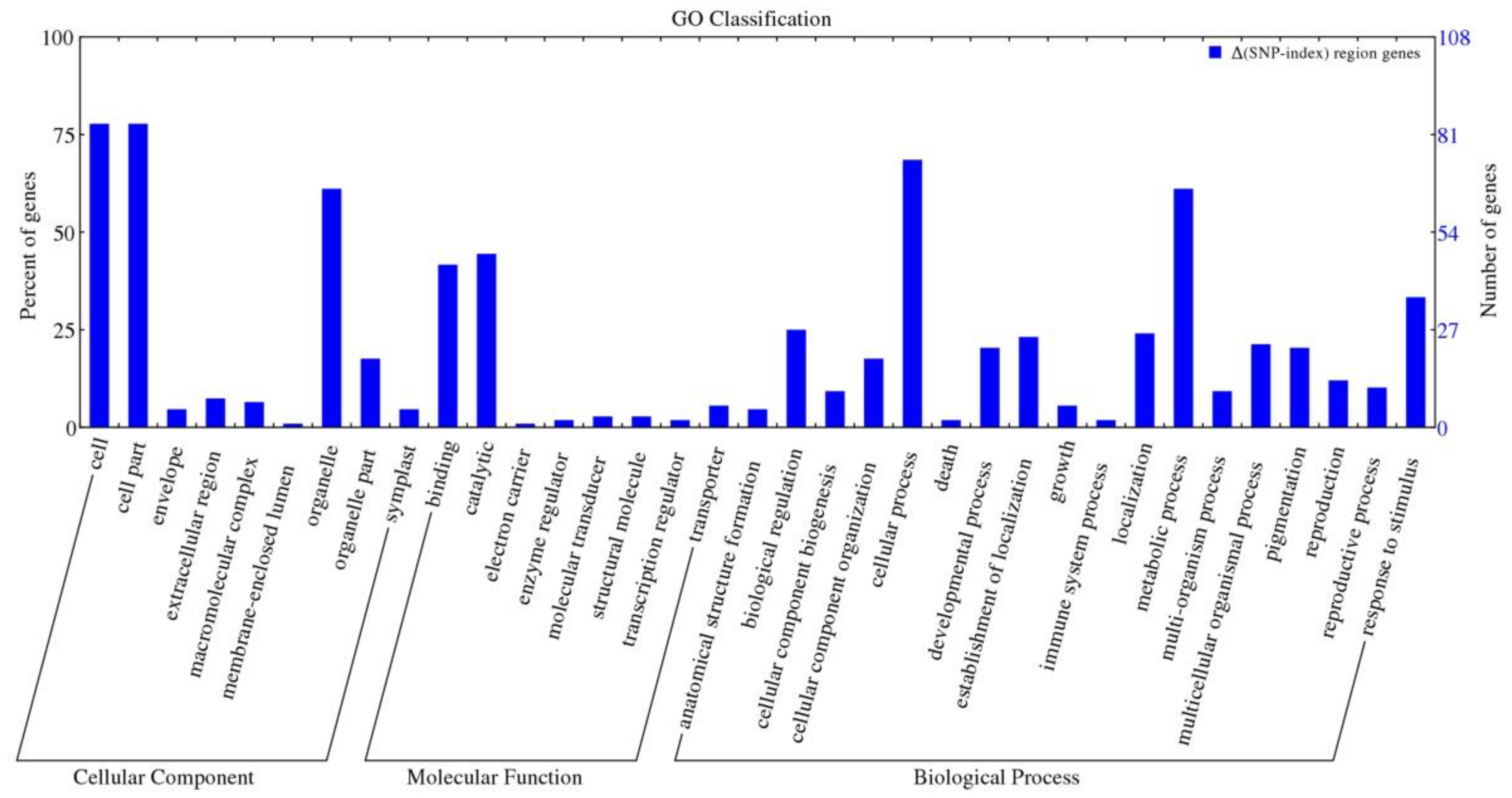
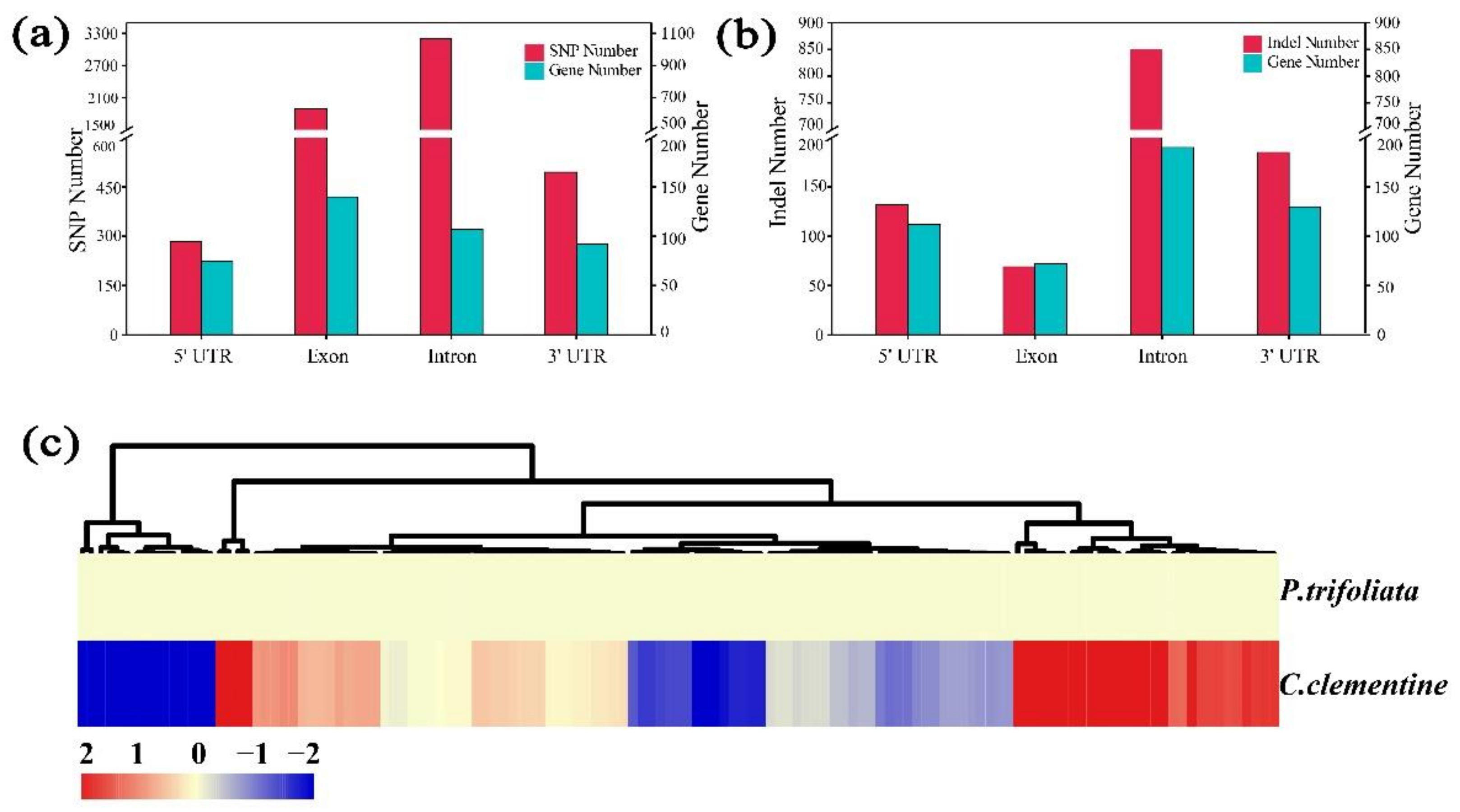
2.8. Analysis and Annotation of the Candidate Genes
2.9. Analysis and Annotation of the Candidate Genes
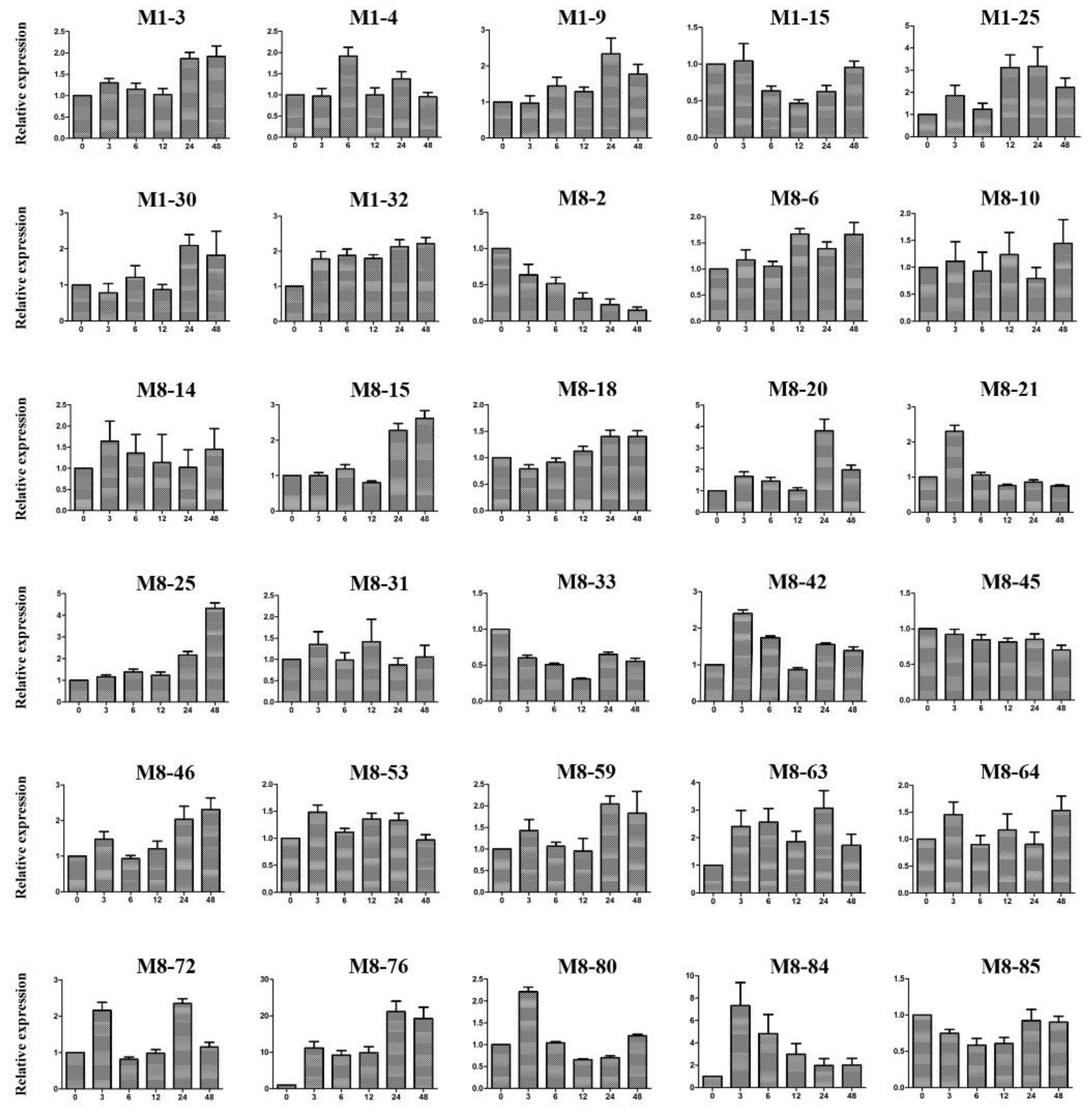
2.10. Expression Patterns of Candidate Genes under Cold Stress Treatment
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Scanning Electron Microscopy (SEM)
4.3. Quantification of ABA and Ethylene
4.4. SSR and InDel Genotyping
4.5. SLAF Library Construction and High-Throughput Sequencing
4.6. SNP Identification and Genotyping
4.7. High-Density Genetic Map Construction
4.8. QTL Mapping for Leaf Abscission Trait in Citrus
4.9. Functional Annotation of Candidate Genes
4.10. Real-Time RT-PCR Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ruiz, C.; Asins, M.J. Comparison between Poncirus and Citrus genetic linkage maps. Theor. Appl. Genet. 2003, 106, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, F.; Huang, Y.; He, J.; Yang, S.; Zeng, J.; Deng, C.; Jiang, X.; Fang, Y.; Wen, S.; et al. Genome of Wild Mandarin and Domestication History of Mandarin. Mol. Plant 2018, 11, 1024–1037. [Google Scholar] [CrossRef]
- Peng, Z.; Bredeson, J.V.; Wu, G.A.; Shu, S.; Rawat, N.; Du, D.; Parajuli, S.; Yu, Q.; You, Q.; Rokhsar, D.S.; et al. A chromosome-scale reference genome of trifoliate orange (Poncirus trifoliata) provides insights into disease resistance, cold tolerance and genome evolution inCitrus. Plant J. 2020, 104, 1215–1232. [Google Scholar] [CrossRef]
- Spiegelroy, P.; Goldschmidt, E.E. The Biology of Citrus; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Chen, C.; Bowman, K.D.; Choi, Y.A.; Dang, P.M.; Rao, M.N.; Huang, S.; Soneji, J.R.; McCollum, T.G.; Gmitter, F.G. EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet. Genomes 2008, 4, 1–10. [Google Scholar] [CrossRef]
- Guo, F.; Yu, H.; Tang, Z.; Jiang, X.; Wang, L.; Wang, X.; Xu, Q.; Deng, X. Construction of a SNP-based high-density genetic map for pummelo using RAD sequencing. Tree Genet. Genomes 2015, 11, 2. [Google Scholar] [CrossRef]
- Shimada, T.; Fujii, H.; Endo, T.; Ueda, T.; Sugiyama, A.; Nakano, M.; Kita, M.; Yoshioka, T.; Shimizu, T.; Nesumi, H.; et al. Construction of a citrus framework genetic map anchored by 708 gene-based markers. Tree Genet. Genomes 2014, 10, 1001–1013. [Google Scholar] [CrossRef]
- Wu, G.A.; Prochnik, S.; Jenkins, J.; Salse, J.; Hellsten, U.; Murat, F.; Perrier, X.; Ruiz, M.; Scalabrin, S.; Terol, J.; et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 2014, 32, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Bernet, G.P.; Fernandez-Ribacoba, J.; Carbonell, E.A.; Asins, M.J. Comparative genome-wide segregation analysis and map construction using a reciprocal cross design to facilitate citrus germplasm utilization. Mol. Breed. 2009, 25, 659–673. [Google Scholar] [CrossRef]
- Gulsen, O.; Uzun, A.; Canan, I.; Seday, U.; Canihos, E. A new citrus linkage map based on SRAP, SSR, ISSR, POGP, RGA and RAPD markers. Euphytica 2010, 173, 265–277. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Cheng, T.; Yang, W.; Pan, H.; Zhong, J.; Huang, L.; Liu, E. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res. 2015, 22, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ollitrault, P.; Terol, J.; Chen, C.; Federici, C.T.; Lotfy, S.; Hippolyte, I.; Ollitrault, F.; Berard, A.; Chauveau, A.; Cuenca, J.; et al. A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping. BMC Genom. 2012, 13, 593. [Google Scholar] [CrossRef]
- Estornell, L.H.; Agusti, J.; Merelo, P.; Talon, M.; Tadeo, F.R. Elucidating mechanisms underlying organ abscission. Plant Sci. 2013, 199–200, 48–60. [Google Scholar] [CrossRef]
- Doebley, J. Genetics, development and plant evolution. Curr. Opin. Genet. Dev. 1993, 3, 865–872. [Google Scholar] [CrossRef]
- Zanne, A.E.; Tank, D.C.; Cornwell, W.K.; Eastman, J.M.; Smith, S.A.; FitzJohn, R.G.; McGlinn, D.J.; O’Meara, B.C.; Moles, A.T.; Reich, P.B.; et al. Three keys to the radiation of angiosperms into freezing environments. Nature 2014, 506, 89–92. [Google Scholar] [CrossRef]
- Antúnez, I.; Villar, R. Relative growth rate in phylogenetically related deciduous and evergreen woody species. Oecologia 2001, 128, 172–180. [Google Scholar] [CrossRef]
- Agustí, J.; Gimeno, J.; Merelo, P.; Serrano, R.; Cercós, M.; Conesa, A.; Talón, M.; Tadeo, F.R. Early gene expression events in the laminar abscission zone of abscission-promoted citrus leaves after a cycle of water stress/rehydration: Involvement of CitbHLH1. J. Exp. Bot. 2012, 63, 6079–6091. [Google Scholar] [CrossRef]
- Agustí, J.; Merelo, P.; Cercos, M.; Tadeo, F.R.; Talon, M. Comparative transcriptional survey between laser-microdissected cells. BMC Plant Biol. 2009, 9, 127. [Google Scholar] [CrossRef]
- Agustí, J.; Merelo, P.; Cercos, M.; Tadeo, F.R.; Talon, M. Ethylene-induced differential gene expression during abscission of citrus leaves. J. Exp. Bot. 2008, 59, 2717–2733. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Meredith, F. Effect of exposure to subfreezing temperatures on ethylene evolution and leaf abscission in citrus. Plant Physiol. 1971, 48, 724–727. [Google Scholar] [CrossRef]
- Gómez-Cadenas, A.; Tadeo, F.R.; Primo-Millo, E.; Talón, M. Involvement of abscisic acid and ethylene in the responses of citrus seedlings to salt shock. Physiol. Plant. 1998, 103, 475–484. [Google Scholar] [CrossRef]
- Malladi, A.; Burns, J.K. CsPLDa1 and CsPLDg1 are differentially induced during leaf and fruit abscission and diurnally regulated in Citrus sinensis. J. Exp. Bot. 2008, 59, 3729–3739. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef]
- Belfield, E.J.; Ruperti, B.; Roberts, J.A.; Mcqueen-Mason, S. Changes in expansin activity and gene expression during ethylene-promoted leaflet abscission in Sambucus nigra. J. Exp. Bot. 2005, 56, 817–823. [Google Scholar] [CrossRef]
- Busov, V.B.; Goldfarb, B. An auxin-inducible gene from loblolly pine (Pinus taeda L.) is differentially expressed in mature and juvenile-phase shoots and encodes a putative transmembrane protein. Planta 2004, 218, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K. Induction of leaf abscission in cotton is a common effect of urea- and Adenine-type cytokinins. Plant Physiol. 1991, 95, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.C.L. Analysis of the auxin control of bean leaf abscission. Plant Physiol. 1963, 38, 262–267. [Google Scholar] [CrossRef]
- Jinn, T.L.; Stone, J.M.; Walker, J.C. HAESA, an Arabidopsis leucine-rich repeat receptor kinase, controls floral organ abscission. Genes Dev. 2000, 14, 108–117. [Google Scholar] [PubMed]
- Saurin, A.T.; Brownlow, N.; Parker, P.J. Protein kinase C epsilon in cell division: Control of abscission. Cell Cycle 2009, 8, 549–555. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Cho, S.K.; Seitz, K.; Walker, J.C. Letting Go is Never Easy: Abscission and Receptor-Like Protein Kinases. J. Integr. Plant Biol. 2013, 55, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, S.; Hu, C. Identifying the genome-wide genetic variation between precocious trifoliate orange and its wild type and developing new markers for genetics research. DNA Res. 2016, 24, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-J.; Zhou, J.-J.; Chen, F.-Y.; Gan, Z.-M.; Li, Y.-P.; Zhang, J.-Z.; Hu, C.-G. Identification of the genetic variation and gene exchange between Citrus trifoliata and Citrus clementina. Biomolecules 2018, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, V.M.; Butrón, A.; Rady, M.O.A.; Soengas, P.; Revilla, P. Identification of quantitative trait loci involved in the response to cold stress in maize (Zea mays L.). Mol. Breed. 2013, 33, 363–371. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Xin, H.; Li, D.; Ma, C.; Ding, X.; Hong, W.; Zhang, X. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplifed fragment (SLAF) sequencing. BMC Plant Biol. 2013, 13, 141. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Liu, J. Deep sequencing-based characterization of transcriptome of trifoliate orange (Poncirus trifoliata (L.) Raf.) in response to cold stress. BMC Genom. 2014, 16, 555. [Google Scholar] [CrossRef]
- Li, B.; Tian, L.; Zhang, J.; Huang, L.; Han, F.; Yan, S.; Wang, L.; Zheng, H.; Sun, J. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Plant Biol. 2014, 15, 1086. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, Y.; Qin, X.; Zhang, Y.; Zhang, Z.; Wang, J.; Li, L.; Lou, Q.; Chen, J. An SNP-based saturated genetic map and QTL analysis of fruit-r533elated traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC Genom. 2014, 15, 1158. [Google Scholar] [CrossRef]
- Xu, X.; Lu, L.; Zhu, B.; Xu, Q.; Qi, X.; Chen, X. QTL mapping of cucumber fruit flesh thickness by SLAF-seq. Sci. Rep. 2015, 5, 15829. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, P.; Choi, Y.A.; Huang, S.; Gmitter, F. Mining and characterizing microsatellites from citrus ESTs. Theor. Appl. Genet. 2006, 112, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Van Os, H.; Andrzejewski, S.; Bakker, E.; Barrena, I.; Bryan, G.J.; Caromel, B.; Ghareeb, B.; Isidore, E.; de Jong, W.; Van Koert, P. Construction of a 10,000-marker ultradense genetic recombination map of potato: Providing a framework for accelerated gene isolation and a genomewide physical map. Genetics 2006, 173, 1075–1087. [Google Scholar] [CrossRef]
- Faris, J.; Laddomada, B.; Gill, B. Molecular mapping of segregation distortion loci in Aegilops tauschii. Genetics 1998, 149, 319–327. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, C.; Zhai, H.; Wan, J. Mapping segregation distortion loci and quantitative trait loci for spikelet sterility in rice (Oryza sativa L.). Genet. Res. 2005, 86, 97–106. [Google Scholar] [CrossRef]
- Xu, S.; Hu, Z. Mapping quantitative trait loci using distorted markers. Int. J. Plant Genom. 2010, 2009, 410825. [Google Scholar] [CrossRef]
- Xu, S. Quantitative trait locus mapping can benefit from segregation distortion. Genetics 2008, 180, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Guo, W.; Yi, H.; Pang, X.; Deng, X. An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol. Biol. Rep. 2003, 21, 177–178. [Google Scholar] [CrossRef]
- Liu, S.; Li, W.; Long, D.; Hu, C.; Zhang, J. Development and characterization of genomic and expressed SSRs in citrus by genome-wide analysis. PLoS ONE 2013, 8, e75149. [Google Scholar] [CrossRef] [PubMed]
- Bernet, G.P.; Margaix, C.; Jacas, J.; Carbonell, E.A.; Asins, M.J. Genetic analysis of citrus leafminer susceptibility. Theor. Appl. Genet. 2005, 110, 1393–1400. [Google Scholar] [CrossRef]
- Pan, X.; Welti, R.; Wang, X. Quantitative analysis of major plant hormones in crude plant extracts by high-performance liquid chromatography–mass spectrometry. Nat. Protoc. 2010, 5, 986–992. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Quan, R.; Wang, X.-C.; Huang, R. Transcriptional regulation of the ethylene response factor LeERF2 in the expression of ethylene biosynthesis genes controls ethylene production in tomato and tobacco. Plant Physiol. 2009, 150, 365–377. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT-the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.; Deng, D.; Xin, H.; Song, J.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Khan, M.R.G.; Li, Y.; Zhang, J.; Hu, C. Comprehensive analysis of CCCH-type zinc finger gene family in citrus (Clementine mandarin) by genome-wide characterization. Mol. Genet. Genom. 2014, 289, 855–872. [Google Scholar] [CrossRef] [PubMed]
- Van Os, H.; Stam, P.; Visser, R.G.; van Eck, H.J. SMOOTH: A statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor. Appl. Genet. 2005, 112, 187–194. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Vuong, T.D.; Sleper, D.A.; Shannon, J.G.; Nguyen, H.T. Novel quantitative trait loci for broad-based resistance to soybean cyst nematode (Heterodera glycines Ichinohe) in soybean PI 567516C. Theor. Appl. Genet. 2010, 121, 1253–1266. [Google Scholar] [CrossRef]
- Schmid, R.; Blaxter, M.L. annot8r: GO, EC and KEGG annotation of EST datasets. BMC Bioinform. 2008, 9, 180. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Reads | 489,334,306 |
|---|---|
| No. of reads (♀) | 7,664,165 |
| No. of reads (♂) | 8,095,110 |
| No. of reads (F1, 1–200) | 473,575,031 |
| No. of high-quality SLAFs | 257,409 |
| Polymorphic SLAFs | 84,525 (32.84%) |
| Non-polymorphic SLAFs | 172,899 (67.16%) |
| Repetitive SLAFs | 73 (0.03%) |
| Average depth in female parent | 37.07× |
| Average depth in male parent | 39.81× |
| Average depth in individuals | 9.69× |
| No. of high-quality SLAF markers | 4307 |
| Linkage Group | Female-Specific Map | Male-Specific Map | ||||||
|---|---|---|---|---|---|---|---|---|
| Mapped Markers | Distinct Positions | Genetic Length (cM) | Average Interval (cM) | Mapped Markers | Distinct Positions | Genetic Length (cM) | Average Interval (cM) | |
| 1 | 106 | 54 | 123.48 | 2.29 | 376 | 125 | 162.89 | 1.02 |
| 2 | 168 | 61 | 160.78 | 2.64 | 447 | 164 | 181.71 | 1.11 |
| 3 | 373 | 69 | 98.64 | 1.43 | 707 | 218 | 233.97 | 1.07 |
| 4 | 193 | 34 | 108.42 | 3.19 | 311 | 107 | 110.31 | 1.03 |
| 5 | 90 | 61 | 89.41 | 1.46 | 71 | 30 | 44.10 | 1.47 |
| 6 | 359 | 102 | 155.96 | 1.53 | 324 | 111 | 114.30 | 1.03 |
| 7 | 94 | 33 | 132.05 | 4.00 | 374 | 116 | 141.15 | 1.22 |
| 8 | 36 | 27 | 120.76 | 4.47 | 39 | 27 | 85.10 | 3.15 |
| 9 | 59 | 36 | 104.40 | 2.90 | 327 | 119 | 153.50 | 1.29 |
| Total | 1478 | 477 | 1093.90 | 2.66 | 2976 | 1017 | 1227.03 | 1.38 |
| Linkage Group | Integrated Linkage Map | ||||||
|---|---|---|---|---|---|---|---|
| Mapped Markers | Distinct Positions | Genetic Length (cM) | Average Interval (cM) | Max Gap (cM) | Gap < 5 cM (%) | Segregation Distortion Ratio (0.001 < p < 0.05) | |
| 1 | 470 | 173 | 146.50 | 0.85 | 20.99 | 99.79 | 26.6 |
| 2 | 589 | 209 | 171.60 | 0.82 | 3.28 | 99.15 | 2.2 |
| 3 | 971 | 263 | 200.58 | 0.76 | 5.59 | 99.18 | 6.0 |
| 4 | 494 | 136 | 110.42 | 0.81 | 3.26 | 99.59 | 2.8 |
| 5 | 158 | 88 | 95.83 | 1.09 | 11.73 | 98.09 | 44.9 |
| 6 | 588 | 183 | 136.41 | 0.75 | 4.96 | 99.32 | 13.1 |
| 7 | 444 | 143 | 137.35 | 0.96 | 14.74 | 99.10 | 20.3 |
| 8 | 74 | 53 | 167.90 | 3.17 | 19.35 | 84.93 | 24.3 |
| 9 | 375 | 145 | 129.71 | 0.89 | 5.59 | 99.73 | 4.5 |
| Average | - | - | - | 1.12 | 9.94 | 97.65 | 16.1 |
| Total | 4163 | 1393 | 1296.3 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.-Y.; Liu, S.-R.; Gan, Z.-M.; Zeng, R.-F.; Zhang, J.-Z.; Hu, C.-G. High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata. Int. J. Mol. Sci. 2021, 22, 5723. https://doi.org/10.3390/ijms22115723
Xu Y-Y, Liu S-R, Gan Z-M, Zeng R-F, Zhang J-Z, Hu C-G. High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata. International Journal of Molecular Sciences. 2021; 22(11):5723. https://doi.org/10.3390/ijms22115723
Chicago/Turabian StyleXu, Yuan-Yuan, Sheng-Rui Liu, Zhi-Meng Gan, Ren-Fang Zeng, Jin-Zhi Zhang, and Chun-Gen Hu. 2021. "High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata" International Journal of Molecular Sciences 22, no. 11: 5723. https://doi.org/10.3390/ijms22115723
APA StyleXu, Y.-Y., Liu, S.-R., Gan, Z.-M., Zeng, R.-F., Zhang, J.-Z., & Hu, C.-G. (2021). High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata. International Journal of Molecular Sciences, 22(11), 5723. https://doi.org/10.3390/ijms22115723