
The Function of KDEL Receptors as UPR Genes in Disease



Abstract
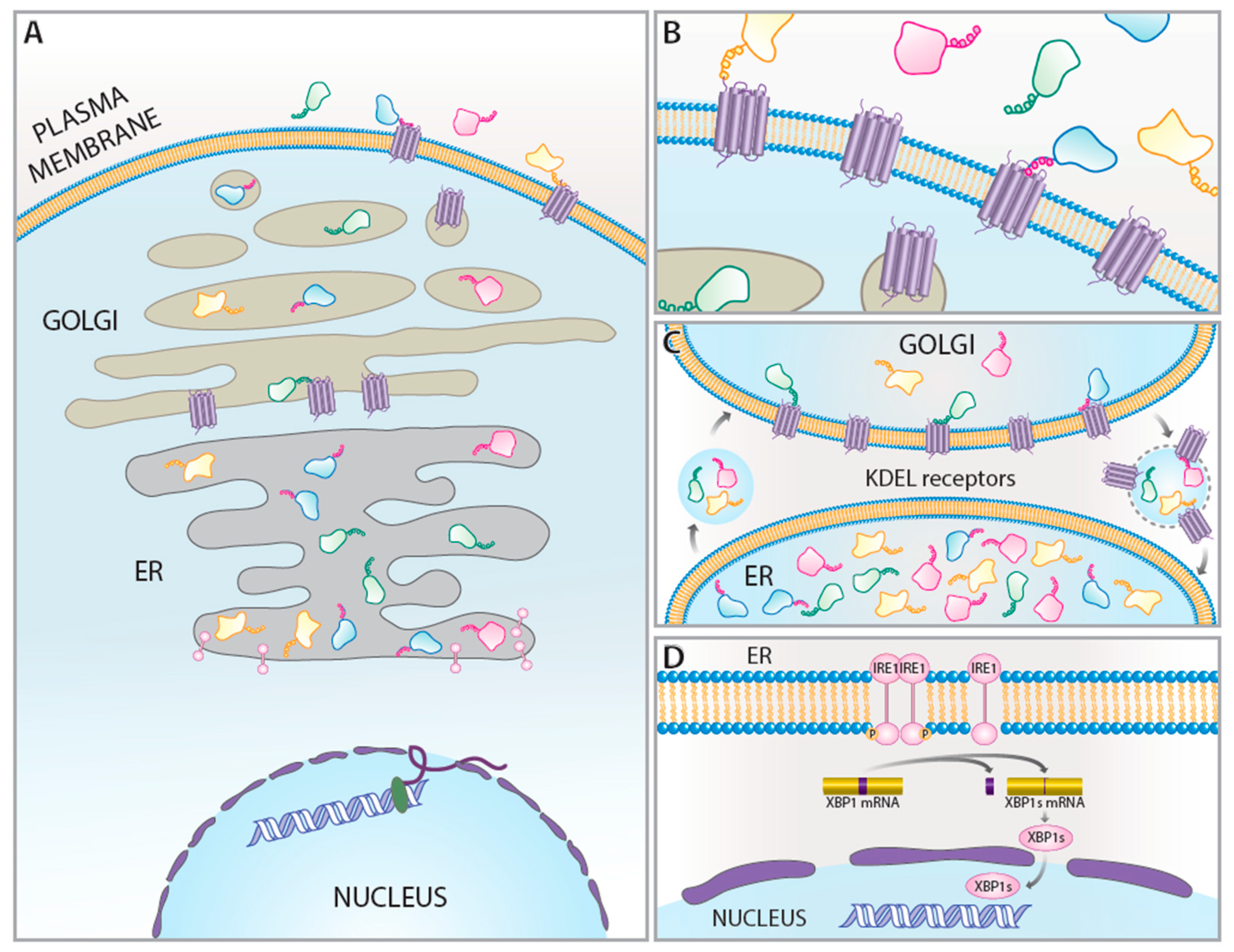
1. Introduction
2. KDEL Retrieval Pathway and KDEL Receptors
3. KDEL Receptors as UPR-Regulated Genes
4. KDEL Receptors, UPR, and Disease
4.1. KDEL Receptor and Autophagy
4.2. Neurodegeneration
4.3. Ischemia
4.4. Cancer
4.5. Diabetes
4.6. Immune Response
4.7. Osteogenesis Imperfecta
4.8. Wolfram’s Syndrome
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Henderson, M.J.; Wires, E.S.; Trychta, K.A.; Richie, C.T.; Harvey, B.K. SERCaMP: A carboxy-terminal protein modification that enables monitoring of ER calcium homeostasis. Mol. Biol. Cell 2014, 25, 2828–2839. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Cribb, A.E.; Peyrou, M.; Muruganandan, S.; Schneider, L. The endoplasmic reticulum in xenobiotic toxicity. Drug Metab Rev. 2005, 37, 405–442. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Ron, D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef]
- Trychta, K.A.; Back, S.; Henderson, M.J.; Harvey, B.K. KDEL Receptors Are Differentially Regulated to Maintain the ER Proteome under Calcium Deficiency. Cell Rep. 2018, 25, 1829–1840.e6. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R. A human homologue of the yeast HDEL receptor. Nature 1990, 348, 162–163. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R. Sequence of a second human KDEL receptor. J. Mol. Biol. 1992, 226, 913–916. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell 1992, 68, 353–364. [Google Scholar] [CrossRef]
- Majoul, I.; Straub, M.; Hell, S.W.; Duden, R.; Söling, H.D. KDEL-cargo regulates interactions between proteins involved in COPI vesicle traffic: Measurements in living cells using FRET. Dev. Cell 2001, 1, 139–153. [Google Scholar] [CrossRef]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef]
- Hulo, N.; Bairoch, A.; Bulliard, V.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Pagni, M.; Sigrist, C.J. The PROSITE database. Nucleic Acids Res. 2006, 34, D227–D230. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.; Zhai, C.; Li, X.; Zhou, Y.; Peng, W.; Ma, L.; Wang, Q.; Iverson, B.L.; Zhang, G.; Yi, L. Characterization of aromatic residue-controlled protein retention in the endoplasmic reticulum of. J. Biol. Chem. 2017, 292, 20707–20719. [Google Scholar] [CrossRef]
- Capitani, M.; Sallese, M. The KDEL receptor: New functions for an old protein. FEBS Lett. 2009, 583, 3863–3871. [Google Scholar] [CrossRef]
- Bräuer, P.; Parker, J.L.; Gerondopoulos, A.; Zimmermann, I.; Seeger, M.A.; Barr, F.A.; Newstead, S. Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 2019, 363, 1103–1107. [Google Scholar] [CrossRef]
- Saudek, V. Cystinosin, MPDU1, SWEETs and KDELR belong to a well-defined protein family with putative function of cargo receptors involved in vesicle trafficking. PLoS ONE 2012, 7, e30876. [Google Scholar] [CrossRef] [PubMed]
- Jäntti, M.; Harvey, B.K. Trophic activities of endoplasmic reticulum proteins CDNF and MANF. Cell Tissue Res. 2020, 382, 83–100. [Google Scholar] [CrossRef]
- Henderson, M.J.; Richie, C.T.; Airavaara, M.; Wang, Y.; Harvey, B.K. Mesencephalic astrocyte-derived neurotrophic factor (MANF) secretion and cell surface binding are modulated by KDEL receptors. J. Biol. Chem. 2013, 288, 4209–4225. [Google Scholar] [CrossRef] [PubMed]
- Maciel, L.; de Oliveira Dahienne, F.; Mesquita, F.; Souza Hercules Antônio da, S.; Oliveira, L.; Christie Michelle Lopes, A.; Palhano Fernando, L.; Campos de Carvalho Antônio, C.; Nascimento José Hamilton, M.; Foguel, D. New Cardiomyokine Reduces Myocardial Ischemia/Reperfusion Injury by PI3K-AKT Pathway Via a Putative KDEL-Receptor Binding. J. Am. Heart Assoc. 2021, 10, e019685. [Google Scholar] [CrossRef]
- Bartels, A.K.; Gottert, S.; Desel, C.; Schafer, M.; Krossa, S.; Scheidig, A.J.; Grotzinger, J.; Lorenzen, I. KDEL Receptor 1 Contributes to Cell Surface Association of Protein Disulfide Isomerases. Cell Physiol. Biochem. 2019, 52, 850–868. [Google Scholar]
- Becker, B.; Shaebani, M.R.; Rammo, D.; Bubel, T.; Santen, L.; Schmitt, M.J. Cargo binding promotes KDEL receptor clustering at the mammalian cell surface. Sci. Rep. 2016, 6, 28940. [Google Scholar] [CrossRef]
- Samy, A.; Kaneyoshi, K.; Omasa, T. Improvement of Intracellular Traffic System by Overexpression of KDEL Receptor 1 in Antibody-Producing CHO Cells. Biotechnol. J. 2020, 15, e1900352. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cells response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Yamamoto, K.; Hamada, H.; Shinkai, H.; Kohno, Y.; Koseki, H.; Aoe, T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 2003, 278, 34525–34532. [Google Scholar] [CrossRef]
- Giannotta, M.; Ruggiero, C.; Grossi, M.; Cancino, J.; Capitani, M.; Pulvirenti, T.; Consoli, G.M.; Geraci, C.; Fanelli, F.; Luini, A.; et al. The KDEL receptor couples to Gαq/11 to activate Src kinases and regulate transport through the Golgi. EMBO J. 2012, 31, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, T.; Giannotta, M.; Capestrano, M.; Capitani, M.; Pisanu, A.; Polishchuk, R.S.; San Pietro, E.; Beznoussenko, G.V.; Mironov, A.A.; Turacchio, G.; et al. A traffic-activated Golgi-based signalling circuit coordinates the secretory pathway. Nat. Cell Biol. 2008, 10, 912–922. [Google Scholar] [CrossRef]
- Wang, P.; Li, B.; Zhou, L.; Fei, E.; Wang, G. The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience 2011, 190, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Tapia, D.; Jimenez, T.; Zamora, C.; Espinoza, J.; Rizzo, R.; Gonzalez-Cardenas, A.; Fuentes, D.; Hernandez, S.; Cavieres, V.A.; Soza, A.; et al. KDEL receptor regulates secretion by lysosome relocation- and autophagy-dependent modulation of lipid-droplet turnover. Nat. Commun. 2019, 10, 735. [Google Scholar] [CrossRef]
- Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Chen, L. Endoplasmic Reticulum Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 167–177. [Google Scholar] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef]
- Jin, H.; Mimura, N.; Kashio, M.; Koseki, H.; Aoe, T. Late-onset of spinal neurodegeneration in knock-in mice expressing a mutant BiP. PLoS ONE 2014, 9, e112837. [Google Scholar] [CrossRef] [PubMed]
- Valencia, M.; Kim, S.R.; Jang, Y.; Lee, S.H. Neuronal Autophagy: Characteristic Features and Roles in Neuronal Pathophysiology. Biomol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Komita, M.; Aoe, T. The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef]
- Paschen, W.; Gissel, C.; Linden, T.; Doutheil, J. Erp72 Expression Activated by Transient Cerebral Ischemia or Disturbance of Neuronal Endoplasmic Reticulum Calcium Stores. Metab. Brain Dis. 1998, 13, 55–68. [Google Scholar] [CrossRef]
- Paschen, W.; Gissel, C.; Linden, T.; Althausen, S.; Doutheil, J. Activation of gadd153 expression through transient cerebral ischemia: Evidence that ischemia causes endoplasmic reticulum dysfunction. Mol. Brain Res. 1998, 60, 115–122. [Google Scholar] [CrossRef]
- Hayashi, T.; Saito, A.; Okuno, S.; Ferrand-Drake, M.; Chan, P.H. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J. Cereb. Blood Flow Metab. 2003, 23, 949–961. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, Y.; Li, Y.; Xie, L.; Zhuang, W.; Liu, J.; Gong, J. Unfolded protein response plays a critical role in heart damage after myocardial ischemia/reperfusion in rats. PLoS ONE 2017, 12, e0179042. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.; Ke, X.; Guo, Y.; Yao, C.; Tang, N.; Pang, P.; Xie, G.; Fang, L.; Zhang, Z.; et al. Transcriptome Sequencing Unravels Potential Biomarkers at Different Stages of Cerebral Ischemic Stroke. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Lukovic, D.; Gugerell, A.; Zlabinger, K.; Winkler, J.; Pavo, N.; Baranyai, T.; Giricz, Z.; Varga, Z.V.; Riesenhuber, M.; Spannbauer, A.; et al. Transcriptional Alterations by Ischaemic Postconditioning in a Pig Infarction Model: Impact on Microvascular Protection. Int. J. Mol. Sci. 2019, 20, 344. [Google Scholar] [CrossRef]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Luo, P.; Gao, Y.; Yang, J.; Lao, K.-H.; Wang, G.; Cockerill, G.; Hu, Y.; Xu, Q.; et al. XBP1 splicing triggers miR-150 transfer from smooth muscle cells to endothelial cells via extracellular vesicles. Sci. Rep. 2016, 6, 28627. [Google Scholar] [CrossRef]
- Kim, H.S.; Kang, S.H.; Park, C.H.; Yang, W.I.; Jeung, H.C.; Chung, H.C.; Roh, J.K.; Ahn, J.B.; Kim, N.K.; Min, B.S.; et al. Genome-wide molecular characterization of mucinous colorectal adenocarcinoma using cDNA microarray analysis. Oncol. Rep. 2011, 25, 717–727. [Google Scholar]
- Kim, T.M.; Jeong, H.J.; Seo, M.Y.; Kim, S.C.; Cho, G.; Park, C.H.; Kim, T.S.; Park, K.H.; Chung, H.C.; Rha, S.Y. Determination of genes related to gastrointestinal tract origin cancer cells using a cDNA microarray. Clin. Cancer Res. 2005, 11, 79–86. [Google Scholar]
- Mao, H.; Nian, J.; Wang, Z.; Li, X.; Huang, C. KDELR2 is an unfavorable prognostic biomarker and regulates CCND1 to promote tumor progression in glioma. Pathol. Res. Pract. 2020, 216, 152996. [Google Scholar] [CrossRef]
- Marie, K.L.; Sassano, A.; Yang, H.H.; Michalowski, A.M.; Michael, H.T.; Guo, T.; Tsai, Y.C.; Weissman, A.M.; Lee, M.P.; Jenkins, L.M.; et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat. Commun. 2020, 11, 333. [Google Scholar] [CrossRef]
- Pan, Y.; Bush, E.C.; Toonen, J.A.; Ma, Y.; Solga, A.C.; Sims, P.A.; Gutmann, D.H. Whole tumor RNA-sequencing and deconvolution reveal a clinically-prognostic PTEN/PI3K-regulated glioma transcriptional signature. Oncotarget 2017, 8, 52474–52487. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; She, C.; Ma, L.; Sun, Z.; Li, P.; Zhang, X.; Wang, P.; Li, W. KDELR2 Promotes Glioblastoma Tumorigenesis Targeted by HIF1a via mTOR Signaling Pathway. Cell. Mol. Neurobiol. 2019, 39, 1207–1215. [Google Scholar] [CrossRef]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Tiwari, P.; Wahi, N.; Soni, A.; Bansiwal, R.C.; Kumar, A.; Sharma, B.; Punjabi, P.; Gupta, N.; Malik, B.; et al. Transcriptome profiling reveals association of peripheral adipose tissue pathology with type-2 diabetes in Asian Indians. Adipocyte 2019, 8, 125–136. [Google Scholar] [CrossRef]
- Neelankal John, A.; Ram, R.; Jiang, F.-X. RNA-Seq Analysis of Islets to Characterise the Dedifferentiation in Type 2 Diabetes Model Mice db/db. Endocr. Pathol. 2018, 29, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Reznichenko, A.; Witasp, A.; Liu, P.; Greasley, P.J.; Sorrentino, A.; Blondal, T.; Zambrano, S.; Nordström, J.; Bruchfeld, A.; et al. Novel insights into the disease transcriptome of human diabetic glomeruli and tubulointerstitium. Nephrol. Dial. Transplant. 2020, 35, 2059–2072. [Google Scholar] [CrossRef]
- Das, S.; Frisk, C.; Eriksson, M.J.; Walentinsson, A.; Corbascio, M.; Hage, C.; Kumar, C.; Asp, M.; Lundeberg, J.; Maret, E.; et al. Transcriptomics of cardiac biopsies reveals differences in patients with or without diagnostic parameters for heart failure with preserved ejection fraction. Sci. Rep. 2019, 9, 3179. [Google Scholar] [CrossRef] [PubMed]
- Haywood, M.E.; Cocciolo, A.; Porter, K.F.; Dobrinskikh, E.; Slavov, D.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome signature of ventricular arrhythmia in dilated cardiomyopathy reveals increased fibrosis and activated TP53. J. Mol. Cell Cardiol. 2020, 139, 124–134. [Google Scholar] [CrossRef]
- Colak, D.; Al-Harazi, O.; Mustafa, O.M.; Meng, F.; Assiri, A.M.; Dhar, D.K.; Broering, D.C. RNA-Seq transcriptome profiling in three liver regeneration models in rats: Comparative analysis of partial hepatectomy, ALLPS, and PVL. Sci. Rep. 2020, 10, 5213. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, A.; Li, Q. Whole transcriptome analyis of human lung tissue to identify COPD-associated genes. Genomics 2020, 112, 3135–3141. [Google Scholar] [CrossRef]
- Ek, W.E.; Reznichenko, A.; Ripke, S.; Niesler, B.; Zucchelli, M.; Rivera, N.V.; Schmidt, P.T.; Pedersen, N.L.; Magnusson, P.; Talley, N.J.; et al. Exploring the genetics of irritable bowel syndrome: A GWA study in the general population and replication in multinational case-control cohorts. Gut 2015, 64, 1774–1782. [Google Scholar] [CrossRef]
- Zhang, W.; Yi, Z.; Keung, K.L.; Shang, H.; Wei, C.; Cravedi, P.; Sun, Z.; Xi, C.; Woytovich, C.; Farouk, S.; et al. A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection. J. Am. Soc. Nephrol. JASN 2019, 30, 1481–1494. [Google Scholar] [CrossRef]
- Direito, I.; Fardilha, M.; Helguero, L.A. Contribution of the unfolded protein response to breast and prostate tissue homeostasis and its significance to cancer endocrine response. Carcinogenesis 2019, 40, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Jin, Z.; Chen, N.-Z.; Lu, M.; Liu, C.-B.; Hu, W.-L.; Zheng, C.-G. Activation of IRE1α-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem. Biophys. Res. Commun. 2016, 470, 75–81. [Google Scholar] [CrossRef]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.A.; Lencioni, A.M.; Russell, R.L.; Madsen, H.; Nega, M.; et al. Induction of the Unfolded Protein Response Drives Enhanced Metabolism and Chemoresistance in Glioma Cells. PLoS ONE 2013, 8, e73267. [Google Scholar] [CrossRef]
- Ghosh, J.; Kapur, R. Role of mTORC1-S6K1 signaling pathway in regulation of hematopoietic stem cell and acute myeloid leukemia. Exp. Hematol. 2017, 50, 13–21. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological Targeting of IRE1 in Cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Hassler, J.R.; Scheuner, D.L.; Wang, S.; Han, J.; Kodali, V.K.; Li, P.; Nguyen, J.; George, J.S.; Davis, C.; Wu, S.P.; et al. The IRE1α/XBP1s Pathway Is Essential for the Glucose Response and Protection of β Cells. PLoS Biol. 2015, 13, e1002277. [Google Scholar] [CrossRef]
- Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. XBP1: A Pivotal Transcriptional Regulator of Glucose and Lipid Metabolism. Trends Endocrinol. Metab. 2016, 27, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Nguyen, K.H.; Mishra, S.; Nyomba, B.L. Prohibitin is expressed in pancreatic beta-cells and protects against oxidative and proapoptotic effects of ethanol. FEBS J. 2010, 277, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Verma, G.; Dixit, A.; Nunemaker, C.S. A Putative Prohibitin-Calcium Nexus in β Cell Mitochondria and Diabetes. J. Diabetes Res. 2020, 2020, 7814628. [Google Scholar] [CrossRef]
- Giannotta, M.; Fragassi, G.; Tamburro, A.; Vanessa, C.; Luini, A.; Sallese, M. Prohibitin: A Novel Molecular Player in KDEL Receptor Signalling. BioMed Res. Int. 2015, 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Xia, L.; Goldberg, H.J.; Lee, K.W.K.; Shah, A.; Stavar, L.; Masson, E.A.Y.; Momen, A.; Shikatani, E.A.; John, R.; et al. Inhibition of Src Kinase Blocks High Glucose–Induced EGFR Transactivation and Collagen Synthesis in Mesangial Cells and Prevents Diabetic Nephropathy in Mice. Diabetes 2013, 62, 3874. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Lee, A.H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Kamimura, D.; Katsunuma, K.; Arima, Y.; Atsumi, T.; Jiang, J.J.; Bando, H.; Meng, J.; Sabharwal, L.; Stofkova, A.; Nishikawa, N.; et al. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat. Commun 2015, 6, 7474. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Arima, Y.; Tsuruoka, M.; Jiang, J.J.; Bando, H.; Meng, J.; Sabharwal, L.; Stofkova, A.; Nishikawa, N.; Higuchi, K.; et al. Strong TCR-mediated signals suppress integrated stress responses induced by KDELR1 deficiency in naive T cells. Int. Immunol. 2016, 28, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Popkin, D.L.; Krebs, P.; Li, X.; Tang, M.; Zhan, X.; Zeng, M.; Lin, P.; Xia, Y.; Oldstone, M.B.; et al. Mutation of the ER retention receptor KDELR1 leads to cell-intrinsic lymphopenia and a failure to control chronic viral infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5706–E5714. [Google Scholar] [CrossRef]
- Van Dijk, F.S.; Semler, O.; Etich, J.; Köhler, A.; Jimenez-Estrada, J.A.; Bravenboer, N.; Claeys, L.; Riesebos, E.; Gegic, S.; Piersma, S.R.; et al. Interaction between KDELR2 and HSP47 as a Key Determinant in Osteogenesis Imperfecta Caused by Bi-allelic Variants in KDELR2. Am. J. Hum. Genet. 2020, 107, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Garibaldi, N.; Leoni, L.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Mottes, M.; Aglan, M.; Otaify, G.A.; Temtamy, S.A.; et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef]
- Samara, A.; Rahn, R.; Neyman, O.; Park, K.Y.; Marshall, B.; Dougherty, J.; Hershey, T. Developmental hypomyelination in Wolfram syndrome: New insights from neuroimaging and gene expression analyses. Orphanet. J. Rare. Dis. 2019, 14, 279. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Tajima, T.; Nakamura, A.; Ishizu, K.; Ariga, T. A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of Wolfram syndrome. Pediatr. Diabetes 2017, 18, 934–941. [Google Scholar] [CrossRef]
- Ruggiero, C.; Fragassi, G.; Grossi, M.; Picciani, B.; Di Martino, R.; Capitani, M.; Buccione, R.; Luini, A.; Sallese, M. A Golgi-based KDELR-dependent signalling pathway controls extracellular matrix degradation. Oncotarget 2015, 6, 3375–3393. [Google Scholar] [CrossRef]
- Jia, J.; Yue, X.; Zhu, L.; Jing, S.; Wang, Y.; Gim, B.; Qian, Y.; Lee, I. KDEL receptor is a cell surface receptor that cycles between the plasma membrane and the Golgi via clathrin-mediated transport carriers. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Santen, L.; Schmitt, M.J.; Shaebani, M.R.; Becker, B. Cell-type-specific differences in KDEL receptor clustering in mammalian cells. PLoS ONE 2020, 15, e0235864. [Google Scholar] [CrossRef]
- Bikard, Y.; Viviano, J.; Orr, M.N.; Brown, L.; Brecker, M.; Jeger, J.L.; Grits, D.; Suaud, L.; Rubenstein, R.C. The KDEL receptor has a role in the biogenesis and trafficking of the epithelial sodium channel (ENaC). J. Biol. Chem. 2019, 294, 18324–18336. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef]
- Kokubun, H.; Jin, H.; Aoe, T. Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. Int. J. Mol. Sci. 2019, 20, 5614. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Model | KDELR1 | KDELR2 | KDELR3 | Reference |
|---|---|---|---|---|
| Ischemia | ||||
| Cerebral ischemic stroke RNAseq | ↑ | ↑ | ↑ | [52] |
| Myocardial infarction RNAseq | ↑ | ↑ | [53] | |
| Myocardial infarction with ischemic postconditioning RNAseq | ↓ | ↓ | [53] | |
| SH-SY5Y oxygen–glucose deprivation RT-qPCR | ↑ | [8] | ||
| Ischemic cardiomyopathy RNAseq | ↑ | [54] | ||
| Cancer | ||||
| Colorectal carcinoma CDNA microarray | ↑ | [56] | ||
| YCC-16 gastric cancer cell line cDNA microarray | ↓ | [57] | ||
| Glioma tissue TCGA, CGGA and GSE16011 database | ↑ | [58] | ||
| Melanoma human tumor RNAseq | ↑ | [59] | ||
| FMPC optic glioma RNAseq | ↑ | [60] | ||
| Glioblastoma tissue qRT-PCR | ↑ | [61] | ||
| Pancreatic Disorders | ||||
| Pancreatic islets modified by palmitate RNAseq | ↓ | [62] | ||
| Type 2 Diabetes GeneChip expression array | ↑ | ↑ | [63] | |
| Type 2 Diabetes db/db mice RNAseq | ↑ | [64] | ||
| Diabetic glomeruli and tubulointerstitium RNAseq | ↑ | ↑ | [65] | |
| Heart Disease | ||||
| Dilated cardiomyopathy RNAseq | ↓ | [54] | ||
| Heart failure RNAseq | ↓ | ↑ | [66] | |
| Dilated cardiomyopathy RNAseq | ↑ | [67] | ||
| Other | ||||
| Liver regeneration following partial hepatectomy RNAseq | ↑ | [68] | ||
| Liver regeneration following portal vein ligation RNAseq | ↑ | [68] | ||
| Liver regeneration following associated liver partition and portal vein ligation for staged hepatectomy RNAseq | ↑ | [68] | ||
| Chronic obstructive pulmonary disease RNAseq | ↑ | [69] | ||
| Irritable bowel syndrome rectal biopsy qPCR | ↑ | [70] | ||
| Subclinical acute rejection of kidney transplant RNAseq | ↓ | [71] |
| Protein | Symbol | C-Terminus | Function |
|---|---|---|---|
| BiP | HSPA5 | TAEKDEL | Chaperone |
| Grp94 | HSP90B1 | TAEKDEL | Chaperone |
| Calreticulin | CALR | GQAKDEL | Chaperone |
| Protein disulfide isomerase | P4HB | KAVKDEL | Isomerase |
| Protein disulfide isomerase A3 | PDIA3 | KKAQEDL | Isomerase |
| Protein disulfide isomerase A4 | PDIA4 | SRTKEEL | Isomerase |
| Protein disulfide isomerase A5 | PDIA5 | GKKKEEL | Isomerase |
| Protein disulfide isomerase A6 | PDIA6 | DLGKDEL | Isomerase |
| Hypoxia up-regulated protein 1 | HYOU1 | PLKNDEL | Chaperone |
| Mesencephalic astrocyte-derived neurotrophic factor | MANF | ASARTDL | Pleiotropic |
| Cerebral dopamine neurotrophic factor | CDNF | THPKTEL | Pleiotropic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wires, E.S.; Trychta, K.A.; Kennedy, L.M.; Harvey, B.K. The Function of KDEL Receptors as UPR Genes in Disease. Int. J. Mol. Sci. 2021, 22, 5436. https://doi.org/10.3390/ijms22115436
Wires ES, Trychta KA, Kennedy LM, Harvey BK. The Function of KDEL Receptors as UPR Genes in Disease. International Journal of Molecular Sciences. 2021; 22(11):5436. https://doi.org/10.3390/ijms22115436
Chicago/Turabian StyleWires, Emily S., Kathleen A. Trychta, Lacey M. Kennedy, and Brandon K. Harvey. 2021. "The Function of KDEL Receptors as UPR Genes in Disease" International Journal of Molecular Sciences 22, no. 11: 5436. https://doi.org/10.3390/ijms22115436
APA StyleWires, E. S., Trychta, K. A., Kennedy, L. M., & Harvey, B. K. (2021). The Function of KDEL Receptors as UPR Genes in Disease. International Journal of Molecular Sciences, 22(11), 5436. https://doi.org/10.3390/ijms22115436