
Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters

 , , , and
, , , and 
Abstract
1. Introduction
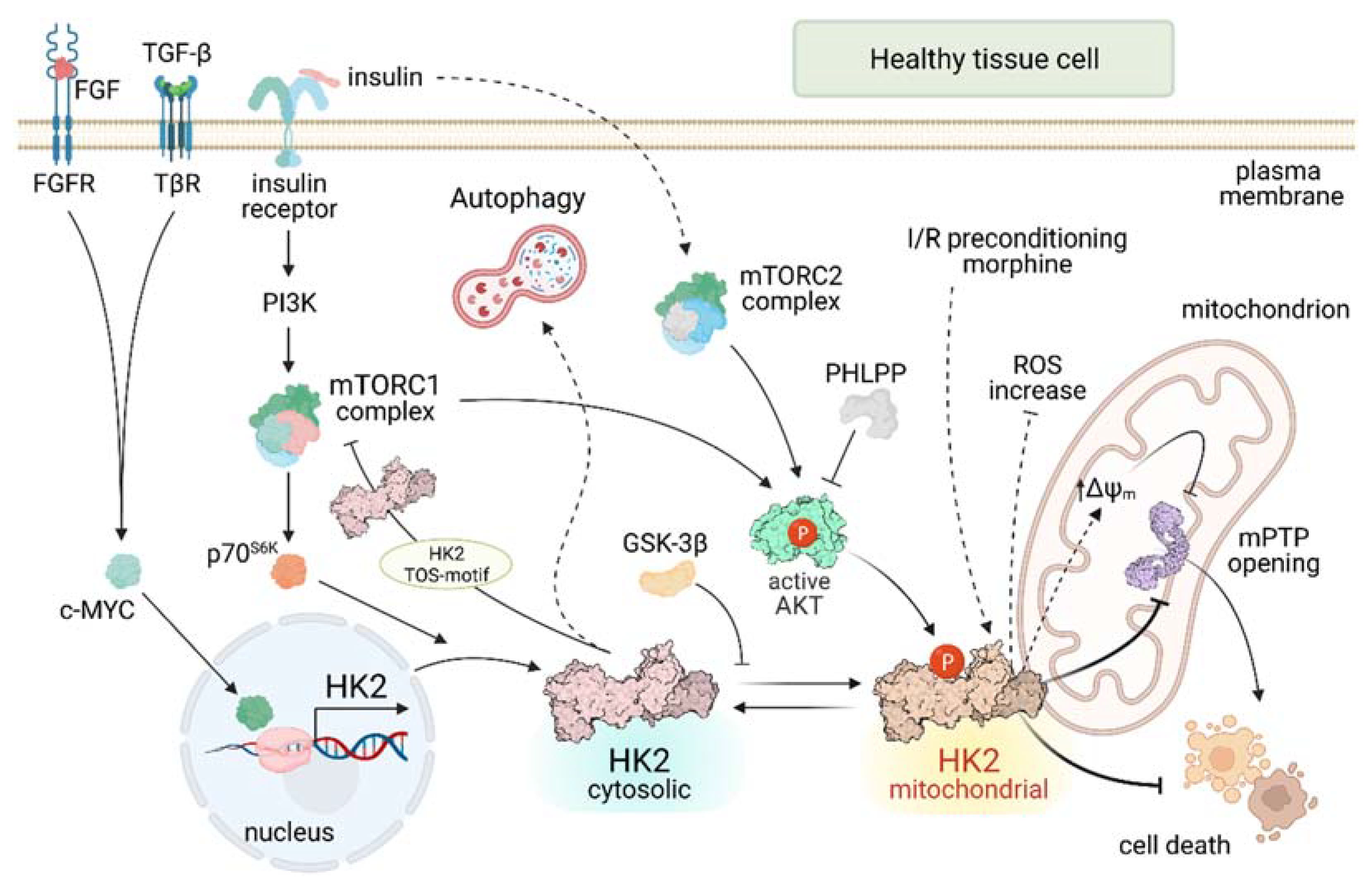
2. HK2 Regulation: Where, How and Why
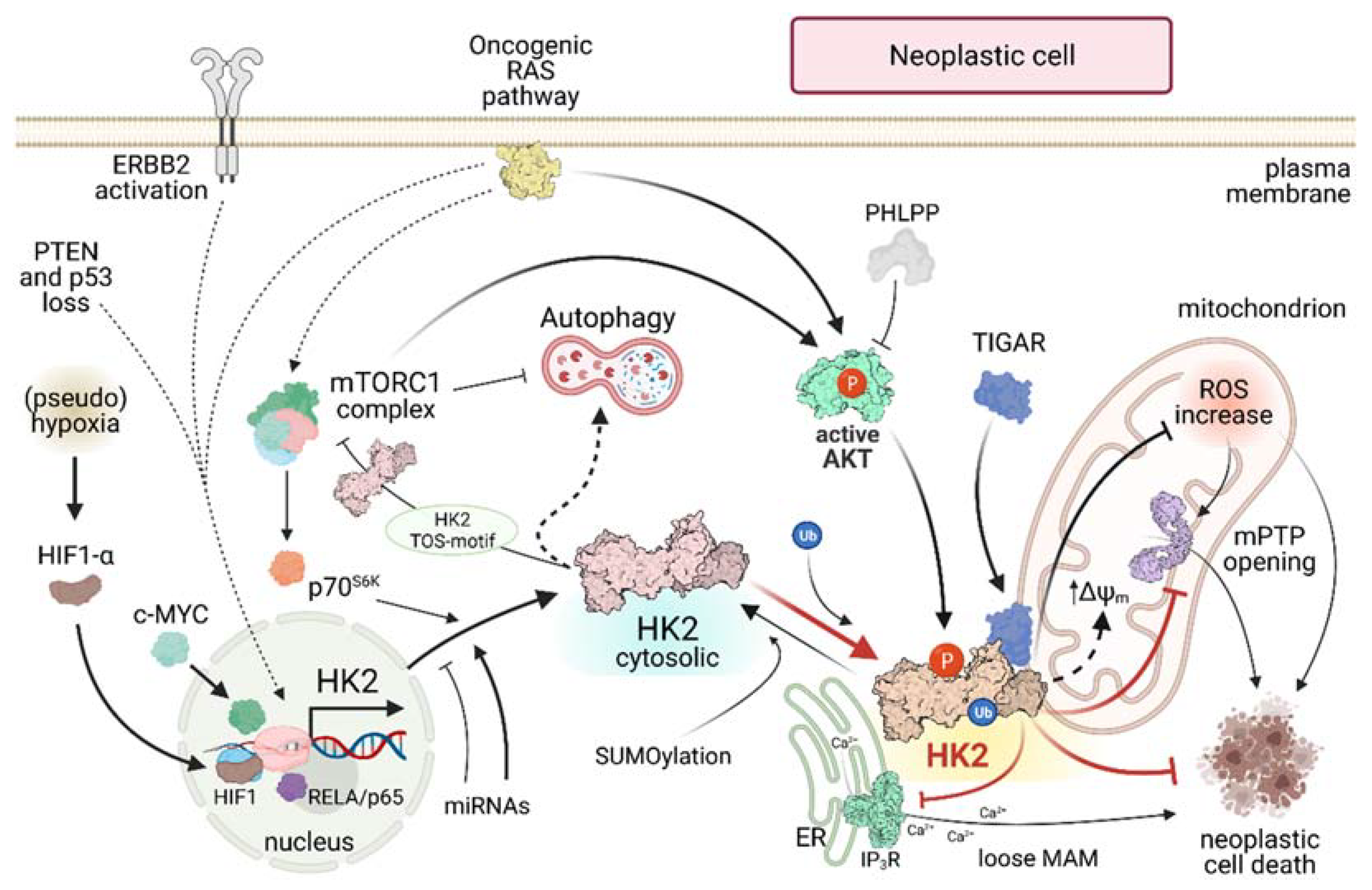
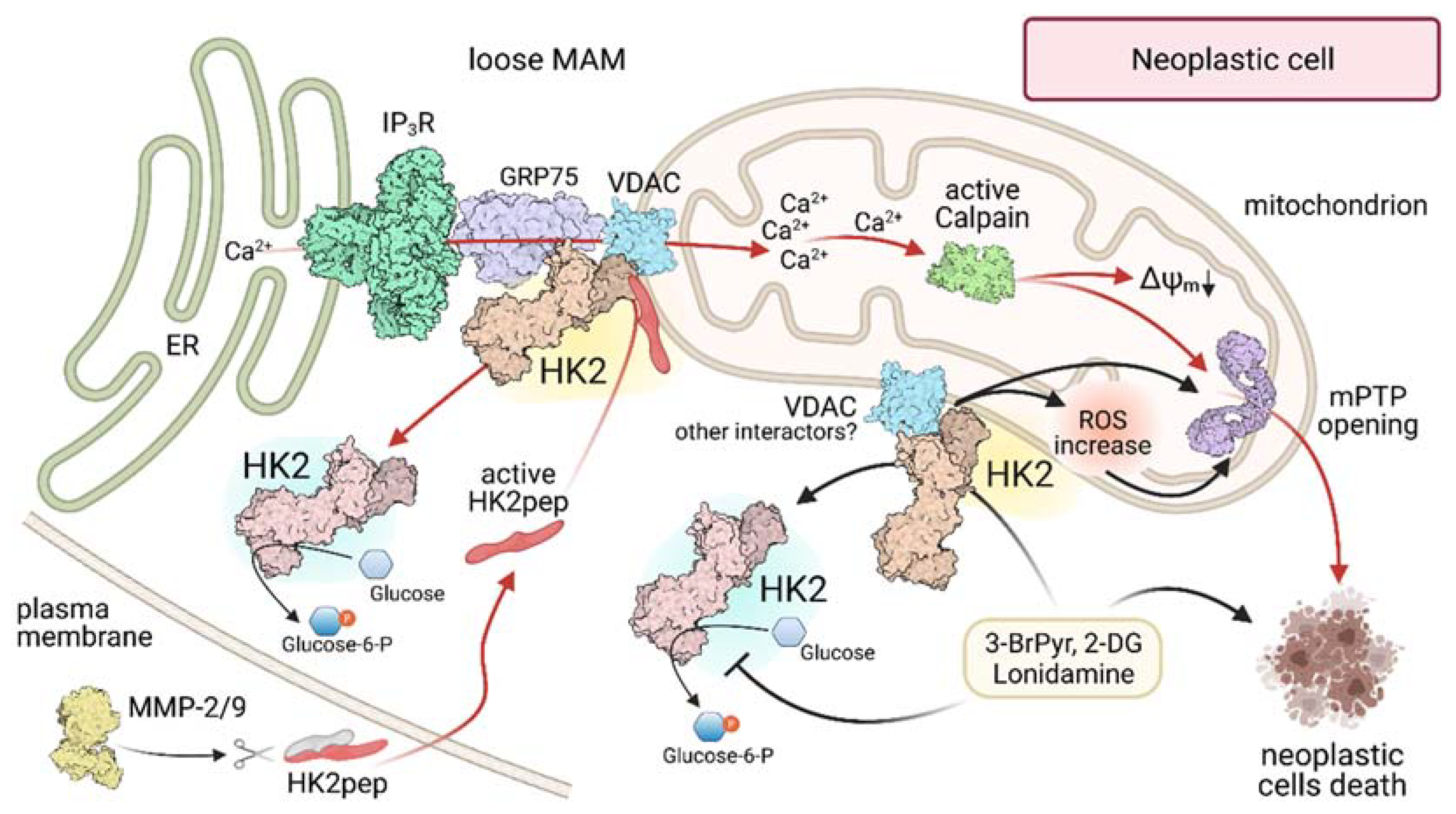
3. HK2 on Mitochondria of Neoplastic Cells: Maintaining the Bad Guy in Perfect Shape
4. Spotting and Picking off HK2 in Tumors: A Matter of Precision
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef]
- Irwin, D.M.; Tan, H. Molecular evolution of the vertebrate hexokinase gene family: Identification of a conserved fifth vertebrate hexokinase gene. Comp. Biochem. Physiol. Part D Genom. Proteom. 2008, 3, 96–107. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: AKTing on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.E.; Mayfield, A.C.; Haas, J.A. Heart muscle hexokinase: Subcellular distribution and inhibition by glucose 6-phosphate. Mol. Pharmacol. 1966, 2, 393–405. [Google Scholar] [PubMed]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar] [CrossRef]
- Yin, X.; Choudhury, M.; Kang, J.H.; Schaefbauer, K.J.; Jung, M.Y.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-beta. Sci. Signal. 2019, 12, eaax4067. [Google Scholar] [CrossRef]
- Podar, K.; Anderson, K.C. A therapeutic role for targeting c-akt/Hif-1-dependent signaling pathways. Cell Cycle 2010, 9, 1722–1728. [Google Scholar] [CrossRef]
- Corona, J.C.; Gimenez-Cassina, A.; Lim, F.; Diaz-Nido, J. Hexokinase II gene transfer protects against neurodegeneration in the rotenone and MPTP mouse models of Parkinson’s disease. J. Neurosci. Res. 2010, 88, 1943–1950. [Google Scholar] [CrossRef]
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2013, 2, e000355. [Google Scholar] [CrossRef]
- Wu, R.; Smeele, K.M.; Wyatt, E.; Ichikawa, Y.; Eerbeek, O.; Sun, L.; Chawla, K.; Hollmann, M.W.; Nagpal, V.; Heikkinen, S.; et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ. Res. 2011, 108, 60–69. [Google Scholar] [CrossRef]
- Wu, R.; Wyatt, E.; Chawla, K.; Tran, M.; Ghanefar, M.; Laakso, M.; Epting, C.L.; Ardehali, H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol. Med. 2012, 4, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Pietila, M.; Halmekyto, M.; Suppola, S.; Pirinen, E.; Deeb, S.S.; Janne, J.; Laakso, M. Hexokinase II-deficient mice. Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J. Biol. Chem. 1999, 274, 22517–22523. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, P.T.; Nogueira, V.; Patra, K.C.; Jeon, S.M.; Park, Y.; Robey, R.B.; Hay, N. mTORC1 hyperactivity inhibits serum deprivation-induced apoptosis via increased hexokinase II and GLUT1 expression, sustained Mcl-1 expression, and glycogen synthase kinase 3beta inhibition. Mol. Cell. Biol. 2009, 29, 5136–5147. [Google Scholar] [CrossRef]
- Osawa, H.; Sutherland, C.; Robey, R.B.; Printz, R.L.; Granner, D.K. Analysis of the signaling pathway involved in the regulation of hexokinase II gene transcription by insulin. J. Biol. Chem. 1996, 271, 16690–16694. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell. Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-AKT signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. AKT mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. AKT phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- Chiara, F.; Rasola, A. GSK-3 and mitochondria in cancer cells. Front. Oncol. 2013, 3, 16. [Google Scholar] [CrossRef]
- Miyamoto, S.; Purcell, N.H.; Smith, J.M.; Gao, T.; Whittaker, R.; Huang, K.; Castillo, R.; Glembotski, C.C.; Sussman, M.A.; Newton, A.C.; et al. PHLPP-1 negatively regulates AKT activity and survival in the heart. Circ. Res. 2010, 107, 476–484. [Google Scholar] [CrossRef]
- Chiara, F.; Castellaro, D.; Marin, O.; Petronilli, V.; Brusilow, W.S.; Juhaszova, M.; Sollott, S.J.; Forte, M.; Bernardi, P.; Rasola, A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 2008, 3, e1852. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833. [Google Scholar] [CrossRef] [PubMed]
- Pantic, B.; Trevisan, E.; Citta, A.; Rigobello, M.P.; Marin, O.; Bernardi, P.; Salvatori, S.; Rasola, A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Eerbeek, O.; Meijer, A.J. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H496–H499. [Google Scholar] [CrossRef]
- Calmettes, G.; Ribalet, B.; John, S.; Korge, P.; Ping, P.; Weiss, J.N. Hexokinases and cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 107–115. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Zorov, S.D.; Kotova, E.A.; Zorov, D.B.; Antonenko, Y.N. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010, 584, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Zhao, Y.; Zhang, J.; Liu, B.; Jiao, S.; Zhang, X. Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic. Biol. Med. 2019, 131, 251–263. [Google Scholar] [CrossRef]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.Y.; Lin, L.; Liu, B.L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef]
- Ishaq, M.; Ojha, R.; Sharma, A.P.; Singh, S.K. Autophagy in cancer: Recent advances and future directions. Semin. Cancer Biol. 2020, 66, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- Ikeda, S.; Abe, F.; Matsuda, Y.; Kitadate, A.; Takahashi, N.; Tagawa, H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci. 2020, 111, 4088–4101. [Google Scholar] [CrossRef]
- Tan, V.P.; Smith, J.M.; Tu, M.; Yu, J.D.; Ding, E.Y.; Miyamoto, S. Dissociation of mitochondrial HK-II elicits mitophagy and confers cardioprotection against ischemia. Cell Death Dis. 2019, 10, 730. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, X.; Zhang, Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819871306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, M.; Cong, Q.; Zhang, M.X.; Zhang, M.Y.; Lu, Y.Y.; Xu, C.J. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef]
- Singh, A.; Sen, E. Reciprocal role of SIRT6 and Hexokinase 2 in the regulation of autophagy driven monocyte differentiation. Exp. Cell Res. 2017, 360, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Vidalain, P.O.; Jacquemin, C.; Aublin-Gex, A.; Olmstead, K.; Panthu, B.; Rautureau, G.J.P.; Andre, P.; Nyczka, P.; Hutt, M.T.; et al. A hexokinase isoenzyme switch in human liver cancer cells promotes lipogenesis and enhances innate immunity. Commun. Biol. 2021, 4, 217. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.M.; Weinberg, S.E.; Steinert, E.M.; Chhiba, K.; Martinez, C.A.; Gao, P.; Perlman, H.R.; Bryce, P.; Hay, N.; Chandel, N.S. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab. 2018, 6, 10. [Google Scholar] [CrossRef]
- Kumar, V. How could we forget immunometabolism in SARS-CoV2 infection or COVID-19? Int. Rev. Immunol. 2020, 40, 72–107. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 2010, 1797, 1225–1230. [Google Scholar] [CrossRef]
- Gwak, G.Y.; Yoon, J.H.; Kim, K.M.; Lee, H.S.; Chung, J.W.; Gores, G.J. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 2005, 42, 358–364. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell. Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Puliga, E.; Cabras, L.; Sulas, P.; Petrelli, A.; Perra, A.; Ledda-Columbano, G.M.; Morandi, A.; Merlin, S.; Orru, C.; et al. Thyroid hormone inhibits hepatocellular carcinoma progression via induction of differentiation and metabolic reprogramming. J. Hepatol. 2020, 72, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, M.; Karasawa, H.; Takagi, K.; Nakayama, S.; Yabuuchi, S.; Fujishima, F.; Naitoh, T.; Watanabe, M.; Suzuki, T.; Unno, M.; et al. Hexokinase 2 in colorectal cancer: A potent prognostic factor associated with glycolysis, proliferation and migration. Histol. Histopathol. 2017, 32, 351–360. [Google Scholar] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 dimerization and interaction with voltage-dependent anion channel promoted resistance to cell apoptosis induced by gemcitabine in pancreatic cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef]
- Huang, X.; Liu, M.; Sun, H.; Wang, F.; Xie, X.; Chen, X.; Su, J.; He, Y.; Dai, Y.; Wu, H.; et al. HK2 is a radiation resistant and independent negative prognostic factor for patients with locally advanced cervical squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 4054–4063. [Google Scholar]
- Lee, H.J.; Li, C.F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Jiang, L.; Ren, X.; Cheng, B.; Xia, J. Prognostic value of glycolysis markers in head and neck squamous cell carcinoma: A meta-analysis. Aging 2021, 13, 7284–7299. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, K.; Shi, L.; Xiang, F.; Tao, K.; Wang, G. Prognostic Significance of the Metabolic Marker Hexokinase-2 in Various Solid Tumors: A Meta-Analysis. PLoS ONE 2016, 11, e0166230. [Google Scholar]
- Wu, J.; Hu, L.; Wu, F.; Zou, L.; He, T. Poor prognosis of hexokinase 2 overexpression in solid tumors of digestive system: A meta-analysis. Oncotarget 2017, 8, 32332–32344. [Google Scholar] [CrossRef] [PubMed]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Patra, K.C.; Hay, N. Selective eradication of cancer displaying hyperactive AKT by exploiting the metabolic consequences of AKT activation. Elife 2018, 7, e32213. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci. Rep. 2018, 8, 744. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, A.; Agnihotri, S.; Wilson, M.R.; Burrell, K.E.; Tonge, P.D.; Alamsahebpour, A.; Jalali, S.; Taccone, M.S.; Mansouri, S.; Golbourn, B.; et al. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget 2016, 7, 69518–69535. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.J.; Yu, S.J.; Na, J.; Kim, K.; Cho, Y.Y.; Lee, Y.B.; Cho, E.J.; Lee, J.H.; Kim, Y.J.; Youn, H.; et al. Hexokinase-II Inhibition Synergistically Augments the Anti-tumor Efficacy of Sorafenib in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1292. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget 2018, 9, 4020–4033. [Google Scholar] [CrossRef]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sanchez-Martin, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Tateishi, K.; Miyake, Y.; Kawazu, M.; Sasaki, N.; Nakamura, T.; Sasame, J.; Yoshii, Y.; Ueno, T.; Miyake, A.; Watanabe, J.; et al. A Hyperactive RelA/p65-Hexokinase 2 Signaling Axis Drives Primary Central Nervous System Lymphoma. Cancer Res. 2020, 80, 5330–5343. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, S.; Schneider, B.K.; Allen, C.B.; Chang, L.Y.; White, C.W. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L573–L584. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Giacobbe, A.; Formosa, A.; Markert, E.K.; Bongiorno-Borbone, L.; Levine, A.J.; Candi, E.; D’Alessandro, A.; Zolla, L.; Finazzi Agro, A.; et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene 2013, 32, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wang, Y.; Zhu, Y.; Li, S.; Liu, Y.; Chen, C.; Wang, X.; Zen, K.; Li, L. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci. Rep. 2017, 7, 3089. [Google Scholar] [CrossRef]
- Liu, S.; Chen, Q.; Wang, Y. MiR-125b-5p suppresses the bladder cancer progression via targeting HK2 and suppressing PI3K/AKT pathway. Hum. Cell. 2020, 33, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huo, Y.; Wang, D.; Tai, Y.; Li, J.; Pang, D.; Zhang, Y.; Zhao, W.; Du, N.; Huang, Y. MiR-216a-5p/Hexokinase 2 axis regulates uveal melanoma growth through modulation of Warburg effect. Biochem. Biophys. Res. Commun. 2018, 501, 885–892. [Google Scholar] [CrossRef]
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996. [Google Scholar] [CrossRef]
- Li, H.; Lu, S.; Chen, Y.; Zheng, L.; Chen, L.; Ding, H.; Ding, J.; Lou, D.; Liu, F.; Zheng, B. AKT2 phosphorylation of hexokinase 2 at T473 promotes tumorigenesis and metastasis in colon cancer cells via NF-kappaB, HIF1alpha, MMP2, and MMP9 upregulation. Cell. Signal. 2019, 58, 99–110. [Google Scholar] [CrossRef]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wen, Y.A.; Mitov, M.I.; Oaks, M.C.; Miyamoto, S.; Gao, T. PHLPP regulates hexokinase 2-dependent glucose metabolism in colon cancer cells. Cell Death Discov. 2017, 3, 16103. [Google Scholar] [CrossRef] [PubMed]
- Baffi, T.R.; Cohen-Katsenelson, K.; Newton, A.C. PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 723–743. [Google Scholar] [CrossRef]
- Tang, J.; Chen, L.; Qin, Z.H.; Sheng, R. Structure, regulation, and biological functions of TIGAR and its role in diseases. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by AKT is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell. Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef]
- Arzoine, L.; Zilberberg, N.; Ben-Romano, R.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 2009, 284, 3946–3955. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and AKT. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Filadi, R.; Masgras, I.; Pizzi, M.; Marin, O.; Damiano, N.; Pizzo, P.; Gori, A.; Frezzato, F.; Chiara, F.; et al. Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+-dependent death of cancer cells. EMBO Rep. 2020, 21, e49117. [Google Scholar] [CrossRef]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Subcell. Biochem. 2007, 45, 481–506. [Google Scholar]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca2+ Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 14, 2313–2324. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Hamada, K.; Mikoshiba, K. IP3 Receptor Plasticity Underlying Diverse Functions. Annu. Rev. Physiol. 2020, 82, 151–176. [Google Scholar] [CrossRef]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef]
- Kim, W.; Yoon, J.H.; Jeong, J.M.; Cheon, G.J.; Lee, T.S.; Yang, J.I.; Park, S.C.; Lee, H.S. Apoptosis-inducing antitumor efficacy of hexokinase II inhibitor in hepatocellular carcinoma. Mol. Cancer Ther. 2007, 6, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Calvino, E.; Estan, M.C.; Sanchez-Martin, C.; Brea, R.; de Blas, E.; Boyano-Adanez Mdel, C.; Rial, E.; Aller, P. Regulation of death induction and chemosensitizing action of 3-bromopyruvate in myeloid leukemia cells: Energy depletion, oxidative stress, and protein kinase activity modulation. J. Pharmacol. Exp. Ther. 2014, 348, 324–335. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.F.; Qiu, Y.Y.; Yu, S.; Clark, S.; Chu, F.; Madonna, M.B. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J. Pediatr. Surg. 2014, 49, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xu, J.; Yuan, W.; Wu, B.; Wang, H.; Liu, G.; Wang, X.; Du, J.; Cai, S. The reversal effects of 3-bromopyruvate on multidrug resistance in vitro and in vivo derived from human breast MCF-7/ADR cells. PLoS ONE 2014, 9, e112132. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, J.; North, P.E.; Yang, S.; Lubet, R.A.; Wang, Y.; You, M. Aerosolized 3-bromopyruvate inhibits lung tumorigenesis without causing liver toxicity. Cancer Prev. Res. 2012, 5, 717–725. [Google Scholar] [CrossRef]
- El Sayed, S.M.; Mohamed, W.G.; Seddik, M.A.; Ahmed, A.S.; Mahmoud, A.G.; Amer, W.H.; Helmy Nabo, M.M.; Hamed, A.R.; Ahmed, N.S.; Abd-Allah, A.A. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: A concise literature review and case study. Chin. J. Cancer 2014, 33, 356–364. [Google Scholar]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef]
- Masgras, I.; Rasola, A.; Bernardi, P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochim. Biophys. Acta 2012, 1817, 1860–1866. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Olson, E.S.; Timmers, M.M.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr. Biol. 2009, 1, 371–381. [Google Scholar] [CrossRef]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. 2009, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | HK2 Expression | Resistance | References |
|---|---|---|---|
| Hepatocellular carcinoma (HCC) | Poor prognosis; tumor progression | Sorafenib | [45,60,61,62,67] |
| Colorectal cancer | Poor prognosis; tumor progression | [54,60,61] | |
| Glioblastoma | Poor prognosis; tumor progression | Radiotherapy | [55,66] |
| Pancreatic cancer | Poor prognosis | Gemcitabine | [56] |
| Cervical squamous cell carcinoma | Poor prognosis | Radiotherapy; 4-hydroxytamoxifen | [39,57] |
| Prostate Cancer | Poor prognosis; tumor progression | [58,63,64] | |
| Head and neck squamous cell carcinoma | Poor prognosis | [59] | |
| Gastric cancer | Poor prognosis | [60,61] | |
| Lung cancer | Tumor progression | [15] | |
| Breast cancer | Tumor progression | Paclitaxel | [15,38] |
| Diffuse large B-cell lymphoma | Tumor progression | R-CHOP regimen: Rituximab + cyclophosphamide, doxorubicin, vincristine and prednisone | [65,68] |
| Ovarian Cancer | Cisplatin | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. https://doi.org/10.3390/ijms22094716
Ciscato F, Ferrone L, Masgras I, Laquatra C, Rasola A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. International Journal of Molecular Sciences. 2021; 22(9):4716. https://doi.org/10.3390/ijms22094716
Chicago/Turabian StyleCiscato, Francesco, Lavinia Ferrone, Ionica Masgras, Claudio Laquatra, and Andrea Rasola. 2021. "Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters" International Journal of Molecular Sciences 22, no. 9: 4716. https://doi.org/10.3390/ijms22094716
APA StyleCiscato, F., Ferrone, L., Masgras, I., Laquatra, C., & Rasola, A. (2021). Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. International Journal of Molecular Sciences, 22(9), 4716. https://doi.org/10.3390/ijms22094716