
Enhanced Anti-Tumor Activity in Mice with Temozolomide-Resistant Human Glioblastoma Cell Line-Derived Xenograft Using SN-38-Incorporated Polymeric Microparticle


 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
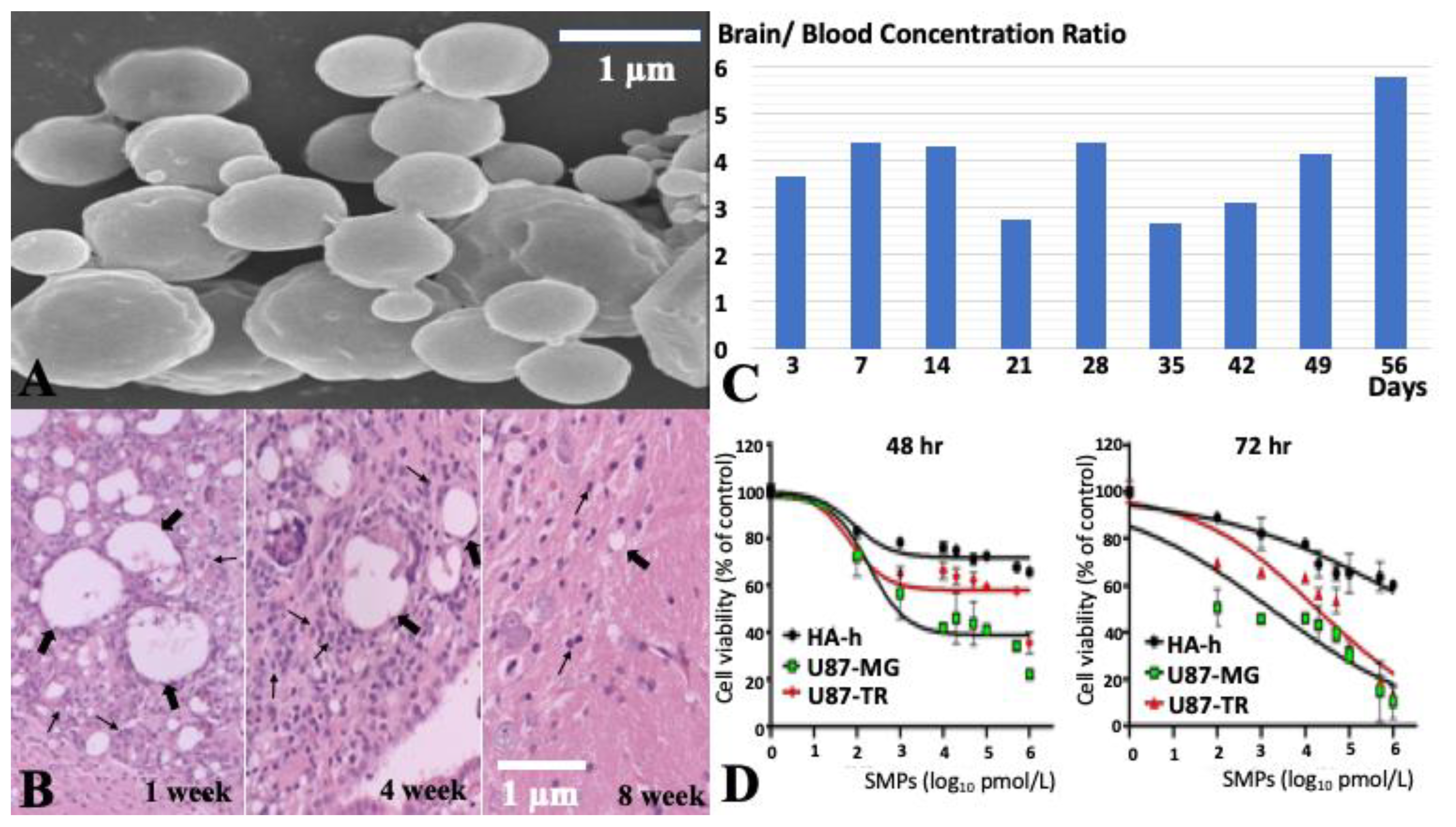
2.1. Characteristics of SN-38-Incorporated Microparticles
2.2. Cytotoxicity of SMPs in Human U87 and Astrocytes-Hippocampal (HA-h) Cell Lines
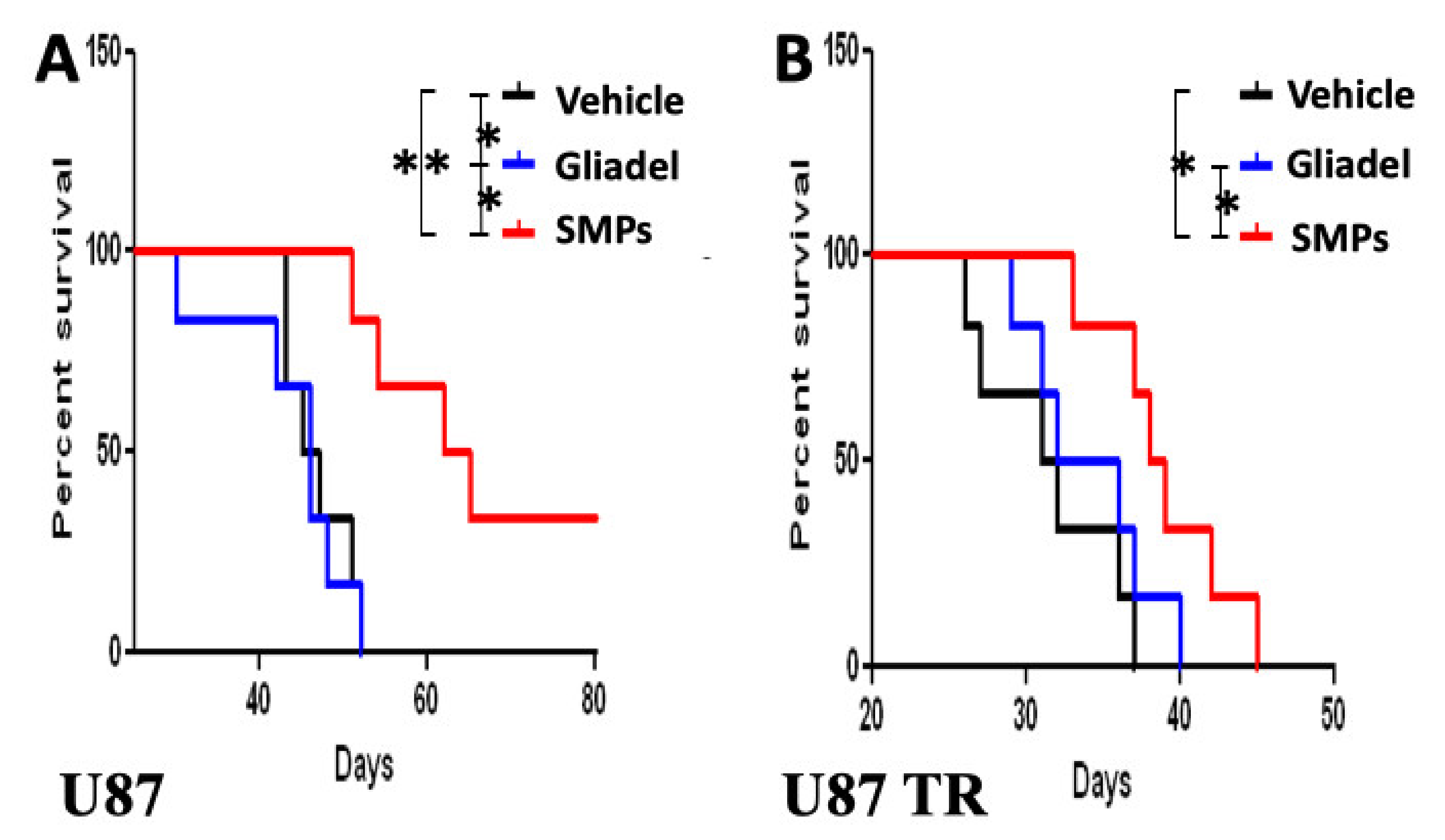
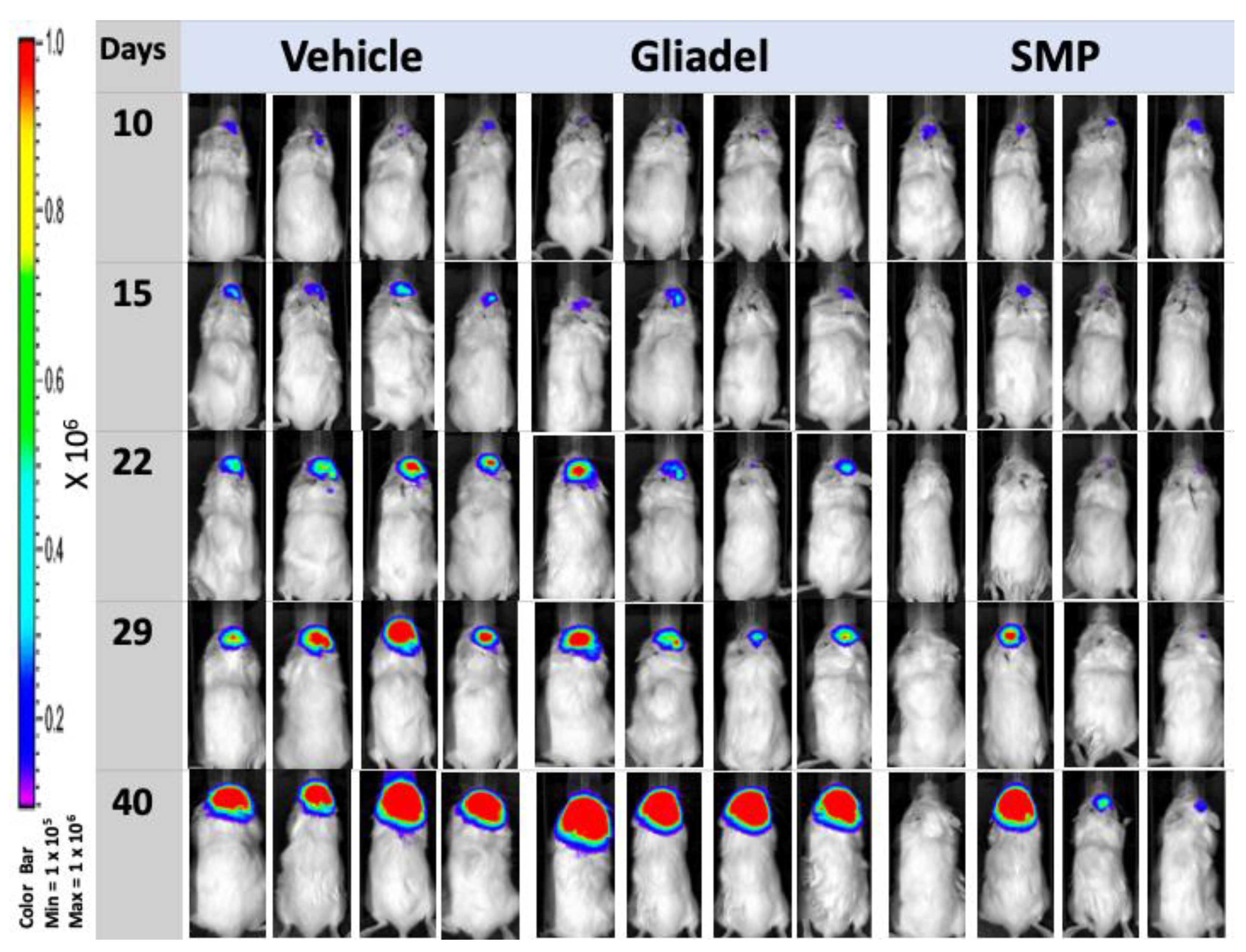
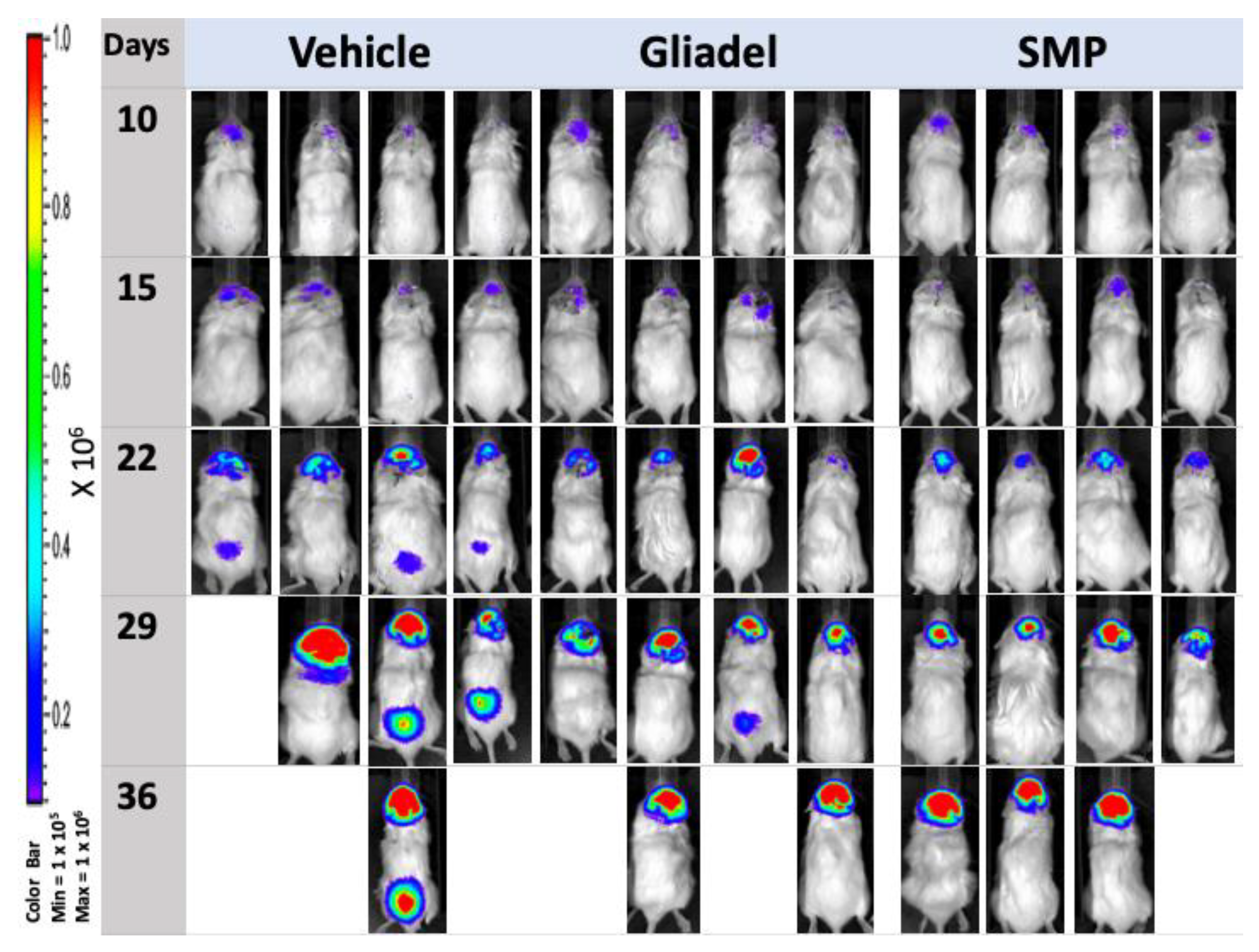
2.3. Combination Treatment of TMZ and SMPs Prolongs the Survival of Mice Bearing U87 or U87-TR Xenografts
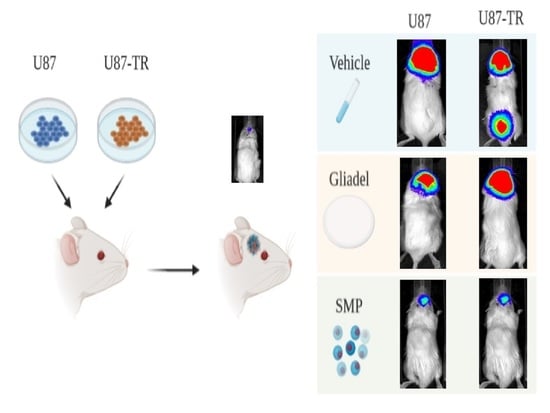
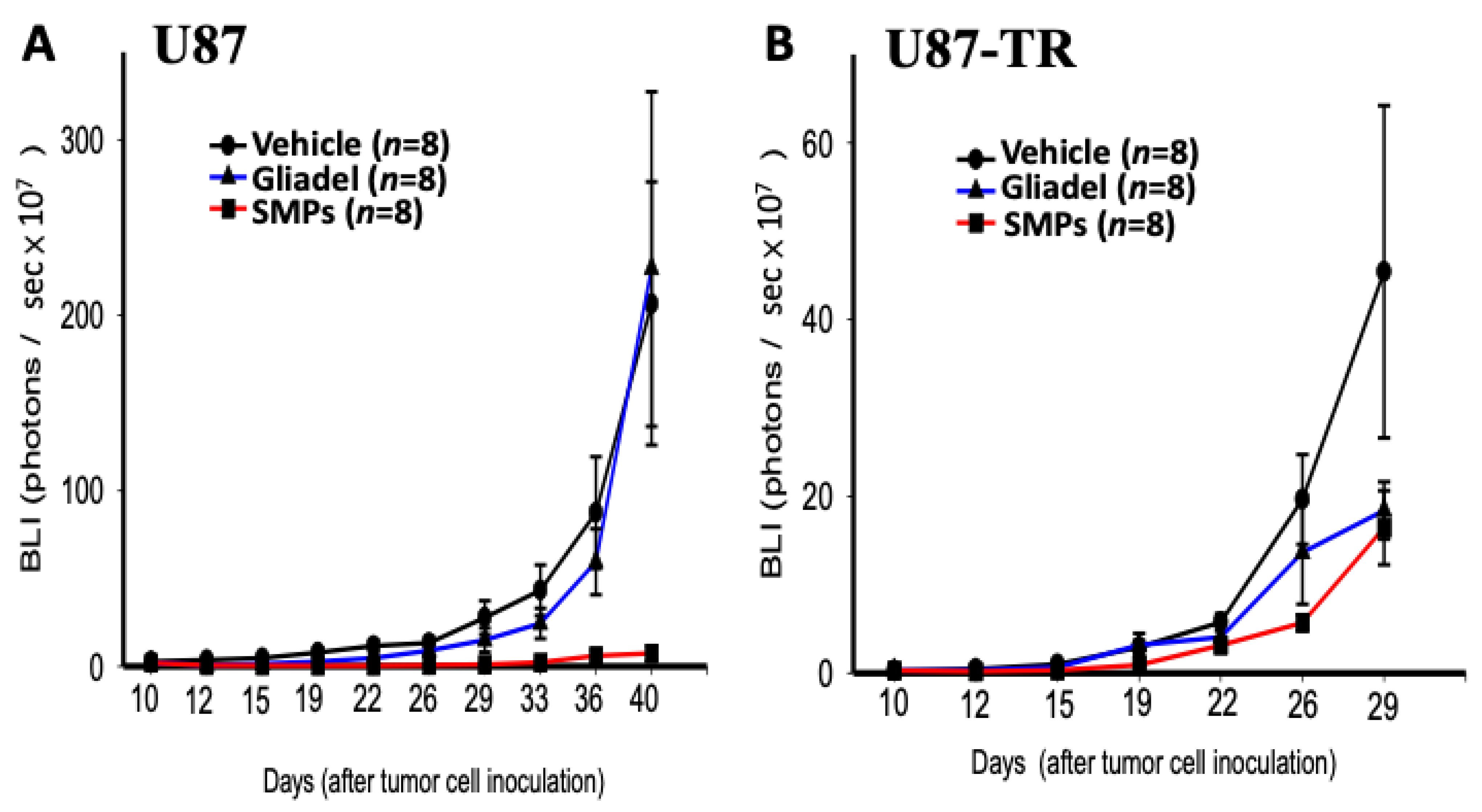
2.4. Combination Treatment of TMZ and SMPs Suppresses Tumor Growth in both TMZ-Sensitive and TMZ-Resistant Xenograft Models
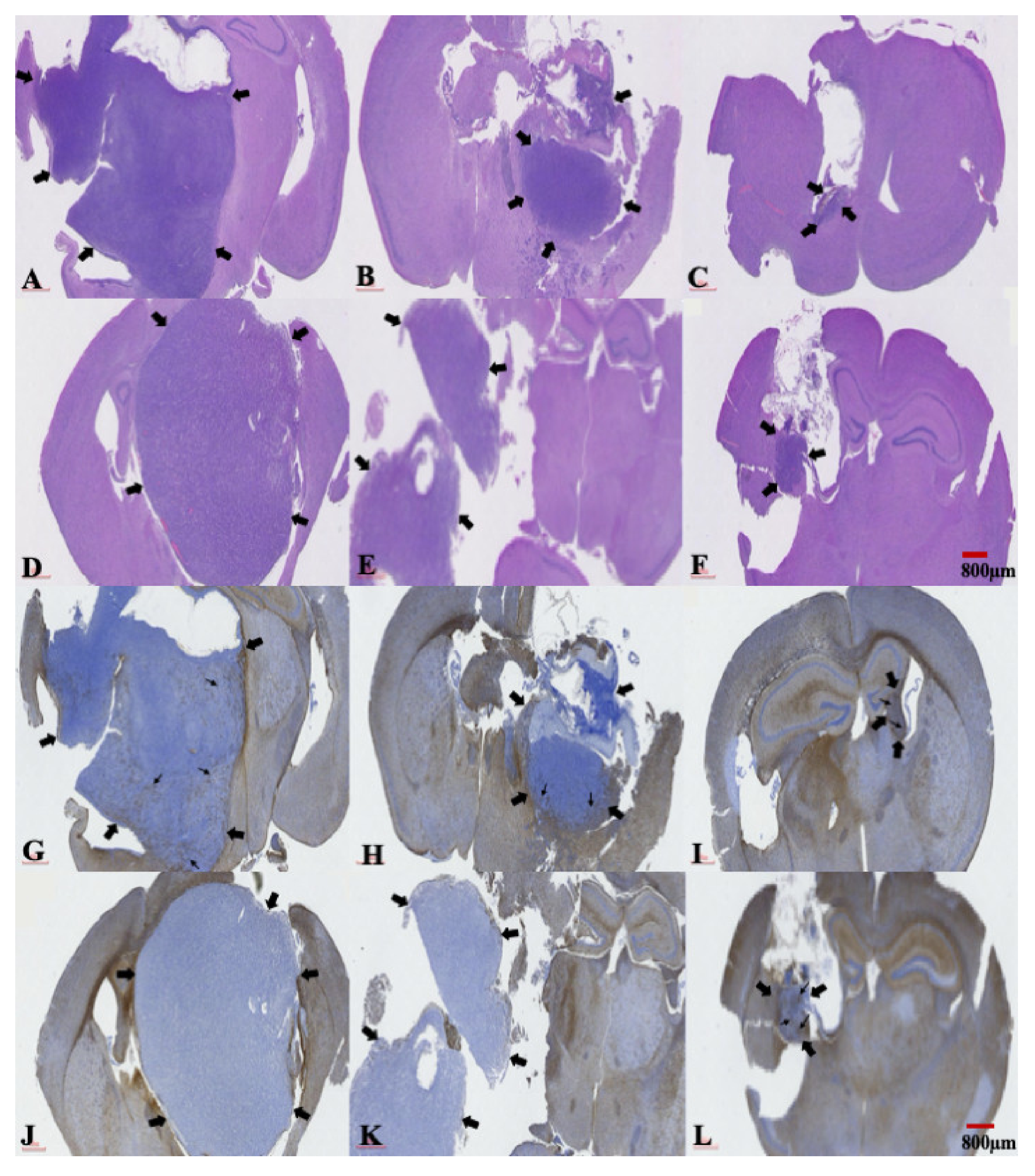
2.5. The Presence of a Central Necrosis Area and GFAP Expression Changes in both TMZ-Sensitive and TMZ-Resistant Xenografted Tumors after a Combination Treatment of TMZ and SMPs
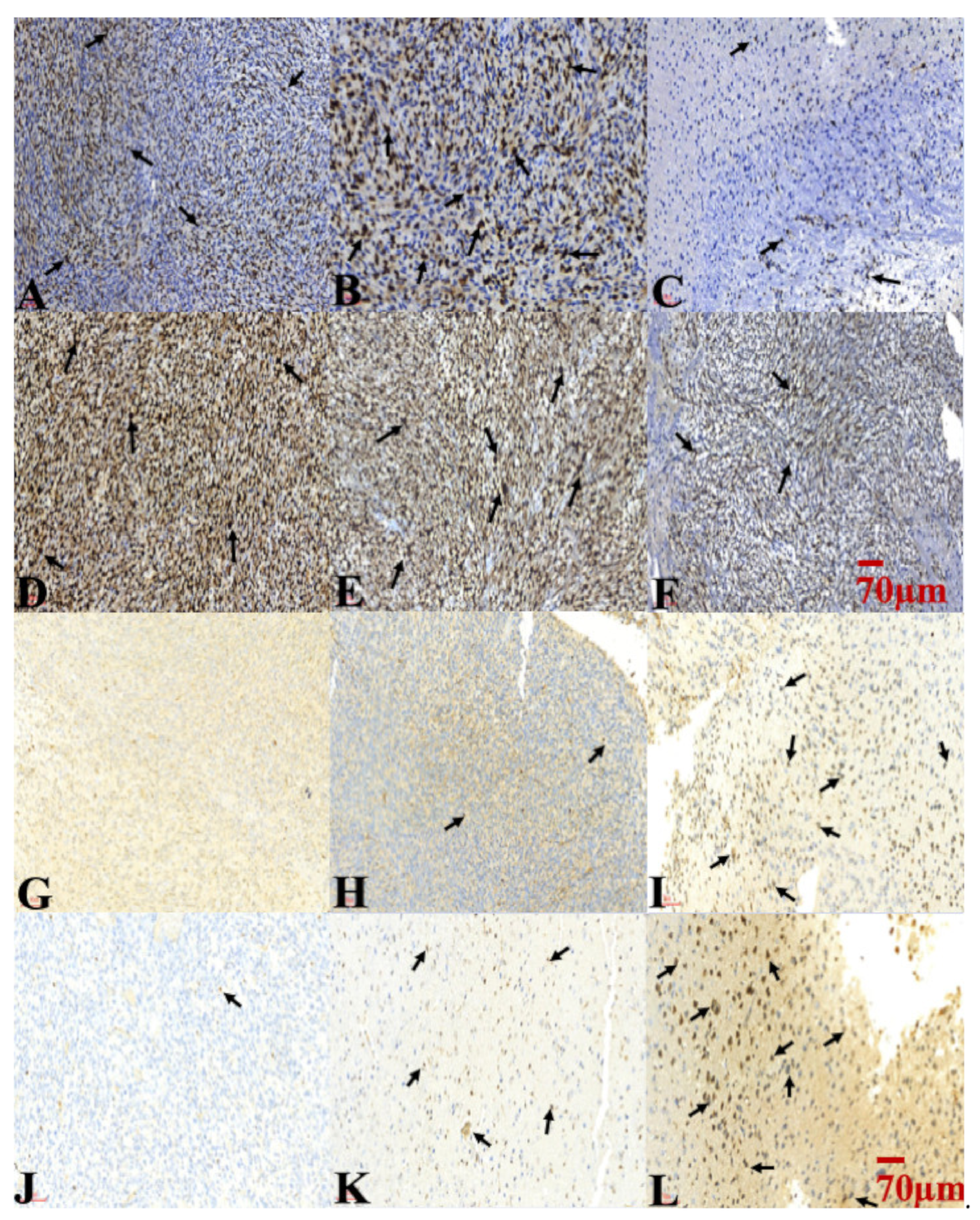
2.6. Combination Treatment of TMZ and SMPs Inhibited Cell Proliferation and Induced Apoptosis in both TMZ-Sensitive and TMZ-Resistant Xenograft Models
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Human Glioma Cell Lines and Cell Cultures
4.3. Experimental Animals
4.4. Preparation of SN-38 Incorporated Polymeric Microparticles (SMPs)
4.5. In Vivo Elution Characteristics of SMPs
4.6. Cell Viability Assay
4.7. Generation of Human Glioma Cell-Derived TMZ-Sensitive and TMZ-Resistant Mouse Xenografts
4.8. Animal Grouping and Drug Treatment Protocols
4.9. Pathology Assessment of Tumor Tissues
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandes, A.A.; Tosoni, A.; Basso, U.; Reni, M.; Valduga, F.; Monfardini, S.; Amistà, P.; Nicolardi, L.; Sotti, G.; Ermani, M. Second-Line Chemotherapy with Irinotecan Plus Carmustine in Glioblastoma Recurrent or Progressive After First-Line Temozolomide Chemotherapy: A Phase II Study of the Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). J. Clin. Oncol. 2004, 22, 4779–4786. [Google Scholar] [CrossRef]
- Liu, S.-J.; Yang, S.-T.; Chen, S.-M.; Huang, Y.-C.; Lee, W.-H.; Ho, J.; Chen, Y.-C.; Tseng, Y.-Y. Novel multi-drugs incorporating hybrid-structured nanofibers enhance alkylating agent activity in malignant gliomas. Ther. Adv. Med Oncol. 2019, 11, 1758835919875555. [Google Scholar] [CrossRef] [PubMed]
- Aberoumandi, S.M.; Mohammadhosseini, M.; Abasi, E.; Saghati, S.; Nikzamir, N.; Akbarzadeh, A.; Panahi, Y.; Davaran, S. An update on applications of nanostructured drug delivery systems in cancer therapy: A review. Artif. Cells Nanomed. Biotechnol. 2016, 45, 1058–1068. [Google Scholar] [CrossRef]
- Affronti, M.L.; Heery, C.R.; Rn, M.M.L.A.; Rich, J.N.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Friedman, A.H.; Bigner, D.D.; Friedman, H.S. Overall survival of newly diagnosed glioblastoma patients receiving carmustine wafers followed by radiation and concurrent temozolomide plus rotational multiagent chemotherapy. Cancer 2009, 115, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-Y.; Hsu, T.-I.; Hsu, C.-C.; Tsai, S.-Y.; Liu, J.-J.; Chou, S.-W.; Liu, M.-S.; Liou, J.-P.; Ko, C.-Y.; Chen, K.-Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O6-methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Thomé, M.P.; e Silva, M.M.B.; Pelegrini, A.L.; Ledur, P.F.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol abrogates the Temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the Temozolomide-induced senescence in glioma cells. BMC Cancer 2013, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Mhaidat, N.M.; Zhang, X.D.; Allen, J.; Avery-Kiejda, K.; Scott, R.J.; Hersey, P. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br. J. Cancer 2007, 97, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Desjardins, A.; A Quinn, J.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel®) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Yi, G.-Z.; Huang, G.; Guo, M.; Zhang, X.; Wang, H.; Deng, S.; Li, Y.; Xiang, W.; Chen, Z.; Pan, J.; et al. Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2. Brain 2019, 142, 2352–2366. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-J.; Yang, T.-C.; Yang, S.-T.; Chen, Y.-C.; Tseng, Y.-Y. Biodegradable hybrid-structured nanofibrous membrane supported chemoprotective gene therapy enhances chemotherapy tolerance and efficacy in malignant glioma rats. Artif. Cells Nanomed. Biotechnol. 2018, 46, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Du, X.L.; Lu, G.; Zhu, J.-J. Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: A population-based study. Oncotarget 2017, 8, 44015–44031. [Google Scholar] [CrossRef] [PubMed]
- Kokkinakis, D.M.; Bocangel, D.B.; Schold, S.C.; Moschel, R.C.; Pegg, A.E. Thresholds of O6-alkylguanine-DNA alkyltrans-ferase which confer significant resistance of human glial tumor xenografts to treatment with 1,3-bis(2-chloroethyl)-1-nitrosourea or temozolomide. Clin. Cancer Res. 2001, 7, 421–428. [Google Scholar]
- Tiek, D.M.; Rone, J.D.; Graham, G.T.; Pannkuk, E.L.; Haddad, B.R.; Riggins, R.B. Alterations in Cell Motility, Proliferation, and Metabolism in Novel Models of Acquired Temozolomide Resistant Glioblastoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Reardon, D.A.; Friedman, H.S.; Powell, J.B.; Gilbert, M.; Yung, W.K. Irinotecan: Promising activity in the treatment of malignant glioma. Oncology 2003, 17, 9–14. [Google Scholar] [PubMed]
- Vejjasilpa, K.; Nasongkla, N.; Manaspon, C.; Larbcharoensub, N.; Boongird, A.; Hongeng, S.; Israsena, N. Antitumor efficacy and intratumoral distribution of SN-38 from polymeric depots in brain tumor model. Exp. Biol. Med. 2015, 240, 1640–1647. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Ii, J.E.H.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab Plus Irinotecan in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Gershenson, D.M. Irinotecan in epithelial ovarian cancer. Oncology 2002, 16, 29–31. [Google Scholar] [PubMed]
- Vanhoefer, U.; Harstrick, A.; Achterrath, W.; Cao, S.; Seeber, S.; Rustum, Y.M. Irinotecan in the Treatment of Colorectal Cancer: Clinical Overview. J. Clin. Oncol. 2001, 19, 1501–1518. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Quinn, J.A.; Vredenburgh, J.; Rich, J.N.; Gururangan, S.; Badruddoja, M.; Herndon, J.E.; Dowell, J.M.; Friedman, A.H.; Friedman, H.S. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer 2005, 103, 329–338. [Google Scholar] [CrossRef]
- Quinn, J.A.; Jiang, X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Friedman, A.H.; Sampson, J.H.; McLendon, R.E.; Ii, J.E.H.; Friedman, H.S. Phase II trial of temozolomide (TMZ) plus irinotecan (CPT-11) in adults with newly diagnosed glioblastoma multiforme before radiotherapy. J. Neuro Oncol. 2009, 95, 393–400. [Google Scholar] [CrossRef]
- Chen, T.C.; Su, S.; Fry, D.; Liebes, L. Combination therapy with irinotecan and protein kinase C inhibitors in malignant glioma. Cancer 2003, 97, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- De Man, F.; Goey, A.K.L.; Van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [PubMed]
- Vredenburgh, J.J.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Experience with irinotecan for the treatment of malignant glioma. Neuro Oncol. 2009, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.P.; Chuang, C.H.; Wu, Y.J.; Lin, H.C.; Lu, Y.C. SN38-loaded <100 nm targeted liposomes for improving poor solubility and minimizing burst release and toxicity: In vitro and in vivo study. Int. J. Nanomed. 2018, 13, 2789–2802. [Google Scholar] [CrossRef]
- Palakurthi, S. Challenges in SN38 drug delivery: Current success and future directions. Expert Opin. Drug Deliv. 2015, 12, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Saito, R.; Mano, Y.; Sumiyoshi, A.; Kanamori, M.; Sonoda, Y.; Kawashima, R.; Tominaga, T. Convection-enhanced delivery of SN-38-loaded polymeric micelles (NK012) enables consistent distribution of SN-38 and is effective against rodent intracranial brain tumor models. Drug Deliv. 2015, 23, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-Y.; Yang, T.-C.; Chen, S.-M.; Yang, S.-T.; Tang, Y.-L.; Liu, S.-J. Injectable SN-38-embedded Polymeric Microparticles Promote Antitumor Efficacy against Malignant Glioma in an Animal Model. Pharmaceutics 2020, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Kamath, A.A.; Leuthardt, E.C.; Kim, A.H. Management of Intracranial Metastatic Disease with Laser Interstitial Thermal Therapy. Front. Oncol. 2018, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Ashby, L.S.; Smith, K.A.; Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: A systematic literature review. World J. Surg. Oncol. 2016, 14, 225. [Google Scholar] [CrossRef] [PubMed]
- Wait, S.D.; Prabhu, R.S.; Burri, S.H.; Atkins, T.G.; Asher, A.L. Polymeric drug delivery for the treatment of glioblastoma. Neuro Oncol. 2015, 17, ii9–ii23. [Google Scholar] [CrossRef] [PubMed]
- Burri, S.H.; Prabhu, R.S.; Sumrall, A.L.; Brick, W.; Blaker, B.D.; Heideman, B.E.; Boltes, P.; Kelly, R.; Symanowski, J.T.; Wiggins, W.F.; et al. BCNU wafer placement with temozolomide (TMZ) in the immediate postoperative period after tumor resection followed by radiation therapy with TMZ in patients with newly diagnosed high grade glioma: Final results of a prospective, multi-institutional, phase II trial. J. Neuro Oncol. 2015, 123, 259–266. [Google Scholar] [CrossRef]
- Noel, G.; Schott, R.; Froelich, S.; Gaub, M.-P.; Boyer, P.; Fischer-Lokou, D.; Dufour, P.; Kehrli, P.; Maitrot, D. Retrospective Comparison of Chemoradiotherapy Followed by Adjuvant Chemotherapy, With or Without Prior Gliadel Implantation (Carmustine) After Initial Surgery in Patients with Newly Diagnosed High-Grade Gliomas. Int. J. Radiat. Oncol. 2012, 82, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Maurya, D.K.; Ayuzawa, R.; Doi, C.; Troyer, D.; Tamura, M. Topoisomerase I inhibitor SN-38 effectively attenuates growth of human non-small cell lung cancer cell lines in vitro and in vivo. J. Environ. Pathol. Toxicol. Oncol. 2011, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.R.; Rosa, S.C.D.S.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Y.; Xu, K.; Wang, Z.; Fan, X.; Zhang, C.; Li, S.; Qiu, X.; Jiang, T. Relationship between necrotic patterns in glioblastoma and patient survival: Fractal dimension and lacunarity analyses using magnetic resonance imaging. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.; Khalili, K. Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 2009, 8, 1791–1797. [Google Scholar] [CrossRef]
- Wojton, J.; Meisen, W.H.; Kaur, B. How to train glioma cells to die: Molecular challenges in cell death. J. Neuro Oncol. 2015, 126, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, U.; Eliasson, C.; Bjerkvig, R.; Pekny, M. Loss of GFAP expression in high-grade astrocytomas does not contribute to tumor development or progression. Oncogene 2003, 22, 3407–3411. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.; Stevenson, M.; McComb, R.D.; Volsky, D.J. Expression of glial fibrillary acidic protein in human glioma cell lines as detected by molecular hybridization. Acta Neuropathol. 1987, 73, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.-H.; Lee, W.-H.; Yang, S.-T.; Han, C.-T.; Tseng, Y.-Y. Spinal metastasis of glioblastoma multiforme before gliosarcomatous transformation: A case report. BMC Neurol. 2020, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-L.; Hsu, T.-I.; Yang, W.-B.; Kao, T.-J.; Wu, M.-H.; Huang, Y.-N.; Yeh, S.-H.; Chuang, J.-Y. Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma. Cancers 2020, 12, 981. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-C.; Liu, S.-J.; Lo, W.-L.; Chen, S.-M.; Tang, Y.-L.; Tseng, Y.-Y. Enhanced Anti-Tumor Activity in Mice with Temozolomide-Resistant Human Glioblastoma Cell Line-Derived Xenograft Using SN-38-Incorporated Polymeric Microparticle. Int. J. Mol. Sci. 2021, 22, 5557. https://doi.org/10.3390/ijms22115557
Yang T-C, Liu S-J, Lo W-L, Chen S-M, Tang Y-L, Tseng Y-Y. Enhanced Anti-Tumor Activity in Mice with Temozolomide-Resistant Human Glioblastoma Cell Line-Derived Xenograft Using SN-38-Incorporated Polymeric Microparticle. International Journal of Molecular Sciences. 2021; 22(11):5557. https://doi.org/10.3390/ijms22115557
Chicago/Turabian StyleYang, Tao-Chieh, Shih-Jung Liu, Wei-Lun Lo, Shu-Mei Chen, Ya-Ling Tang, and Yuan-Yun Tseng. 2021. "Enhanced Anti-Tumor Activity in Mice with Temozolomide-Resistant Human Glioblastoma Cell Line-Derived Xenograft Using SN-38-Incorporated Polymeric Microparticle" International Journal of Molecular Sciences 22, no. 11: 5557. https://doi.org/10.3390/ijms22115557
APA StyleYang, T.-C., Liu, S.-J., Lo, W.-L., Chen, S.-M., Tang, Y.-L., & Tseng, Y.-Y. (2021). Enhanced Anti-Tumor Activity in Mice with Temozolomide-Resistant Human Glioblastoma Cell Line-Derived Xenograft Using SN-38-Incorporated Polymeric Microparticle. International Journal of Molecular Sciences, 22(11), 5557. https://doi.org/10.3390/ijms22115557