
Mesoporous Silica-Bioglass Composite Pellets as Bone Drug Delivery System with Mineralization Potential

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. DOX Adsorption onto MCM-41
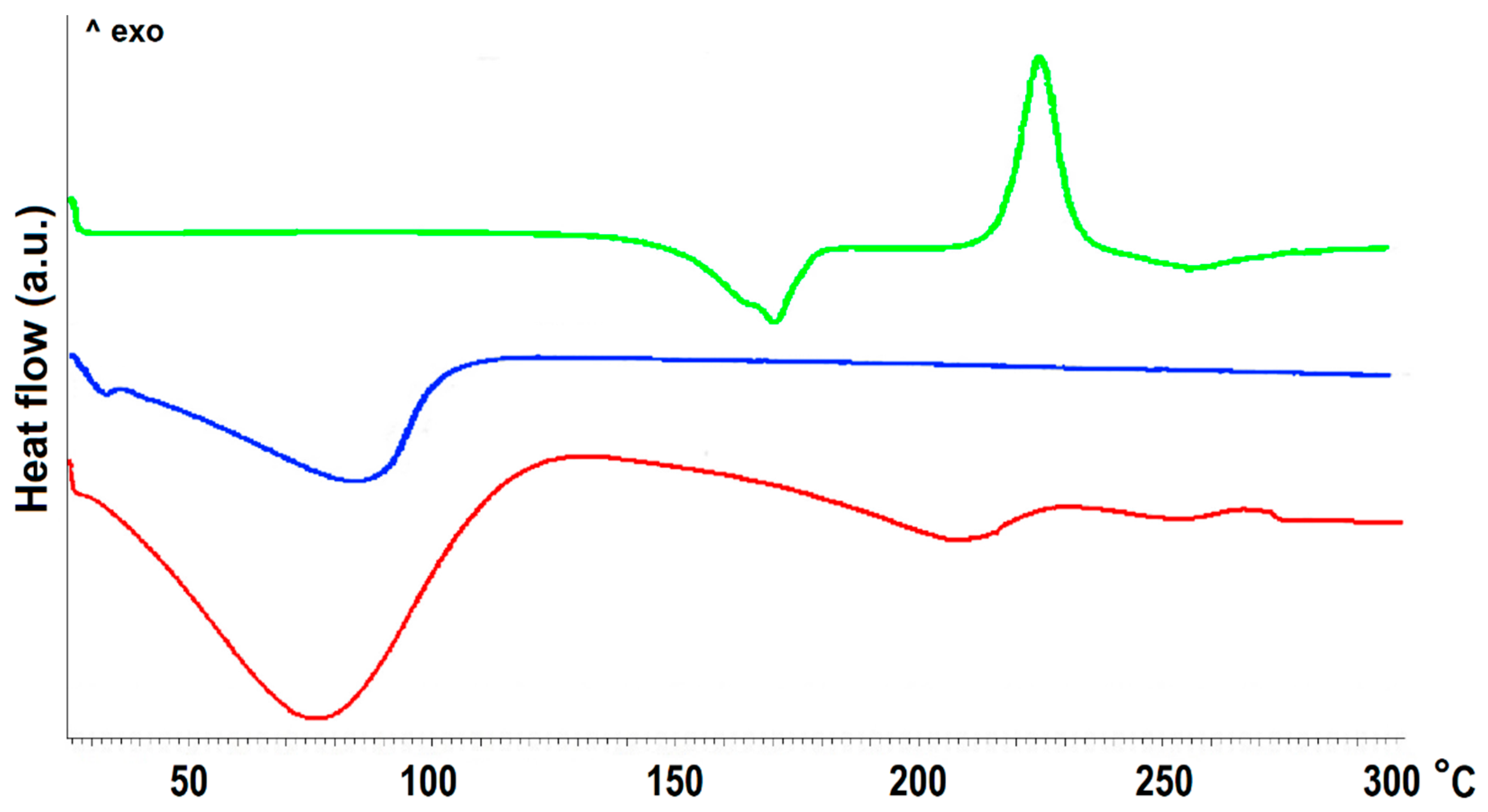
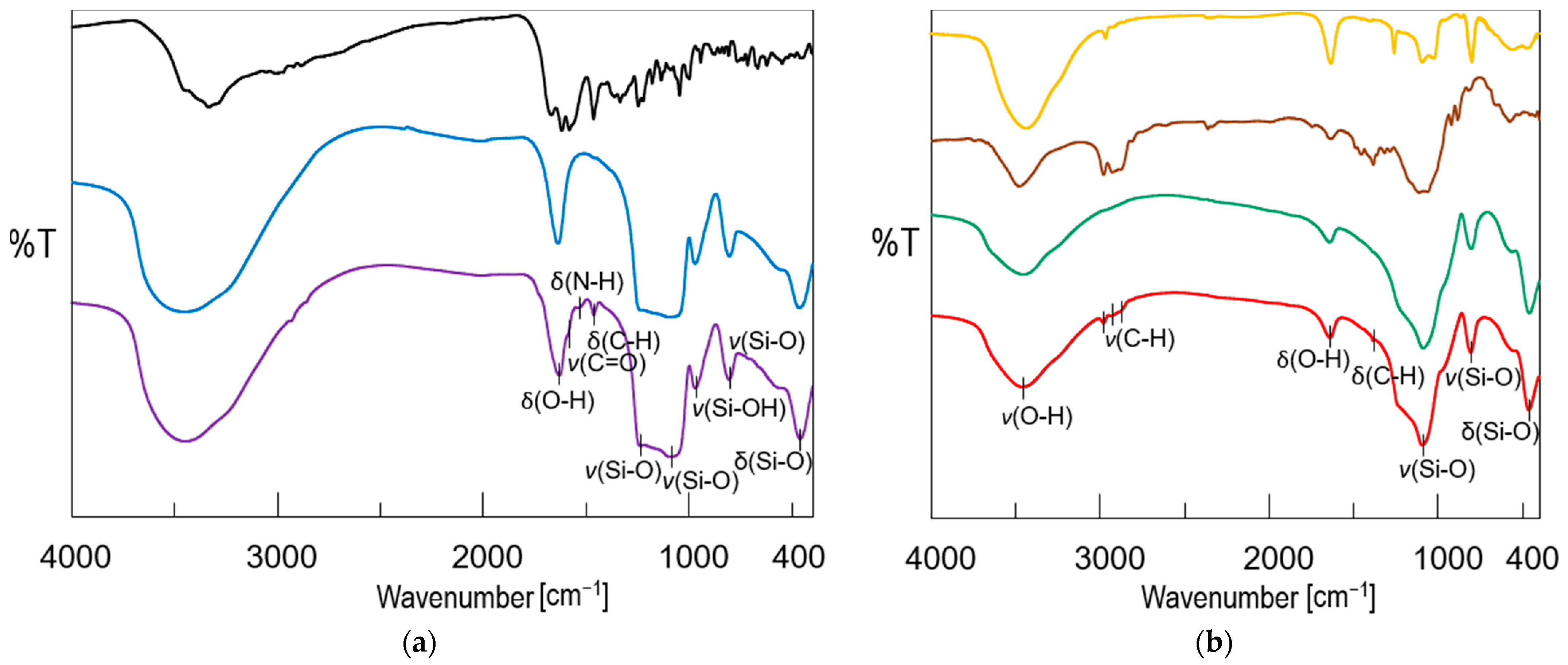
2.2. Physicochemical Characterization of Pellets
2.3. Drug Release Profiles
2.4. Antimicrobial Activity of Pellets
2.5. Mineralization Potential Assay
2.6. Cytotoxicity Assay of Pellets
3. Materials and Methods
3.1. Synthesis of Bioglass
3.2. Synthesis of Mesoporous Silica MCM-41
3.3. Adsorption of Doxycycline Hydrochloride onto the MCM-41
3.4. Preparation of Pellets Composed of BG and MCM-DOX
3.5. Drug Release Studies
3.6. Antimicrobial Activity Assay
3.7. Mineralization Potential Assay
3.8. Cytotoxicity Assay
3.9. Physicochemical Characterization Methods
3.9.1. Fourier Transform Infrared Spectroscopy Analysis
3.9.2. Differential Scanning Calorimetry Analysis
3.9.3. Powder X-ray Diffraction Analysis
3.9.4. Stereoscopic Microscopy
3.9.5. Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy
3.9.6. Yield of Pelletization Process
3.9.7. Size Analysis
3.9.8. Friability Test
3.9.9. Hardness Test
3.9.10. Drug Content Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sayed, E.; Haj-Ahmad, R.; Ruparelia, K.; Arshad, M.S.; Chang, M.W.; Ahmad, Z. Porous inorganic drug delivery systems—A review. AAPS PharmSciTech 2017, 18, 1507–1525. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Lozano, D.; González, B.; Izquierdo-Barba, I. Biomaterials against bone infection. Adv. Healthc. Mater. 2020, 7, 2000310. [Google Scholar] [CrossRef]
- Lazzarini, L.; Mader, J.T.; Calhoun, J.H. Osteomyelitis in long bones. JBJS 2004, 86, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Harrison, W.J.; Esterhai, J.L. Osteomyelitis and septic arthritis in adults. In Global Orthopedics; Gosselin, R., Spiegel, D., Foltz, M., Eds.; Springer International Publishing: New York, NY, USA, 2020; pp. 351–360. [Google Scholar]
- Rosenberg, A.E.; Kattapuram, S.V.; Nielsen, G.P. Bone infections. In Diagnostic Pathology of Infectious Disease; Elsevier: Philadelphia, PA, USA, 2010; pp. 341–375. ISBN 9781416034292. [Google Scholar]
- Arcos, D.; Vallet-Regí, M. Sol–gel silica-based biomaterials and bone tissue regeneration. Acta Biomater. 2010, 6, 2874–2888. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, N.; Mohammed, H.; Al-Hadeethi, Y.; Bakry, A.S.; Umar, A.; Hussein, M.A.; Abbassy, M.A.; Vaidya, K.G.; Al Berakdar, G.; Mkawi, E.M.; et al. Silica-based bioactive glasses and their applications in hard tissue regeneration: A review. Pharmaceuticals 2021, 14, 75. [Google Scholar] [CrossRef]
- Fruijtier-Pölloth, C. The toxicological mode of action and the safety of synthetic amorphous silica-a nanostructured material. Toxicology 2012, 294, 61–79. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Q.; Ding, X.; Shen, Q.; Wu, C.; Zhang, L.; Yang, H. Synthesis and application of several sol–gel-derived materials via sol–gel process combining with other technologies: A review. J. Sol-Gel Sci. Technol. 2016, 79, 328–358. [Google Scholar] [CrossRef]
- Terracciano, M.; De Stefano, L.; Rea, I. Diatoms green nanotechnology for biosilica-based drug delivery systems. Pharmaceutics 2018, 10, 242. [Google Scholar] [CrossRef]
- Bakar, R.A.; Yahya, R.; Gan, S.N. Production of high purity amorphous silica from rice husk. Procedia Chem. 2016, 19, 189–195. [Google Scholar] [CrossRef]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef]
- Jones, J.R. Review of bioactive glass: From Hench to hybrids. Acta Biomater. 2013, 9, 4457–4486. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Gun’ko, Y.; Vallet-Regí, M. Mesoporous silica materials as drug delivery: “The nightmare” of bacterial infection. Pharmaceutics 2018, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Clark, A.E.; Hench, L.L. An investigation of bioactive glass powders by sol-gel processing. J. Appl. Biomater. 1991, 2, 231–239. [Google Scholar] [CrossRef]
- Liang, W.; Wu, X.; Dong, Y.; Shao, R.; Chen, X.; Zhou, P.; Xu, F. In vivo behavior of bioactive glass-based composites in animal models for bone regeneration. Biomater. Sci. 2021, 9, 1924–1944. [Google Scholar] [CrossRef] [PubMed]
- Sergi, R.; Bellucci, D.; Cannillo, V. A review of bioactive glass/natural polymer composites: State of the art. Materials 2020, 13, 5560. [Google Scholar] [CrossRef] [PubMed]
- Hum, J.; Boccaccini, A. Collagen as coating material for 45S5 bioactive glass-based scaffolds for bone tissue engineering. Int. J. Mol. Sci. 2018, 19, 1807. [Google Scholar] [CrossRef] [PubMed]
- Bakry, A.S.; Marghalani, H.Y.; Amin, O.A.; Tagami, J. The effect of a bioglass paste on enamel exposed to erosive challenge. J. Dent. 2014, 42, 1458–1463. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Szewczyk, A.; Skwira, A.; Sądej, R.; Walker, G. Biphasic composite of calcium phosphate-based mesoporous silica as a novel bone drug delivery system. Drug Deliv. Transl. Res. 2020, 10, 455–470. [Google Scholar] [CrossRef]
- Skwira, A.; Szewczyk, A.; Konopacka, A.; Górska, M.; Majda, D.; Sądej, R.; Prokopowicz, M.; Sadej, R.; Prokopowicz, M. Silica-polymer composites as the novel antibiotic delivery systems for bone tissue infection. Pharmaceutics 2020, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M. Ordered mesoporous materials in the context of drug delivery systems and bone tissue engineering. Chem. A Eur. J. 2006, 12, 5934–5943. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Ruiz-González, L.; Izquierdo-Barba, I.; González-Calbet, J.M. Revisiting silica based ordered mesoporous materials: Medical applications. J. Mater. Chem. 2006, 16, 26–31. [Google Scholar] [CrossRef]
- Boccardi, E.; Philippart, A.; Juhasz-Bortuzzo, J.A.; Beltrán, A.M.; Novajra, G.; Vitale-Brovarone, C.; Spiecker, E.; Boccaccini, A.R. Uniform surface modification of 3D Bioglass®-based scaffolds with mesoporous silica particles (MCM-41) for enhancing drug delivery capability. Front. Bioeng. Biotechnol. 2015, 3. [Google Scholar] [CrossRef]
- Slane, J.; Vivanco, J.; Ebenstein, D.; Squire, M.; Ploeg, H.-L. Multiscale characterization of acrylic bone cement modified with functionalized mesoporous silica nanoparticles. J. Mech. Behav. Biomed. Mater. 2014, 37, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Lipsky, B.A. Systemic antibiotic therapy for chronic osteomyelitis in adults. Clin. Infect. Dis. 2012, 54, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Skwira, A.; Szewczyk, A.; Prokopowicz, M. The effect of polydimethylsiloxane-ethylcellulose coating blends on the surface characterization and drug release of ciprofloxacin-loaded mesoporous silica. Polymers 2019, 11, 1450. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Aina, V.; Areán, C.O.; Balas, F.; Cauda, V.; Colilla, M.; Delgado, M.R.; Vallet-Regí, M. Studies on MCM-41 mesoporous silica for drug delivery: Effect of particle morphology and amine functionalization. Chem. Eng. J. 2008, 137, 30–37. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, X.; Xu, H.; Hua, F.; Hu, X.; Xie, Q.; Wang, W.; Jia, J. Development of a doxycycline hydrochloride-loaded electrospun nanofibrous membrane for GTR/GBR applications. J. Nanomater. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Kogawa, A.C.; Zoppi, A.; Quevedo, M.A.; Nunes Salgado, H.R.; Longhi, M.R. Increasing doxycycline hyclate photostability by complexation with β-cyclodextrin. AAPS PharmSciTech 2014, 15, 1209–1217. [Google Scholar] [CrossRef]
- Christoforidou, T.; Giasafaki, D.; Andriotis, E.G.; Bouropoulos, N.; Theodoroula, N.F.; Vizirianakis, I.S.; Steriotis, T.; Charalambopoulou, G.; Fatouros, D.G. Oral drug delivery systems based on ordered mesoporous silica nanoparticles for modulating the release of aprepitant. Int. J. Mol. Sci. 2021, 22, 1896. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elrahman, A.A.; El Nabarawi, M.A.; Hassan, D.H.; Taha, A.A. Ketoprofen mesoporous silica nanoparticles SBA-15 hard gelatin capsules: Preparation and in vitro/in vivo characterization. Drug Deliv. 2016, 23, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Moritz, M.; Geszke-Moritz, M. Sulfonic acid derivative-modified SBA-15, PHTS and MCM-41 mesoporous silicas as carriers for a new antiplatelet drug: Ticagrelor adsorption and release Studies. Materials 2020, 13, 2913. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Prokopowicz, M. Mesoporous silica pellets—A promising oral drug delivery system? J. Drug Deliv. Sci. Technol. 2020, 56, 101491. [Google Scholar] [CrossRef]
- Li, Z.; Kolb, V.M.K.; Jiang, W.-T.; Hong, H. FTIR and XRD investigations of tetracycline intercalation in smectites. Clays Clay Miner. 2010, 58, 462–474. [Google Scholar] [CrossRef]
- Deaconu, M.; Nicu, I.; Tincu, R.; Brezoiu, A.-M.; Mitran, R.-A.; Vasile, E.; Matei, C.; Berger, D. Tailored doxycycline delivery from MCM-41-type silica carriers. Chem. Pap. 2018, 72, 1869–1880. [Google Scholar] [CrossRef]
- Petrescu, M.; Mitran, R.-A.; Luchian, A.-M.; Matei, C.; Berger, D. Mesoporous ceria-silica composites as carriers for doxycycline. UPB Sci. Bull. Ser. B Chem. Mater. Sci. 2015, 77, 13–24. [Google Scholar]
- Ratier, A.; Gibson, I.; Best, S.; Freche, M.; Lacout, J.; Rodriguez, F. Setting characteristics and mechanical behaviour of a calcium phosphate bone cement containing tetracycline. Biomaterials 2001, 22, 897–901. [Google Scholar] [CrossRef]
- Nandi, S.K.; Mukherjee, P.; Roy, S.; Kundu, B.; De, D.K.; Basu, D. Local antibiotic delivery systems for the treatment of osteomyelitis—A review. Mater. Sci. Eng. C 2009, 29, 2478–2485. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Kundu, B.; Nandi, S.K.; Basu, D. Systematic approach to treat chronic osteomyelitis through localized drug delivery system: Bench to bed side. Mater. Sci. Eng. C 2013, 33, 3986–3993. [Google Scholar] [CrossRef]
- Szewczyk, A.; Skwira, A.; Konopacka, A.; Sądej, R.; Walker, G.; Prokopowicz, M. Mesoporous silica pellets as bifunctional bone drug delivery system for cefazolin. Int. J. Pharm. 2020, 588, 119718. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.; Pinczowski, P.; Pérez, M.; Giorello, A.; Martínez, M.Á.; Santamaría, J.; Arruebo, M.; Luján, L. A controlled antibiotic release system to prevent orthopedic-implant associated infections: An in vitro study. Eur. J. Pharm. Biopharm. 2015, 96, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Takadama, H.; Matsushita, T.; Nakamura, T.; Kokubo, T. Cross-sectional analysis of the surface ceramic layer developed on Ti metal by NaOH-heat treatment and soaking in SBF. J. Ceram. Soc. Jpn. 2009, 117, 1126–1130. [Google Scholar] [CrossRef]
- Fraga, A.F.; Filho, E.D.A.; Rigo, E.C.D.S.; Boschi, A.O. Synthesis of chitosan/hydroxyapatite membranes coated with hydroxycarbonate apatite for guided tissue regeneration purposes. Appl. Surf. Sci. 2011, 257, 3888–3892. [Google Scholar] [CrossRef]
- Sarma, B.K.; Barman, P.; Sarma, B.; Das, A.; Pal, A.R. Biomimetic deposition of carbonate apatite and role of carbonate substitution on mechanical properties at nanoscale. Mater. Lett. 2016, 185, 387–390. [Google Scholar] [CrossRef]
- Sahai, N.; Anseau, M. Cyclic silicate active site and stereochemical match for apatite nucleation on pseudowollastonite bioceramic–bone interfaces. Biomaterials 2005, 26, 5763–5770. [Google Scholar] [CrossRef]
- Gheisari, H.; Karamian, E.; Abdellahi, M. A novel hydroxyapatite –Hardystonite nanocomposite ceramic. Ceram. Int. 2015, 41, 5967–5975. [Google Scholar] [CrossRef]
- Yin, N.; Chen, S.; Ouyang, Y.; Tang, L.; Yang, J.; Wang, H. Biomimetic mineralization synthesis of hydroxyapatite bacterial cellulose nanocomposites. Prog. Nat. Sci. Mater. Int. 2011, 21, 472–477. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Szewczyk, A.; Sawicki, W. The bioactivity studies of drug-loaded mesoporous silica-polydimethylsiloxane xerogels using FTIR and SEM/XEDS. J. Mol. Struct. 2014, 1056–1057, 262–266. [Google Scholar] [CrossRef]
- Graziani, G.; Berni, M.; Gambardella, A.; De Carolis, M.; Maltarello, M.C.; Boi, M.; Carnevale, G.; Bianchi, M. Fabrication and characterization of biomimetic hydroxyapatite thin films for bone implants by direct ablation of a biogenic source. Mater. Sci. Eng. C 2019, 99, 853–862. [Google Scholar] [CrossRef]
- Rigo, E.C.S.; Boschi, A.O.; Yoshimoto, M.; Allegrini, S.; Konig, B.; Carbonari, M.J. Evaluation in vitro and in vivo of biomimetic hydroxyapatite coated on titanium dental implants. Mater. Sci. Eng. C 2004, 24, 647–651. [Google Scholar] [CrossRef]
- Szewczyk, A.; Skwira, A.; Ginter, M.; Tajer, D.; Prokopowicz, M. Microwave-assisted fabrication of mesoporous silica-calcium phosphate composites for dental application. Polymers 2020, 13, 53. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Łukasiak, J. Synthesis and in vitro characterization of freeze-dried doxorubicin-loaded silica/PEG composite. J. Non. Cryst. Solids 2010, 356, 1711–1720. [Google Scholar] [CrossRef]
- Prokopowicz, M. Synthesis and in vitro characterization of freeze-dried doxorubicin-loaded silica xerogels. J. Sol-Gel Sci. Technol. 2010, 53, 525–533. [Google Scholar] [CrossRef]
- Szewczyk, A.; Prokopowicz, M. Amino-modified mesoporous silica SBA-15 as bifunctional drug delivery system for cefazolin: Release profile and mineralization potential. Mater. Lett. 2018, 227, 136–140. [Google Scholar] [CrossRef]
- Czarnobaj, K.; Prokopowicz, M.; Greber, K. Use of materials based on polymeric silica as bone-targeted drug delivery systems for metronidazole. Int. J. Mol. Sci. 2019, 20, 1311. [Google Scholar] [CrossRef]
- Gomes, K.d.N.; Alves, A.P.N.N.; Dutra, P.G.P.; Viana, G.S.d.B. Doxycycline induces bone repair and changes in Wnt signalling. Int. J. Oral Sci. 2017, 9, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.S.; Fernandes, M.H. Effect of therapeutic levels of doxycycline and minocycline in the proliferation and differentiation of human bone marrow osteoblastic cells. Arch. Oral Biol. 2007, 52, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Prokopowicz, M.; Szewczyk, A.; Łunio, R.; Sawicki, W. Monolithic polydimethylsiloxane-modified silica composites prepared by a low-temperature sol–gel micromolding technique for controlled drug release. React. Funct. Polym. 2017, 114, 136–145. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Szewczyk, A.; Sawicki, W. Bioactive monolithic composites of silica/polydimethylsiloxane/calcium phosphate obtained at room temperature in sol–gel micromolding technique. Mater. Lett. 2016, 184, 239–242. [Google Scholar] [CrossRef]
- Szewczyk, A.; Prokopowicz, M.; Sawicki, W.; Majda, D.; Walker, G. Aminopropyl-functionalized mesoporous silica SBA-15 as drug carrier for cefazolin: Adsorption profiles, release studies, and mineralization potential. Microporous Mesoporous Mater. 2019, 274, 113–126. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Żeglinski, J.; Szewczyk, A.; Skwira, A.; Walker, G. Surface-activated fibre-like SBA-15 as drug carriers for bone diseases. AAPS PharmSciTech 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Kavanagh, N.; Ryan, E.J.; Widaa, A.; Sexton, G.; Fennell, J.; O’Rourke, S.; Cahill, K.C.; Kearney, C.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcal osteomyelitis: Disease progression, treatment challenges, and future directions. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- ISO 10993-5:2009(en). Biological Evaluation of Medical Devices—Part 5: Tests for in vitro Cytotoxicity. Available online: https://www.iso.org/obp/ui#iso:std:iso:10993:-5:ed-3:v1:en (accessed on 3 June 2020).
- Miller, F.; Hinze, U.; Chichkov, B.; Leibold, W.; Lenarz, T.; Paasche, G. Validation of eGFP fluorescence intensity for testing in vitro cytotoxicity according to ISO 10993-5. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 715–722. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield (%) | Hardness (N) | Friability (%) | Drug Content (%) |
|---|---|---|---|
| 78 ± 3 | 5.5 ± 1.3 | 1.1 ± 0.3 | 91.2 ± 3.5 |
| Higuchi Model | Korsmeyer–Peppas Model | Zero-Order Kinetics * | |||
|---|---|---|---|---|---|
| kH | R2 | n | R2 | k0 | R2 |
| 7.3 | 0.991 | 0.36 | 0.987 | 2.4 | 0.972 |
| Day | CFU/mL | Log10 Difference | Percent of Killed Bacteria | |
|---|---|---|---|---|
| Control | Pellets | |||
| 1 | 1.4 × 109 | 1.4 × 105 | 4.0 | >99.9% |
| 2 | 1.8 × 109 | 6.2 × 103 | 5.5 | |
| 3 | 4.7 × 109 | 4.5 × 104 | 5.0 | |
| 4 | 5.4 × 109 | 8.0 × 104 | 4.8 | |
| 5 | 6.5 × 109 | 1.5 × 105 | 4.6 | |
| 6 | 2.0 × 1010 | 1.0 × 105 | 5.3 | |
| 7 | 1.0 × 1010 | 3.5 × 105 | 4.5 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szewczyk, A.; Skwira, A.; Konopacka, A.; Sądej, R.; Prokopowicz, M. Mesoporous Silica-Bioglass Composite Pellets as Bone Drug Delivery System with Mineralization Potential. Int. J. Mol. Sci. 2021, 22, 4708. https://doi.org/10.3390/ijms22094708
Szewczyk A, Skwira A, Konopacka A, Sądej R, Prokopowicz M. Mesoporous Silica-Bioglass Composite Pellets as Bone Drug Delivery System with Mineralization Potential. International Journal of Molecular Sciences. 2021; 22(9):4708. https://doi.org/10.3390/ijms22094708
Chicago/Turabian StyleSzewczyk, Adrian, Adrianna Skwira, Agnieszka Konopacka, Rafał Sądej, and Magdalena Prokopowicz. 2021. "Mesoporous Silica-Bioglass Composite Pellets as Bone Drug Delivery System with Mineralization Potential" International Journal of Molecular Sciences 22, no. 9: 4708. https://doi.org/10.3390/ijms22094708
APA StyleSzewczyk, A., Skwira, A., Konopacka, A., Sądej, R., & Prokopowicz, M. (2021). Mesoporous Silica-Bioglass Composite Pellets as Bone Drug Delivery System with Mineralization Potential. International Journal of Molecular Sciences, 22(9), 4708. https://doi.org/10.3390/ijms22094708