
Proteomics: A Tool to Study Platelet Function

 ,
, 
 , and
, and 
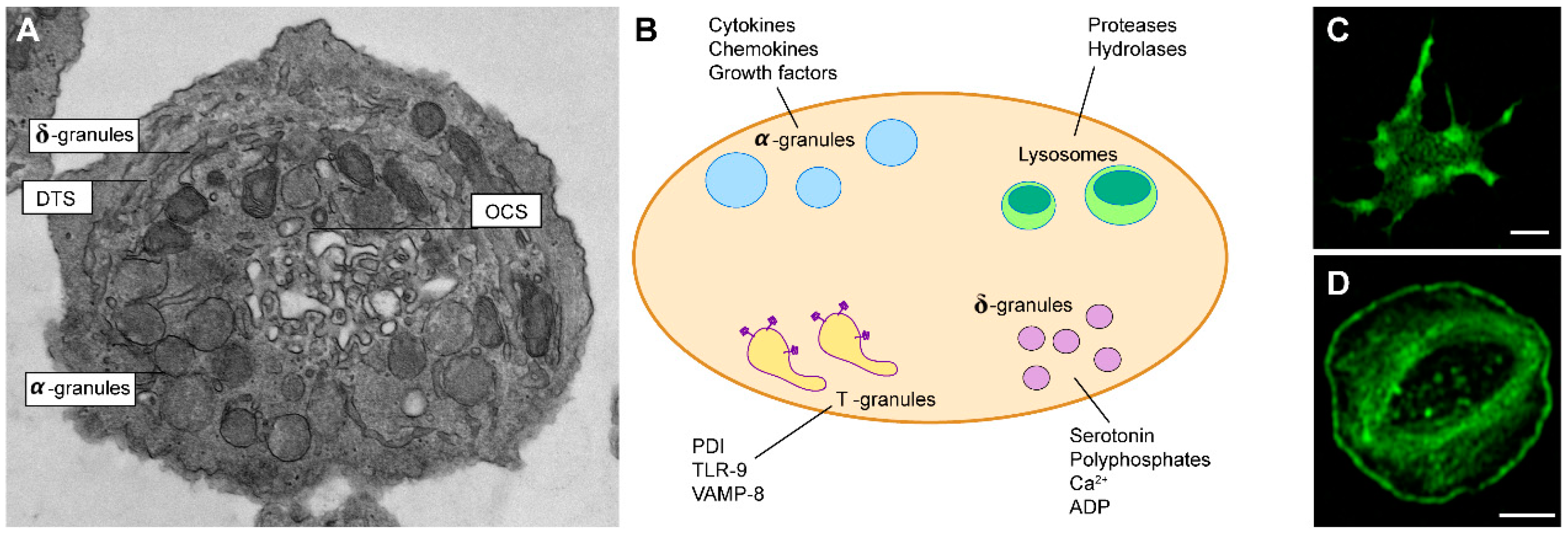{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Platelets Biology and Functions
1.1. The Platelet’s Origin
1.2. Platelets in Hemostasis
1.3. Platelets Activation and Inhibition
1.4. Immunoregulatory Function of Platelets
2. Platelets Proteomics
2.1. Basics of LC-MS-Based Proteomics
2.2. Sample Preparation as a Crucial Step for Accurate Platelets Proteome Research
2.3. Strategies to Study Protein Post-Translational Modifications in Platelet Biology
2.3.1. Phosphorylation
Identification of Key Players of Platelet Activation Pathways by Proteomics Approaches
Identification of Key Players of Platelet Inhibitory Pathways by Proteomic Approaches
2.3.2. Ubiquitylation and Proteolysis in Platelet Biology
3. Platelets Proteomics in Health and Disease
4. Platelets Proteomics in Transfusion Medicine
5. Future Perspective: Platelet Proteomics in Precision Medicine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PTMs | Post-translational modifications |
| OCS | open canalicular system |
| DTS | dense tubular system |
| TLRs | toll-like receptors |
| PDI | protein disulfide isomerase |
| vWF | von Willebrand factor |
| TxA2 | thromboxane A2 |
| FcRγ | Fc receptor γ |
| ITAM | tyrosine-based activation motif |
| PLC | phospholipase C |
| IP3 | inositol 1,4,5,-triphosphate |
| PRRs | pattern recognition receptors |
| NLRs | nod-like receptors |
| CLRs | C-type lectin receptors |
| DAMPs | damage-associated molecular patterns |
| MALDI | matrix-assisted laser desorption/ionization |
| ESI | electrospray ionization |
| HPLC | high-performance liquid chromatography |
| MS | mass spectrometry |
| DDA | data-dependent acquisition |
| DIA | data-independent acquisition |
| MRM | multiple reaction monitoring |
| PRM | parallel reaction monitoring |
| SRM | selected reaction monitoring |
| TMT | tandem mass tags |
| iTRAQ | isobaric tags for relative and absolute quantitation |
| XIC | extracted ion chromatogram |
| TIMS | trapped ion mobility spectrometry coupled to time-of-flight |
| TOF | time-of-flight mass analyzer |
| PASEF | parallel accumulation–aerial fragmentation |
| IMAC | immobilized metal ion affinity chromatography |
| ERLIC | electrostatic repulsion–hydrophilic interaction chromatography |
| SCX | strong cation exchange chromatography |
| SAX | strong anion exchange chromatography |
| CRP | collagen-related-peptide |
| GAPS | GTPase activating proteins |
| ChaFRADIC | charge-based fractional diagonal chromatography |
References
- Humphrey, J.H. Origin of Blood Platelets. Nat. Cell Biol. 1955, 176, 38. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, T.D.S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nat. Cell Biol. 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Lefrançais, E.; Looney, M.R. Platelet Biogenesis in the Lung Circulation. Physiology 2019, 34, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K. Lineage-Specific Hematopoietic Growth Factors. N. Engl. J. Med. 2006, 354, 2034–2045. [Google Scholar] [CrossRef] [PubMed]
- de Sauvage, F.J.; Hass, P.E.; Spencer, S.D.; Malloy, B.E.; Gurney, A.L.; Spencer, S.A.; Darbonne, W.C.; Henzel, W.J.; Wong, S.C.; Kuang, W.-J.; et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 1994, 369, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Bhat, F.A.; Advani, J.; Khan, A.A.; Mohan, S.; Pal, A.; Gowda, H.; Chakrabarti, P.; Prasad, T.S.K.; Chatterjee, A. A network map of thrombopoietin signaling. J. Cell Commun. Signal. 2018, 12, 737–743. [Google Scholar] [CrossRef]
- Nakamura-Ishizu, A.; Matsumura, T.; Stumpf, P.S.; Umemoto, T.; Takizawa, H.; Takihara, Y.; O’Neil, A.; Majeed, A.B.B.A.; MacArthur, B.D.; Suda, T. Thrombopoietin Metabolically Primes Hematopoietic Stem Cells to Megakaryocyte-Lineage Differentiation. Cell Rep. 2018, 25, 1772–1785.e6. [Google Scholar] [CrossRef] [PubMed]
- Pagel, O.; Walter, E.; Jurk, K.; Zahedi, R.P. Taking the stock of granule cargo: Platelet releasate proteomics. Platelets 2016, 28, 119–128. [Google Scholar] [CrossRef]
- Heijnen, H.; Van Der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Escolar, G.; White, J.G. The platelet open canalicular system: A final common pathway. Blood Cells 1991, 17, 467; discussion 486–495. [Google Scholar]
- Heijnen, H.F.G.; Korporaal, S.J.A. Platelet Morphology and Ultrastructure. In Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics: An Update; Gresele, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 21–37. [Google Scholar]
- Gerrard, J.M.; White, J.G.; Peterson, D.A. The platelet dense tubular system: Its relationship to prostaglandin synthesis and calcium flux. Thromb. Haemost. 1978, 40, 224–231. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; MacLeod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Platelets, 3rd ed.; Academic Press: London, UK; Waltham, MA, USA, 2013; p. 1353. [Google Scholar]
- Ghoshal, K.; Bhattacharyya, M. Overview of Platelet Physiology: Its Hemostatic and Nonhemostatic Role in Disease Pathogenesis. Sci. World J. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nuyttens, B.P.; Thijs, T.; Deckmyn, H.; Broos, K. Platelet adhesion to collagen. Thromb. Res. 2011, 127, S26–S29. [Google Scholar] [CrossRef]
- Toonstra, C.; Hu, Y.; Zhang, H. Deciphering the Roles of N-Glycans on Collagen–Platelet Interactions. J. Proteome Res. 2019, 18, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Matzko, C.N.; Poventud-Fuentes, I.; Weisel, J.W.; Brass, L.F.; Stalker, T.J. Interrelationships between structure and function during the hemostatic response to injury. Proc. Natl. Acad. Sci. USA 2019, 116, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Gkaliagkousi, E.; Passacquale, G.; Douma, S.; Zamboulis, C.; Ferro, A. Platelet Activation in Essential Hypertension: Implications for Antiplatelet Treatment. Am. J. Hypertens. 2010, 23, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- Schmaier, A.A.; Zou, Z.; Kazlauskas, A.; Emert-Sedlak, L.; Fong, K.P.; Neeves, K.B.; Maloney, S.F.; Diamond, S.L.; Kunapuli, S.P.; Ware, J.; et al. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc. Natl. Acad. Sci. USA 2009, 106, 21167–21172. [Google Scholar] [CrossRef]
- Heemskerk, J.W.; Harper, M.T.; Cosemans, J.M.; Poole, A.W. Unravelling the different functions of protein kinase C isoforms in platelets. FEBS Lett. 2011, 585, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Bergmeier, W.; Zhang, Y.; Piffath, C.L.; Liang, Y.; Wagner, D.D.; Housman, D.E.; Graybiel, A.M. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004, 10, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin alphaIIbbeta3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L.S.; Branco, L.M.; Franklin, B.S. Regulation of Innate Immune Responses by Platelets. Front. Immunol. 2019, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhong, H.; Zhao, Y.; Luo, X.; Gao, W. Role of platelet biomarkers in inflammatory response. Biomark. Res. 2020, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Ni, H. Crosstalk between Platelets and Microbial Pathogens. Front. Immunol. 2020, 11, 1962. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, C.P.M.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Pietto, M.C.B.; Moiraghi, B.; Ríos, M.A.C.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients With Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casado, C.; Villaseñor, A.; Rodriguez-Nogales, A.; Bueno, J.L.; Barber, D.; Escribese, M.M. Understanding Platelets in Infectious and Allergic Lung Diseases. Int. J. Mol. Sci. 2019, 20, 1730. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef]
- Marcoux, G.; Magron, A.; Sut, C.; Laroche, A.; Laradi, S.; Hamzeh-Cognasse, H.; Allaeys, I.; Cabon, O.; Julien, A.; Garraud, O.; et al. Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. Transfusion 2019, 59, 2403–2414. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-Like Receptor 2 in Human Platelets Induces a Thromboinflammatory Response Through Activation of Phosphoinositide 3-Kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Martyanov, A.A.; Maiorov, A.S.; Filkova, A.A.; Ryabykh, A.A.; Svidelskaya, G.S.; Artemenko, E.O.; Gambaryan, S.P.; Panteleev, M.A.; Sveshnikova, A.N. Effects of bacterial lipopolysaccharides on platelet function: Inhibition of weak platelet activation. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.J.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Anabel, A.-S.; Eduardo, P.-C.; Antonio, H.-C.P.; Carlos, S.-M.; Juana, N.-M.; Honorio, T.-A.; Nicolás, V.-S.; Roberto, A.-R.S. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Avcilar, H.; Eken, A. Could imiquimod (Aldara 5% cream) or other TLR7 agonists be used in the treatment of COVID-19? Med. Hypotheses 2020, 144, 110202. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M. Potential role of platelets in COVID-19: Implications for thrombosis. Res. Pract. Thromb. Haemost. 2020, 4, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, M.; Moser, M.; Mann, M. Copy Number Analysis of the Murine Platelet Proteome Spanning the Complete Abundance Range. Mol. Cell. Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L.G. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.J.; Campos-de-Magalhães, M.; Brandao-Mello, C.E.; de Oliveira, R.V.; do Espirito-Santo, M.P.; Yoshida, C.F.; Lampe, E. Detection of hepatitis C virus in platelets: Evaluating its relationship to antiviral therapy outcome. Hepatogastroenterology 2009, 56, 429–436. [Google Scholar]
- Zucker-Franklin, D.; Seremetis, S.; Zheng, Z.Y. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood 1990, 75, 1920–1923. [Google Scholar] [CrossRef]
- Hoylaerts, M.F.; Vanassche, T.; Verhamme, P. Bacterial killing by platelets: Making sense of (H)IT. J. Thromb. Haemost. 2018, 16, 1182–1186. [Google Scholar] [CrossRef]
- Krauel, K.; Weber, C.; Brandt, S.; Zähringer, U.; Mamat, U.; Greinacher, A.; Hammerschmidt, S. Platelet factor 4 binding to lipid A of Gram-negative bacteria exposes PF4/heparin-like epitopes. Blood 2012, 120, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-S.; Huang, T.-F.; Hsieh, S.-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Mailer, R.K.; Allende, M.; Heestermans, M.; Schweizer, M.; Deppermann, C.; Frye, M.; Pula, G.; Odeberg, J.; Gelderblom, M.; Rose-John, S.; et al. Xenotropic and polytropic retrovirus receptor 1 regulates procoagulant platelet polyphosphate. Blood 2021, 137, 1392–1405. [Google Scholar] [CrossRef]
- Xu, X.; Gnatenko, D.V.; Ju, J.; Hitchcock, I.S.; Martin, D.W.; Zhu, W.; Bahou, W.F. Systematic analysis of microRNA fingerprints in thrombocythemic platelets using integrated platforms. Blood 2012, 120, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M.; Morrell, C.N. Platelets Present Antigen in the Context of MHC Class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef]
- Green, S.A.; Smith, M.; Hasley, R.B.; Stephany, D.; Harned, A.; Nagashima, K.; Abdullah, S.; Pittaluga, S.; Imamichi, T.; Qin, J.; et al. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: Coupling coagulation/inflammation and T cells. AIDS 2015, 29, 1297–1308. [Google Scholar] [CrossRef]
- Trugilho, M.R.D.O.; Hottz, E.D.; Brunoro, G.V.F.; Teixeira-Ferreira, A.; Carvalho, P.C.; Salazar, G.A.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T.; Perales, J. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog. 2017, 13, e1006385. [Google Scholar] [CrossRef]
- Qiu, J.; Ma, J.; Zhang, S.; Han, J.; Liu, S. Promoting platelets is a therapeutic option to combat severe viral infection of the lung. Blood Adv. 2020, 4, 1640–1642. [Google Scholar] [CrossRef]
- Zufferey, A.; Speck, E.R.; Machlus, K.R.; Aslam, R.; Guo, L.; McVey, M.J.; Kim, M.; Kapur, R.; Boilard, E.; Italiano, J.E.; et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. 2017, 1, 1773–1785. [Google Scholar] [CrossRef]
- Colberg, L.; Cammann, C.; Greinacher, A.; Seifert, U. Structure and function of the ubiquitin-proteasome system in platelets. J. Thromb. Haemost. 2020, 18, 771–780. [Google Scholar] [CrossRef]
- Angénieux, C.; Dupuis, A.; Gachet, C.; de la Salle, H.; Maître, B. Cell surface expression of HLA I molecules as a marker of young platelets. J. Thromb. Haemost. 2019, 17, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Klose, J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik 1975, 26, 231–243. [Google Scholar] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Michel, P.E.; Reymond, F.; Arnaud, I.L.; Josserand, J.; Girault, H.H.; Rossier, J.S. Protein fractionation in a multicompartment device using Off-Gel™ isoelectric focusing. Electrophoresis 2003, 24, 3–11. [Google Scholar] [CrossRef]
- Gevaert, K.; Eggermont, L.; Demol, H.; Vandekerckhove, J. A fast and convenient MALDI-MS based proteomic approach: Identification of components scaffolded by the actin cytoskeleton of activated human thrombocytes. J. Biotechnol. 2000, 78, 259–269. [Google Scholar] [CrossRef]
- Marcus, K.; Immler, D.; Sternberger, J.; Meyer, H.E. Identification of platelet proteins separated by two-dimensional gel electrophoresis and analyzed by matrix assisted laser desorption/ionization-time of flight-mass spectrometry and detection of tyrosine-phosphorylated proteins. Electrophoresis 2000, 21, 2622–2636. [Google Scholar] [CrossRef]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Smalley, D.M.; Cho, H.; Shabanowitz, J.; Ley, K.; Hunt, D.F. The Platelet Microparticle Proteome. J. Proteome Res. 2005, 4, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, R.P.; Lewandrowski, U.; Wiesner, J.; Wortelkamp, S.; Moebius, J.; Schütz, C.; Walter, U.; Gambaryan, S.; Sickmann, A. Phosphoproteome of Resting Human Platelets. J. Proteome Res. 2008, 7, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.; Zellner, M.; Diestinger, M.; Babeluk, R.; Marchetti, M.; Goll, A.; Zehetmayer, S.; Bauer, P.; Rappold, E.; Miller, I.; et al. Biological Variation of the Platelet Proteome in the Elderly Population and Its Implication for Biomarker Research. Mol. Cell. Proteom. 2008, 7, 193–203. [Google Scholar] [CrossRef]
- García, A.; Prabhakar, S.; Brock, C.J.; Pearce, A.C.; Dwek, R.A.; Watson, S.P.; Hebestreit, H.F.; Zitzmann, N. Extensive analysis of the human platelet proteome by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 656–668. [Google Scholar] [CrossRef]
- Arias-Salgado, E.G.; Larrucea, S.; Butta, N.; Fernández, D.; García-Muñoz, S.; Parrilla, R.; Ayuso, M.S. Variations in platelet protein associated with arterial thrombosis. Thromb. Res. 2008, 122, 640–647. [Google Scholar] [CrossRef][Green Version]
- Zufferey, A.; Fontana, P.; Reny, J.-L.; Nolli, S.; Sanchez, J.-C. Platelet proteomics. Mass Spectrom. Rev. 2012, 31, 331–351. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Brioschi, M.; Baetta, R.; Mallia, A.; Banfi, C.; Tremoli, E. Platelets in Healthy and Disease States: From Biomarkers Discovery to Drug Targets Identification by Proteomics. Int. J. Mol. Sci. 2020, 21, 4541. [Google Scholar] [CrossRef] [PubMed]
- Vélez, P.; García, Á. Platelet proteomics in cardiovascular diseases. Transl. Proteom. 2015, 7, 15–29. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Gambaryan, S.; Watson, S.P.; Jurk, K.; Walter, U.; Sickmann, A.; Heemskerk, J.W.M.; Zahedi, R.P. What Can Proteomics Tell Us About Platelets? Circ. Res. 2014, 114, 1204–1219. [Google Scholar] [CrossRef]
- Hillenkamp, F.; Karas, M.; Beavis, R.C.; Chait, B.T. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal. Chem. 1991, 63, 1193–1203. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Marx, V. Targeted proteomics. Nat. Methods 2013, 10, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Mann, M. The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef]
- Lee, H.; Chae, S.; Park, J.; Bae, J.; Go, E.-B.; Kim, S.-J.; Kim, H.; Hwang, D.; Lee, S.-W.; Lee, S.-Y. Comprehensive Proteome Profiling of Platelet Identified a Protein Profile Predictive of Responses to An Antiplatelet Agent Sarpogrelate. Mol. Cell. Proteom. 2016, 15, 3461–3472. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.; Burkhart, J.M.; Geiger, J.; Zahedi, R.P.; Sickmann, A. Robust workflow for iTRAQ-based peptide and protein quantification. Methods Mol. Biol. 2012, 893, 101–113. [Google Scholar]
- Bąchor, R.; Waliczek, M.; Stefanowicz, P.; Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules 2019, 24, 701. [Google Scholar] [CrossRef]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Pötz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Schweigel, H.; Geiger, J.; Beck, F.; Buhs, S.; Gerull, H.; Walter, U.; Sickmann, A.; Nollau, P.; Geiger, J. Deciphering of ADP-induced, phosphotyrosine-dependent signaling networks in human platelets by Src-homology 2 region (SH2)-profiling. Proteomics 2013, 13, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.; Geiger, J.; Gambaryan, S.; Veit, J.; Vaudel, M.; Nollau, P.; Kohlbacher, O.; Martens, L.; Walter, U.; Sickmann, A.; et al. Time-resolved characterization of cAMP/PKA-dependent signaling reveals that platelet inhibition is a concerted process involving multiple signaling pathways. Blood 2014, 123, e1–e10. [Google Scholar] [CrossRef]
- Solari, F.A.; Mattheij, N.J.; Burkhart, J.M.; Swieringa, F.; Collins, P.W.; Cosemans, J.M.; Sickmann, A.; Heemskerk, J.W.; Zahedi, R.P. Combined Quantification of the Global Proteome, Phosphoproteome, and Proteolytic Cleavage to Characterize Altered Platelet Functions in the Human Scott Syndrome. Mol. Cell. Proteom. 2016, 15, 3154–3169. [Google Scholar] [CrossRef] [PubMed]
- Sims, M.C.; Mayer, L.; Collins, J.H.; Bariana, T.K.; Megy, K.; Lavenu-Bombled, C.; Seyres, D.; Kollipara, L.; Burden, F.S.; Greene, D.; et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood 2020, 136, 1956–1967. [Google Scholar] [CrossRef]
- Swieringa, F.; Solari, F.A.; Pagel, O.; Beck, F.; Huang, J.; Feijge, M.A.H.; Jurk, K.; Körver-Keularts, I.M.L.W.; Mattheij, N.J.A.; Faber, J.; et al. Impaired iloprost-induced platelet inhibition and phosphoproteome changes in patients with confirmed pseudohypoparathyroidism type Ia, linked to genetic mutations in GNAS. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Loroch, S.; Trabold, K.; Gambaryan, S.; Reiß, C.; Schwierczek, K.; Fleming, I.; Sickmann, A.; Behnisch, W.; Zieger, B.; Zahedi, R.P.; et al. Alterations of the platelet proteome in type I Glanzmann thrombasthenia caused by different homozygous delG frameshift mutations in ITGA2B. Thromb. Haemost. 2017, 117, 556–569. [Google Scholar] [CrossRef]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC−MS/MS. J. Proteome Res. 2011, 10, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mun, D.-G.; Renuse, S.; Saraswat, M.; Madugundu, A.; Udainiya, S.; Kim, H.; Park, S.-K.R.; Zhao, H.; Nirujogi, R.S.; Na, C.H.; et al. PASS-DIA: A Data-Independent Acquisition Approach for Discovery Studies. Anal. Chem. 2020, 92, 14466–14475. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, 016717. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. [Google Scholar] [CrossRef]
- Zhang, C.-C.; Li, R.; Jiang, H.; Lin, S.; Rogalski, J.C.; Liu, K.; Kast, J. Development and Application of a Quantitative Multiplexed Small GTPase Activity Assay Using Targeted Proteomics. J. Proteome Res. 2014, 14, 967–976. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Yuan, D.; Sun, R.-J.; Zhu, J.-J.; Shan, N.-N. Significant reductions in apoptosis-related proteins (HSPA6, HSPA8, ITGB3, YWHAH, and PRDX6) are involved in immune thrombocytopenia. J. Thromb. Thrombolysis 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Malchow, S.; Loosse, C.; Sickmann, A.; Lorenz, C. Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry. Proteomes 2017, 5, 31. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Q.; Weng, L.; Hauser, L.A.; Strawser, C.J.; Rocha, A.G.; Dancis, A.; Mesaros, C.A.; Lynch, D.R.; Blair, I.A. Liquid Chromatography-High Resolution Mass Spectrometry Analysis of Platelet Frataxin as a Protein Biomarker for the Rare Disease Friedreich’s Ataxia. Anal. Chem. 2018, 90, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation–Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.T.; Franks, A.; Slavov, N. DART-ID increases single-cell proteome coverage. PLoS Comput. Biol. 2019, 15, e1007082. [Google Scholar] [CrossRef]
- Levy, E.; Slavov, N. Single cell protein analysis for systems biology. Essays Biochem. 2018, 62, 595–605. [Google Scholar] [CrossRef]
- Marx, R.E. Platelet-Rich Plasma (PRP): What Is PRP and What Is Not PRP? Implant. Dent. 2001, 10, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Sparrow, R.L.; Simpson, R.J. Preparation of platelet concentrates. Methods Mol. Biol. 2011, 728, 267–278. [Google Scholar]
- Wrzyszcz, A.; Urbaniak, J.; Sapa, A.; Woźniak, M. An efficient method for isolation of representative and contamination-free population of blood platelets for proteomic studies. Platelets 2016, 28, 43–53. [Google Scholar] [CrossRef]
- Trichler, S.A.; Bulla, S.C.; Thomason, J.; Lunsford, K.V.; Bulla, C. Ultra-pure platelet isolation from canine whole blood. BMC Vet. Res. 2013, 9, 144. [Google Scholar] [CrossRef]
- Kim, C.-J.; Ki, D.Y.; Park, J.; Sunkara, V.; Kim, T.-H.; Min, Y.; Cho, Y.-K. Fully automated platelet isolation on a centrifugal microfluidic device for molecular diagnostics. Lab Chip 2020, 20, 949–957. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Schumbrutzki, C.; Wortelkamp, S.; Sickmann, A.; Zahedi, R.P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteom. 2012, 75, 1454–1462. [Google Scholar] [CrossRef]
- Maia, T.M.; Staes, A.; Plasman, K.; Pauwels, J.; Boucher, K.; Argentini, A.; Martens, L.; Montoye, T.; Gevaert, K.; Impens, F. Simple Peptide Quantification Approach for MS-Based Proteomics Quality Control. ACS Omega 2020, 5, 6754–6762. [Google Scholar] [CrossRef]
- Fu, Q.; Johnson, C.W.; Wijayawardena, B.K.; Kowalski, M.P.; Kheradmand, M.; Van Eyk, J.E. A Plasma Sample Preparation for Mass Spectrometry using an Automated Workstation. J. Vis. Exp. 2020, 2020, e59842. [Google Scholar] [CrossRef]
- Chiva, C.; Olivella, R.; Borràs, E.; Espadas, G.; Pastor, O.; Solé, A.; Sabido, E. QCloud: A cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS ONE 2018, 13, e0189209. [Google Scholar] [CrossRef]
- Kim, T.; Chen, I.R.; Parker, B.L.; Humphrey, S.J.; Crossett, B.; Cordwell, S.J.; Yang, P.; Yang, J.Y.H. QCMAP: An Interactive Web-Tool for Performance Diagnosis and Prediction of LC-MS Systems. Proteomics 2019, 19, e1900068. [Google Scholar] [CrossRef]
- Van Houtven, J.; Agten, A.; Boonen, K.; Baggerman, G.; Hooyberghs, J.; Laukens, K.; Valkenborg, D.; Hooybergs, J. QCQuan: A Web Tool for the Automated Assessment of Protein Expression and Data Quality of Labeled Mass Spectrometry Experiments. J. Proteome Res. 2019, 18, 2221–2227. [Google Scholar] [CrossRef]
- Mnatsakanyan, R.; Shema, G.; Basik, M.; Batist, G.; Borchers, C.H.; Sickmann, A.; Zahedi, R.P. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev. Proteom. 2018, 15, 515–535. [Google Scholar] [CrossRef]
- Sharma, S.; Toledo, O.; Hedden, M.; Lyon, K.F.; Brooks, S.B.; David, R.P.; Limtong, J.; Newsome, J.M.; Novakovic, N.; Rajasekaran, S.; et al. The Functional Human C-Terminome. PLoS ONE 2016, 11, e0152731. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Schiller, M.R. The carboxy-terminus, a key regulator of protein function. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Looße, C.; Swieringa, F.; Heemskerk, J.W.; Sickmann, A.; Lorenz, C. Platelet proteomics: From discovery to diagnosis. Expert Rev. Proteom. 2018, 15, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Virág, D.; Dalmadi-Kiss, B.; Vékey, K.; Drahos, L.; Klebovich, I.; Antal, I.; Ludányi, K. Current Trends in the Analysis of Post-translational Modifications. Chromatographia 2019, 83, 1–10. [Google Scholar] [CrossRef]
- Dickhut, C.; Radau, S.; Zahedi, R.P. Fast, efficient, and quality-controlled phosphopeptide enrichment from minute sample amounts using titanium dioxide. Methods Mol. Biol. 2014, 1156, 417–430. [Google Scholar] [PubMed]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2011, 43, 1025–1047. [Google Scholar] [CrossRef] [PubMed]
- Loroch, S.; Schommartz, T.; Brune, W.; Zahedi, R.P.; Sickmann, A. Multidimensional electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) for quantitative analysis of the proteome and phosphoproteome in clinical and biomedical research. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 460–468. [Google Scholar] [CrossRef]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef]
- Stolze, S.C.; Nakagami, H. Targeted Quantification of Phosphopeptides by Parallel Reaction Monitoring (PRM). Methods Mol. Biol. 2020, 2139, 213–224. [Google Scholar] [CrossRef]
- Maguire, P.B.; Wynne, K.J.; Harney, D.F.; O’Donoghue, N.M.; Stephens, G.; Fitzgerald, D.J. Identification of the phosphotyrosine proteome from thrombin activated platelets. Proteomics 2002, 2, 642–648. [Google Scholar] [CrossRef]
- García, Á.; Senis, Y.A.; Antrobus, R.; Hughes, C.E.; Dwek, R.A.; Watson, S.P.; Zitzmann, N. A global proteomics approach identifies novel phosphorylated signaling proteins in GPVI-activated platelets: Involvement of G6f, a novel platelet Grb2-binding membrane adapter. Proteomics 2006, 6, 5332–5343. [Google Scholar] [CrossRef]
- Schulz, C.; Leuschen, N.V.; Fröhlich, T.; Lorenz, M.; Pfeiler, S.; Gleissner, C.A.; Kremmer, E.; Kessler, M.; Khandoga, A.G.; Engelmann, B.; et al. Identification of novel downstream targets of platelet glycoprotein VI activation by differential proteome analysis: Implications for thrombus formation. Blood 2010, 115, 4102–4110. [Google Scholar] [CrossRef] [PubMed]
- Bleijerveld, O.B.; Van Holten, T.C.; Preisinger, C.; Van Der Smagt, J.J.; Farndale, R.W.; Kleefstra, T.; Willemsen, M.H.; Urbanus, R.T.; De Groot, P.G.; Heck, A.J.; et al. Targeted Phosphotyrosine Profiling of Glycoprotein VI Signaling Implicates Oligophrenin-1 in Platelet Filopodia Formation. Arter. Thromb. Vasc. Biol. 2013, 33, 1538–1543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- García, Á.; Prabhakar, S.; Hughan, S.; Anderson, T.W.; Brock, C.J.; Pearce, A.C.; Dwek, R.A.; Watson, S.P.; Hebestreit, H.F.; Zitzmann, N. Differential proteome analysis of TRAP-activated platelets: Involvement of DOK-2 and phosphorylation of RGS proteins. Blood 2004, 103, 2088–2095. [Google Scholar] [CrossRef]
- Gachet, C. Antiplatelet drugs: Which targets for which treatments? J. Thromb. Haemost. 2015, 13, S313–S322. [Google Scholar] [CrossRef]
- Schmidt, G.J.; Reumiller, C.M.; Ercan, H.; Resch, U.; Butt, E.; Heber, S.; Liutkevičiūte, Z.; Basílio, J.; Schmid, J.A.; Assinger, A.; et al. Comparative proteomics reveals unexpected quantitative phosphorylation differences linked to platelet activation state. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Makhoul, S.; Walter, E.; Pagel, O.; Walter, U.; Sickmann, A.; Gambaryan, S.; Smolenski, A.; Zahedi, R.P.; Jurk, K. Effects of the NO/soluble guanylate cyclase/cGMP system on the functions of human platelets. Nitric Oxide 2018, 76, 71–80. [Google Scholar] [CrossRef]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef]
- Gambaryan, S.; Tsikas, D. A review and discussion of platelet nitric oxide and nitric oxide synthase: Do blood platelets produce nitric oxide from l-arginine or nitrite? Amino Acids 2015, 47, 1779–1793. [Google Scholar] [CrossRef]
- Gambaryan, S.; Kobsar, A.; Hartmann, S.; Birschmann, I.; Kuhlencordt, P.J.; Müller-Esterl, W.; Lohmann, S.M.; Walter, U. NO-synthase-NO-independent regulation of human and murine platelet soluble guanylyl cyclase activity. J. Thromb. Haemost. 2008, 6, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Rukoyatkina, N.; Herterich, S.; Walter, U.; Gambaryan, S. Soluble guanylyl cyclase is the only enzyme responsible for cyclic guanosine monophosphate synthesis in human platelets. Thromb. Haemost. 2013, 109, 973–975. [Google Scholar] [CrossRef]
- El-Daher, S.S.; Patel, Y.; Siddiqua, A.; Hassock, S.; Edmunds, S.; Maddison, B.; Patel, G.; Goulding, D.; Lupu, F.; Wojcikiewicz, R.J.; et al. Distinct localization and function of (1,4,5)IP(3) receptor subtypes and the (1,3,4,5)IP(4) receptor GAP1(IP4BP) in highly purified human platelet membranes. Blood 2000, 95, 3412–3422. [Google Scholar] [CrossRef]
- Antl, M.; von Brühl, M.L.; Eiglsperger, C.; Werner, M.; Konrad, I.; Kocher, T.; Wilm, M.; Hofmann, F.; Massberg, S.; Schlossmann, J. IRAG mediates NO/cGMP-dependent inhibition of platelet aggregation and thrombus formation. Blood 2007, 109, 552–559. [Google Scholar] [CrossRef]
- Volz, J.; Kusch, C.; Beck, S.; Popp, M.; Vögtle, T.; Meub, M.; Scheller, I.; Heil, H.S.; Preu, J.; Schuhmann, M.K.; et al. BIN2 orchestrates platelet calcium signaling in thrombosis and thrombo-inflammation. J. Clin. Investig. 2020, 130, 6064–6079. [Google Scholar] [CrossRef]
- Aburima, A.; Walladbegi, K.; Wake, J.D.; Naseem, K.M. cGMP signaling inhibits platelet shape change through regulation of the RhoA-Rho Kinase-MLC phosphatase signaling pathway. J. Thromb. Haemost. 2017, 15, 1668–1678. [Google Scholar] [CrossRef]
- Aburima, A.; Wraith, K.S.; Raslan, Z.; Law, R.; Magwenzi, S.; Naseem, K.M. cAMP signaling regulates platelet myosin light chain (MLC) phosphorylation and shape change through targeting the RhoA-Rho kinase-MLC phosphatase signaling pathway. Blood 2013, 122, 3533–3545. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Wennerberg, K.; Burridge, K. Serine Phosphorylation Negatively Regulates RhoA in Vivo. J. Biol. Chem. 2003, 278, 19023–19031. [Google Scholar] [CrossRef] [PubMed]
- Comer, S.; Nagy, Z.; Bolado, A.; Von Kriegsheim, A.; Gambaryan, S.; Walter, U.; Pagel, O.; Zahedi, R.P.; Jurk, K.; Smolenski, A. The RhoA regulators Myo9b and GEF-H1 are targets of cyclic nucleotide-dependent kinases in platelets. J. Thromb. Haemost. 2020, 18, 3002–3012. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Wynne, K.; von Kriegsheim, A.; Gambaryan, S.; Smolenski, A. Cyclic Nucleotide-dependent Protein Kinases Target ARHGAP17 and ARHGEF6 Complexes in Platelets. J. Biol. Chem. 2015, 290, 29974–29983. [Google Scholar] [CrossRef] [PubMed]
- Siess, W.; Winegar, D.A.; Lapetina, E.G. Rap1-b is phosphorylated by protein kinase a in intact human platelets. Biochem. Biophys. Res. Commun. 1990, 170, 944–950. [Google Scholar] [CrossRef]
- Hoffmeister, K.M.; Felbinger, T.W.; Falet, H.; Denis, C.V.; Bergmeier, W.; Mayadas, T.N.; Von Andrian, U.H.; Wagner, D.D.; Stossel, T.P.; Hartwig, J.H. The Clearance Mechanism of Chilled Blood Platelets. Cell 2003, 112, 87–97. [Google Scholar] [CrossRef]
- Subramanian, H.; Zahedi, R.P.; Sickmann, A.; Walter, U.; Gambaryan, S. Phosphorylation of CalDAG-GEFI by protein kinase A prevents Rap1b activation. J. Thromb. Haemost. 2013, 11, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, M.; Riha, P.; Neumüller, O.; Danielewski, O.; Schultess, J.; Smolenski, A.P. Cyclic Nucleotide-dependent Protein Kinases Inhibit Binding of 14-3-3 to the GTPase-activating Protein Rap1GAP2 in Platelets. J. Biol. Chem. 2008, 283, 2297–2306. [Google Scholar] [CrossRef]
- Jones, C.I.; Barrett, N.E.; Moraes, L.A.; Gibbins, J.M.; Jackson, D.E. Endogenous inhibitory mechanisms and the regulation of platelet function. Methods Mol. Biol. 2012, 788, 341–366. [Google Scholar] [PubMed]
- Unsworth, A.J.; Flora, G.D.; Sasikumar, P.; Bye, A.P.; Sage, T.; Kriek, N.; Crescente, M.; Gibbins, J.M. RXR Ligands Negatively Regulate Thrombosis and Hemostasis. Arter. Thromb. Vasc. Biol. 2017, 37, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Bergmeier, W. Negative regulators of platelet activation and adhesion. J. Thromb. Haemost. 2018, 16, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Klockenbusch, C.; Walsh, G.M.; Brown, L.M.; Hoffman, M.D.; Ignatchenko, V.; Kislinger, T.; Kast, J. Global Proteome Analysis Identifies Active Immunoproteasome Subunits in Human Platelets. Mol. Cell. Proteom. 2014, 13, 3308–3319. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.T.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef]
- Dangelmaier, C.A.; Quinter, P.G.; Jin, J.; Tsygankov, A.Y.; Kunapuli, S.P.; Daniel, J.L. Rapid ubiquitination of Syk following GPVI activation in platelets. Blood 2005, 105, 3918–3924. [Google Scholar] [CrossRef] [PubMed]
- Unsworth, A.J.; Bombik, I.; Pinto-Fernandez, A.; McGouran, J.F.; Konietzny, R.; Zahedi, R.P.; Watson, S.P.; Kessler, B.M.; Pears, C.J. Human Platelet Protein Ubiquitylation and Changes following GPVI Activation. Thromb. Haemost. 2018, 119, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.D.; Overall, C.M. Proteolytic Post-translational Modification of Proteins: Proteomic Tools and Methodology. Mol. Cell. Proteom. 2013, 12, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Shema, G.; Nguyen, M.T.N.; Solari, F.A.; Loroch, S.; Venne, A.S.; Kollipara, L.; Sickmann, A.; Verhelst, S.H.; Zahedi, R.P. Simple, scalable, and ultrasensitive tip-based identification of protease substrates. Mol. Cell. Proteom. 2018, 17, 826–834. [Google Scholar] [CrossRef]
- Gutmann, C.; Joshi, A.; Mayr, M. Platelet “-omics” in health and cardiovascular disease. Atherosclerosis 2020, 307, 87–96. [Google Scholar] [CrossRef]
- Parguiña, A.F.; Grigorian-Shamajian, L.; Agra, R.M.; Teijeira-Fernández, E.; Rosa, I.; Alonso, J.; Viñuela-Roldán, J.E.; Seoane, A.; González-Juanatey, J.R.; García, A. Proteins involved in platelet signaling are differentially regulated in acute coronary syndrome: A proteomic study. PLoS ONE 2010, 5, 13404. [Google Scholar]
- Maguire, P.B.; Parsons, M.E.; Szklanna, P.B.; Zdanyte, M.; Münzer, P.; Chatterjee, M.; Wynne, K.; Rath, D.; Comer, S.P.; Hayden, M.; et al. Comparative Platelet Releasate Proteomic Profiling of Acute Coronary Syndrome versus Stable Coronary Artery Disease. Front. Cardiovasc. Med. 2020, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Kuijpers, M.J.E.; Knol, J.C.; Olde Damink, S.W.M.; Dingemans, A.C.; Verheul, H.M.; Piersma, S.R.; Pham, T.V.; Griffioen, A.W.; Oude Egbrink, M.G.A.; et al. Exploration of the platelet proteome in patients with early-stage cancer. J. Proteom. 2018, 177, 65–74. [Google Scholar] [CrossRef]
- Sereni, L.; Castiello, M.C.; Marangoni, F.; Anselmo, A.; di Silvestre, D.; Motta, S.; Draghici, E.; Mantero, S.; Thrasher, A.J.; Giliani, S.; et al. Autonomous role of Wiskott-Aldrich syndrome platelet deficiency in inducing autoimmunity and inflammation. J. Allergy Clin. Immunol. 2018, 142, 1272–1284. [Google Scholar] [CrossRef]
- Jalal, D.I.; Chonchol, M.; Targher, G. Disorders of hemostasis associated with chronic kidney disease. Semin. Thromb. Hemost. 2010, 36, 34–40. [Google Scholar] [CrossRef]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, S.; Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 29–40. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Centron, P.; Barrows, I.; Dwivedi, R.; Raj, D.S. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins (Basel) 2018, 10, 287. [Google Scholar] [CrossRef]
- Fryc, J.; Naumnik, B. Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins (Basel) 2021, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Ravid, J.D.; Chitalia, V.V. Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes. Cells 2020, 9, 2024. [Google Scholar] [CrossRef]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- da Cunha, R.S.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How do Uremic Toxins Affect the Endothelium? Toxins (Basel) 2020, 12, 412. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.S.Y.; Tung, J.-P.; Fraser, J.F. Platelet Storage Lesions: What More Do We Know Now? Transfus. Med. Rev. 2018, 32, 144–154. [Google Scholar] [CrossRef]
- Shea, S.M.; Thomas, K.A.; Spinella, P.C. The effect of platelet storage temperature on haemostatic, immune, and endothelial function: Potential for personalised medicine. Blood Transfus. 2019, 17, 321–330. [Google Scholar] [PubMed]
- Holme, S. Storage and quality assessment of platelets. Vox Sang. 1998, 74, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Aubron, C.; Flint, A.W.J.; Ozier, Y.; McQuilten, Z. Platelet storage duration and its clinical and transfusion outcomes: A systematic review. Crit. Care 2018, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Handigund, M.; Cho, Y.G. Insights into Platelet Storage and the Need for Multiple Approaches. Ann. Clin. Lab. Sci. 2015, 45, 713–719. [Google Scholar] [PubMed]
- Thiele, T.; Steil, L.; Gebhard, S.; Scharf, C.; Hammer, E.; Brigulla, M.; Lubenow, N.; Clemetson, K.J.; Völker, U.; Greinacher, A. Profiling of alterations in platelet proteins during storage of platelet concentrates. Transfusion 2007, 47, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, M.; Eshof, B.L.V.D.; Van Der Meer, P.F.; Van Alphen, F.P.J.; De Korte, D.; Leebeek, F.W.G.; Meijer, A.B.; Voorberg, J.; Jansen, A.J.G. Monitoring storage induced changes in the platelet proteome employing label free quantitative mass spectrometry. Sci. Rep. 2017, 7, 11045. [Google Scholar] [CrossRef]
- Jansen, A.J.; Josefsson, E.C.; Rumjantseva, V.; Liu, Q.P.; Falet, H.; Bergmeier, W.; Cifuni, S.M.; Sackstein, R.; von Andrian, U.H.; Wagner, D.D.; et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIbalpha metalloproteinase-mediated cleavage in mice. Blood 2012, 119, 1263–1273. [Google Scholar] [CrossRef]
- Rumjantseva, V.; Grewal, P.K.; Wandall, H.H.; Josefsson, E.C.; Sørensen, A.L.; Larson, G.; Marth, J.D.; Hartwig, J.H.; Hoffmeister, K.M. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat. Med. 2009, 15, 1273–1280. [Google Scholar] [CrossRef]
- Kleinveld, D.J.; Sloos, P.H.; Noorman, F.; Maas, M.A.W.; Kers, J.; Rijnhout, T.W.; Zoodsma, M.; Hoencamp, R.; Hollmann, M.W.; Juffermans, N.P. The use of cryopreserved platelets in a trauma-induced hemorrhage model. Transfusion 2020, 60, 2079–2089. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, T.; Fan, Y.; Zhao, S. A proteomic approach reveals the variation in human platelet protein composition after storage at different temperatures. Platelets 2018, 30, 403–412. [Google Scholar] [CrossRef]
- Kaiser-Guignard, J.; Canellini, G.; Lion, N.; Abonnenc, M.; Osselaer, J.-C.; Tissot, J.-D. The clinical and biological impact of new pathogen inactivation technologies on platelet concentrates. Blood Rev. 2014, 28, 235–241. [Google Scholar] [CrossRef]
- Sonego, G.; Abonnenc, M.; Crettaz, D.; Lion, N.; Tissot, J.-D.; Prudent, M. Irreversible oxidations of platelet proteins after riboflavin-UVB pathogen inactivation. Transfus. Clin. Biol. 2020, 27, 36–42. [Google Scholar] [CrossRef]
- Hermida-Nogueira, L.; Barrachina, M.N.; Izquierdo, I.; García-Vence, M.; Lacerenza, S.; Bravo, S.; Castrillo, A.; García, Á. Proteomic analysis of extracellular vesicles derived from platelet concentrates treated with Mirasol(R) identifies biomarkers of platelet storage lesion. J. Proteom. 2020, 210, 103529. [Google Scholar] [CrossRef]
- Salunkhe, V.; De Cuyper, I.M.; Papadopoulos, P.; Van Der Meer, P.F.; Daal, B.B.; Villa-Fajardo, M.; De Korte, D.; Berg, T.K.V.D.; Gutiérrez, L. A comprehensive proteomics study on platelet concentrates: Platelet proteome, storage time and Mirasol pathogen reduction technology. Platelets 2018, 30, 368–379. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shevchuk, O.; Begonja, A.J.; Gambaryan, S.; Totzeck, M.; Rassaf, T.; Huber, T.B.; Greinacher, A.; Renne, T.; Sickmann, A. Proteomics: A Tool to Study Platelet Function. Int. J. Mol. Sci. 2021, 22, 4776. https://doi.org/10.3390/ijms22094776
Shevchuk O, Begonja AJ, Gambaryan S, Totzeck M, Rassaf T, Huber TB, Greinacher A, Renne T, Sickmann A. Proteomics: A Tool to Study Platelet Function. International Journal of Molecular Sciences. 2021; 22(9):4776. https://doi.org/10.3390/ijms22094776
Chicago/Turabian StyleShevchuk, Olga, Antonija Jurak Begonja, Stepan Gambaryan, Matthias Totzeck, Tienush Rassaf, Tobias B. Huber, Andreas Greinacher, Thomas Renne, and Albert Sickmann. 2021. "Proteomics: A Tool to Study Platelet Function" International Journal of Molecular Sciences 22, no. 9: 4776. https://doi.org/10.3390/ijms22094776
APA StyleShevchuk, O., Begonja, A. J., Gambaryan, S., Totzeck, M., Rassaf, T., Huber, T. B., Greinacher, A., Renne, T., & Sickmann, A. (2021). Proteomics: A Tool to Study Platelet Function. International Journal of Molecular Sciences, 22(9), 4776. https://doi.org/10.3390/ijms22094776