
AQP3 Increases Intercellular Cohesion in NSCLC A549 Cell Spheroids through Exploratory Cell Protrusions


Abstract
1. Introduction
2. Results and Discussion
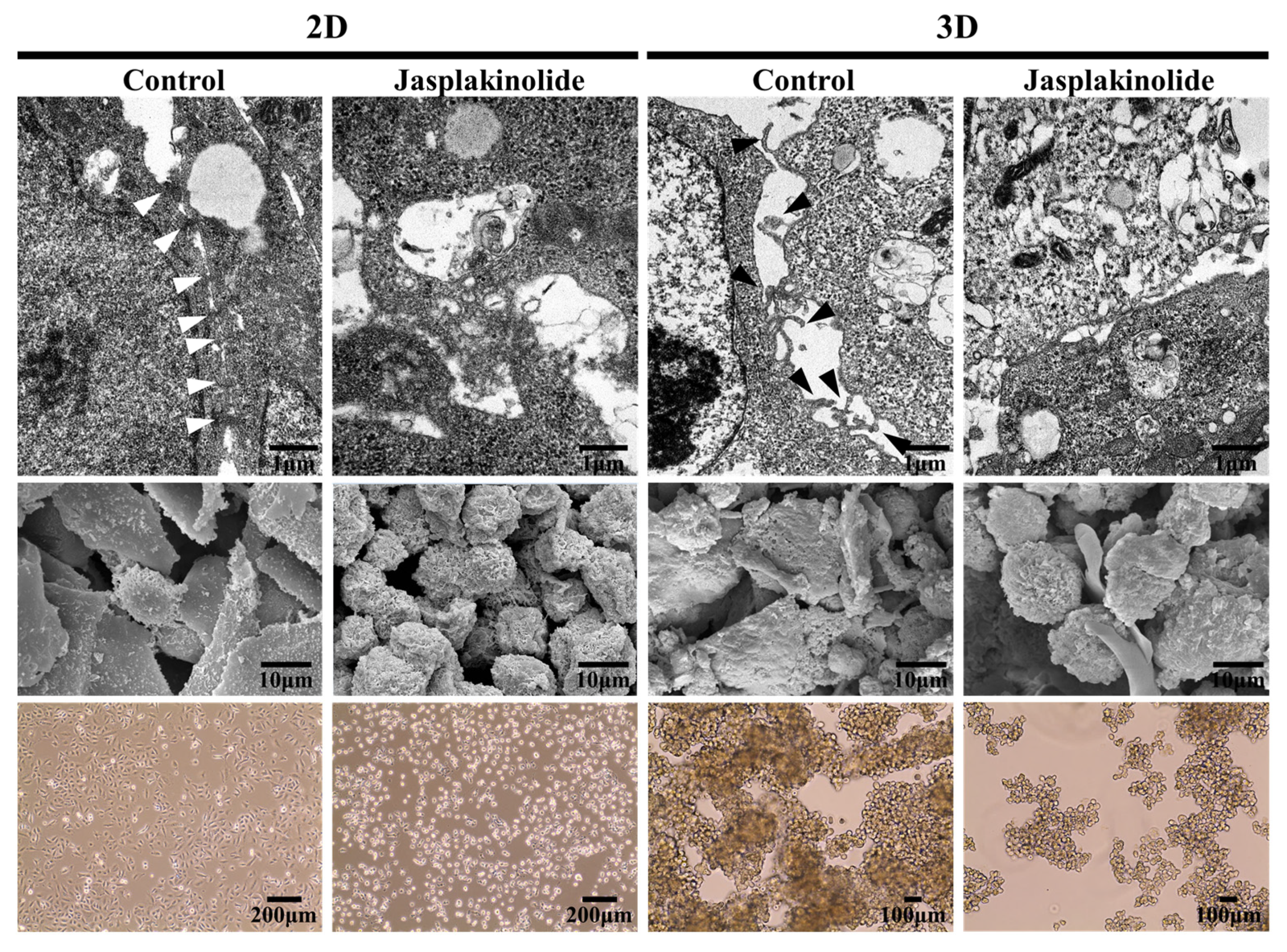
2.1. Detachment of NSCLC A549 Cells Leads to Protrusion Formation
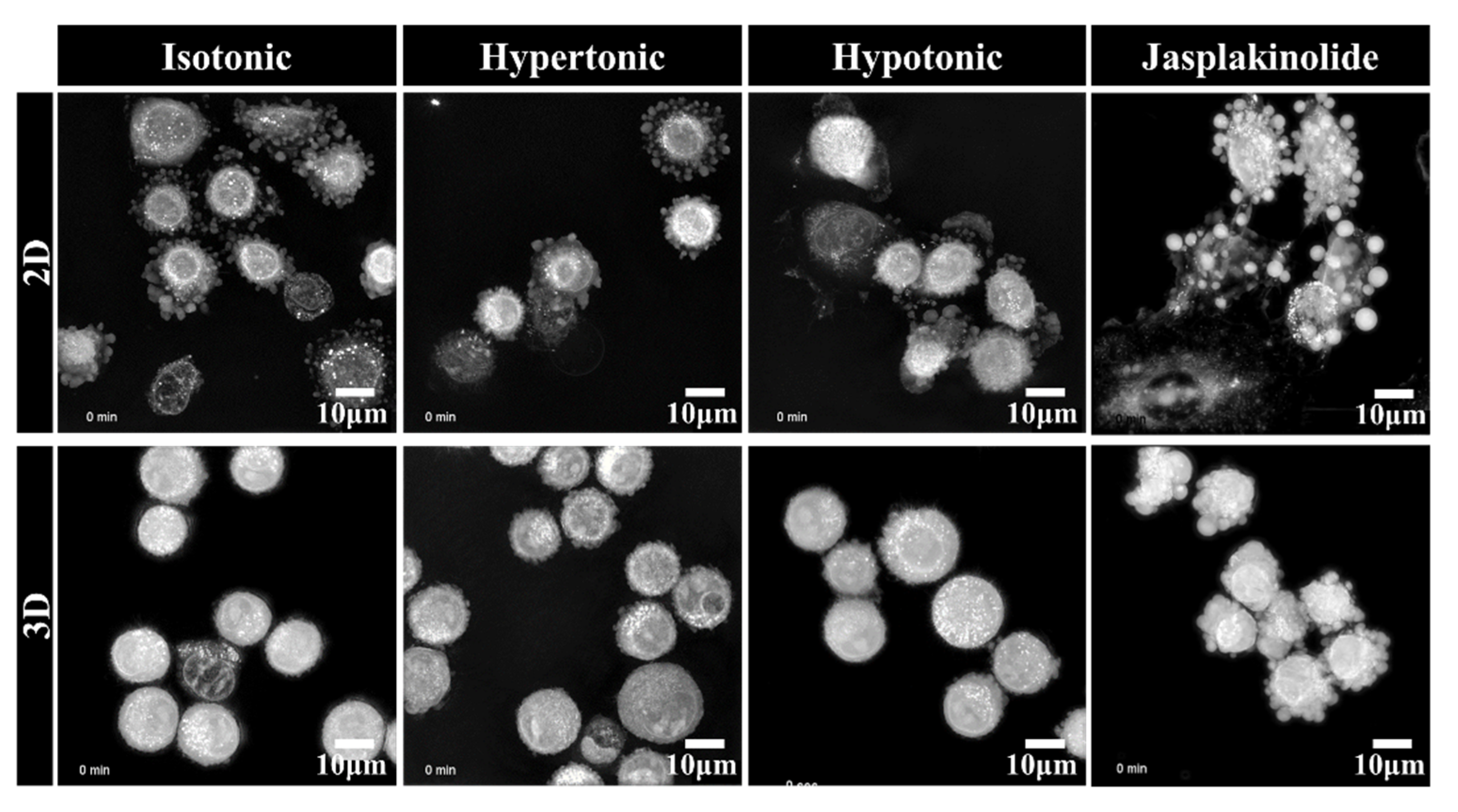
2.2. Clustering of Solitary Human NSCLC A549 Cells Is an Active Process
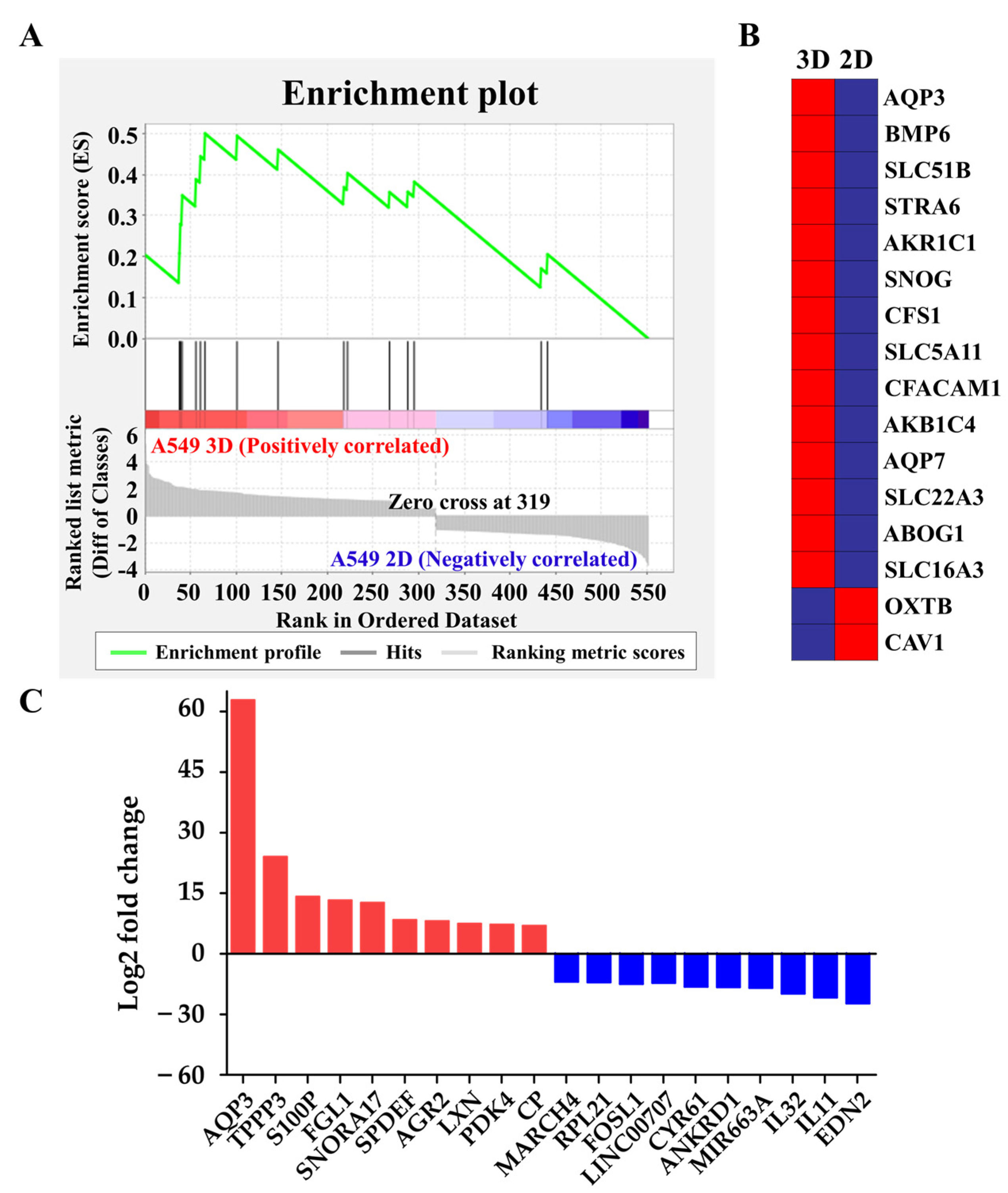
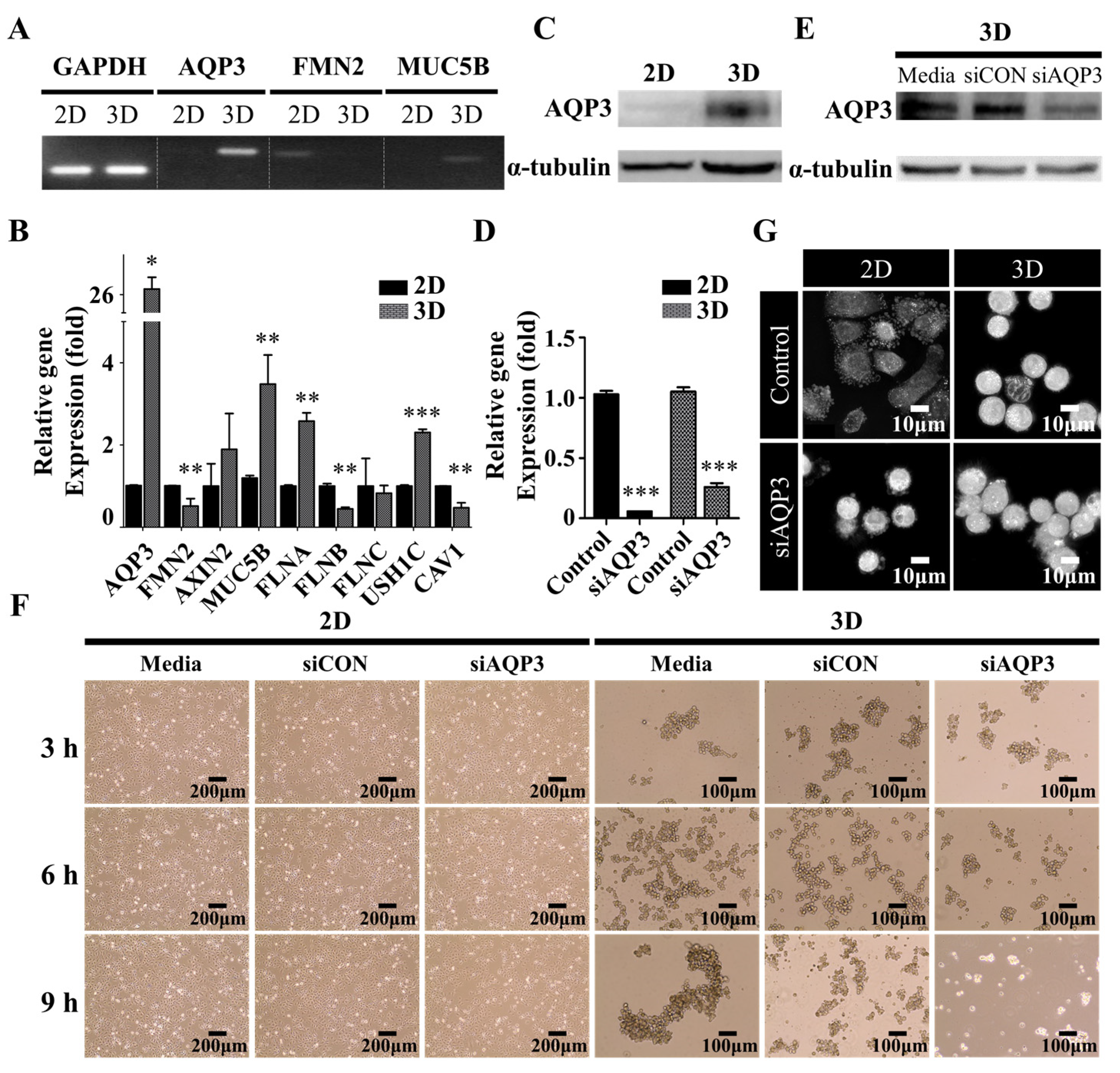
2.3. Upregulation of Hydrostatic Pressure-Regulated Genes in NSCLC A549 Spheroids
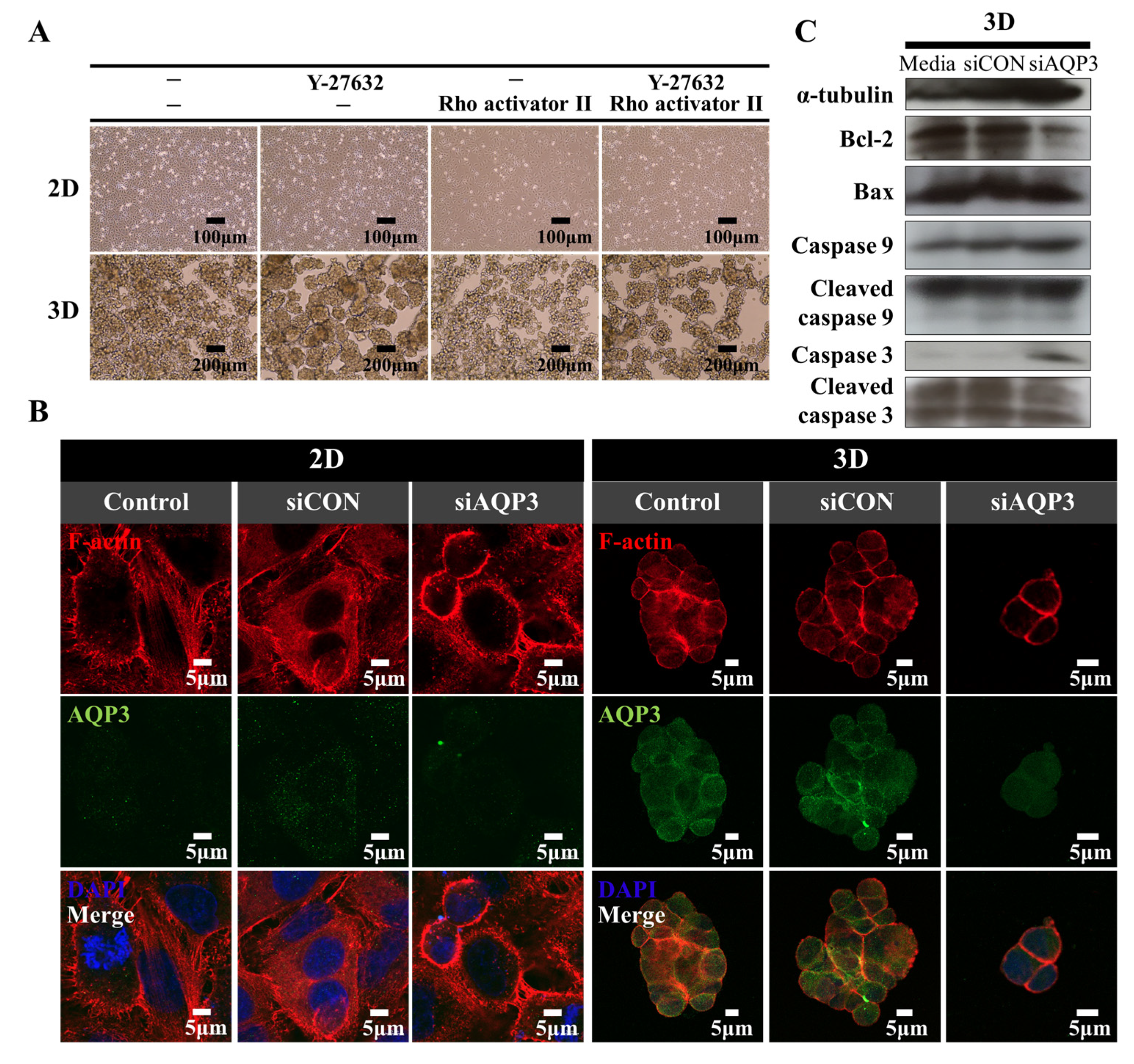
2.4. Downregulation of AQP3 Gene Expression Results in Cortical Actomyosin Remodeling
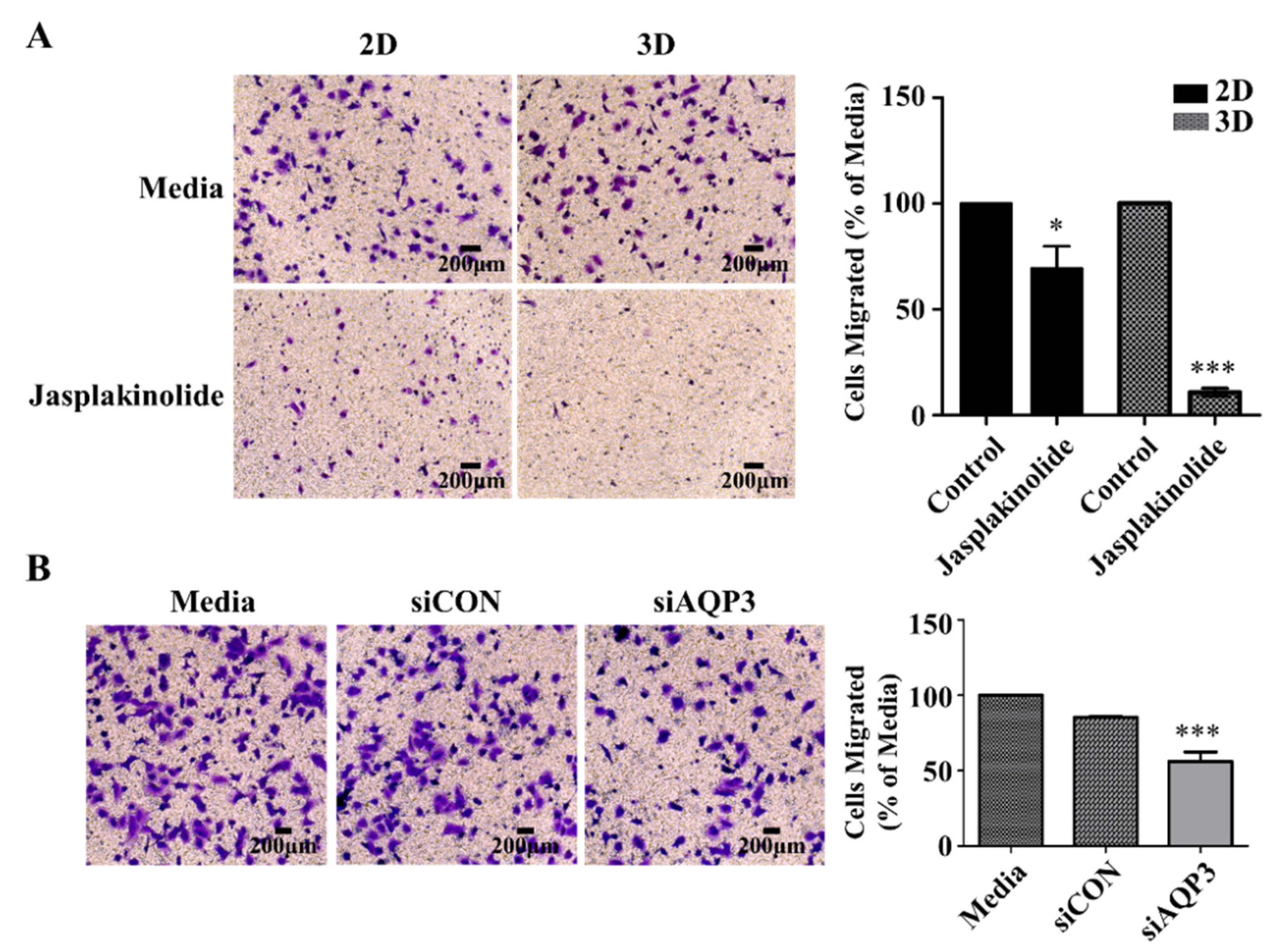
2.5. Protrusion Controls Invasion in A549 Cancer Cells
3. Materials and Methods
3.1. Cell Culture and Reagents
3.2. Imaging
3.3. Time-Lapse Imaging
3.4. Image Analysis
3.5. Poly-HEMA Coating
3.6. RNA Sequencing
3.7. siRNA-Mediated Knockdown of AQP3
3.8. qRT-PCR
3.9. Western Blot Analysis
3.10. Immunocytochemistry
3.11. Scanning Electron Microscopy of Spheroids
3.12. Transmission Electron Microscopy of Spheroids
3.13. Boyden Chamber Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AQP | Aquaporin |
| AQP3 | Aquaporin 3 |
| CAV1 | Caveolin 1 |
| CTC | Circulating tumor cell |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| ES | Enrichment score |
| FLNA | Filamin A |
| FLNB | Filamin B |
| FLNC | Filamin C |
| FMN2 | Formin 2 |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GSEA | Gene set enrichment analysis |
| MUC5B | Mucin 5B |
| NSCLC | Non-small cell lung cancers |
| PBS | Phosphate buffered saline |
| PCR | Polymerase chain reaction |
| poly-HEMA | poly-2-hydroxyethyl methacrylate |
| qRT-PCR | Quantitative real-time reverse transcription-polymerase chain reaction |
| ROCK | Rho-associated protein kinases |
| RPMI | Roswell Park Memorial Institute |
| SEM | Scanning electron microscope |
| TEM | Transmission electron microscope |
| TGF-β | Transforming growth factor-β |
| USH1C | Usher Syndrome 1C |
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Walder, D.; O’Brien, M. Looking back and to the future: Are we improving ‘cure’ in non-small cell lung cancer? Eur. J. Cancer 2017, 75, 192–194. [Google Scholar] [CrossRef]
- Yano, T.; Okamoto, T.; Fukuyama, S.; Maehara, Y. Therapeutic strategy for postoperative recurrence in patients with non-small cell lung cancer. World J. Clin. Oncol. 2014, 5, 1048–1054. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, C.L.; Weigel, K.J.; Schafer, Z.T. Cancer cell survival during detachment from the ECM: Multiple barriers to tumour progression. Nat. Rev. Cancer 2014, 14, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.C.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734.e725. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, E.D.; Yamamoto, A.; Moore, B.M.; Huang, Y.; McBirney, M.; Thomas, A.J.; Greenwood, E.; Rabena, Y.F.; Rahbar, H.; Partridge, S.C.; et al. Regulation of Collective Metastasis by Nanolumenal Signaling. Cell 2020, 183, 395–410.e319. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat. Commun. 2015, 6, 7998. [Google Scholar] [CrossRef]
- Folkman, J.; Moscona, A. Role of cell shape in growth control. Nature 1978, 273, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Raz, A.; Ben-Ze’ev, A. Modulation of the metastatic capability in B16 melanoma by cell shape. Science 1983, 221, 1307–1310. [Google Scholar] [CrossRef]
- Choe, C.; Kim, H.; Min, S.; Park, S.; Seo, J.; Roh, S. SOX2, a stemness gene, induces progression of NSCLC A549 cells toward anchorage-independent growth and chemoresistance to vinblastine. OncoTargets Ther. 2018, 11, 6197–6207. [Google Scholar] [CrossRef]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- King, L.S.; Kozono, D.; Agre, P. From structure to disease: The evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, D.; Guevorkian, K.; Douezan, S.; Brochard-Wyart, F. Soft matter models of developing tissues and tumors. Science 2012, 338, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sebbagh, M.; Renvoize, C.; Hamelin, J.; Riche, N.; Bertoglio, J.; Breard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E.K. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- Ramanathan, S.P.; Helenius, J.; Stewart, M.P.; Cattin, C.J.; Hyman, A.A.; Muller, D.J. Cdk1-dependent mitotic enrichment of cortical myosin II promotes cell rounding against confinement. Nat. Cell Biol. 2015, 17, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Salbreux, G.; Charras, G.; Paluch, E. Actin cortex mechanics and cellular morphogenesis. Trends. Cell Biol. 2012, 22, 536–545. [Google Scholar] [CrossRef]
- Isogai, T.; van der Kammen, R.; Innocenti, M. SMIFH2 has effects on Formins and p53 that perturb the cell cytoskeleton. Sci. Rep. 2015, 5, 9802. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Q.; Hu, Z.; Zhou, Z.; Wang, G.; Li, C.; Xie, W.; Meng, G.; Xiang, Y.; Wu, N.; et al. Long non-coding RNA MUC5B-AS1 promotes metastasis through mutually regulating MUC5B expression in lung adenocarcinoma. Cell Death Dis. 2018, 9, 450. [Google Scholar] [CrossRef]
- Stroka, K.M.; Jiang, H.; Chen, S.H.; Tong, Z.; Wirtz, D.; Sun, S.X.; Konstantopoulos, K. Water permeation drives tumor cell migration in confined microenvironments. Cell 2014, 157, 611–623. [Google Scholar] [CrossRef]
- Leverrier, Y.; Ridley, A.J. Apoptosis: Caspases orchestrate the ROCK ‘n’ bleb. Nat. Cell Biol. 2001, 3, E91–E92. [Google Scholar] [CrossRef] [PubMed]
- Puliafito, A.; De Simone, A.; Seano, G.; Gagliardi, P.A.; Di Blasio, L.; Chianale, F.; Gamba, A.; Primo, L.; Celani, A. Three-dimensional chemotaxis-driven aggregation of tumor cells. Sci. Rep. 2015, 5, 15205. [Google Scholar] [CrossRef]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Beaune, G.; Blanch-Mercader, C.; Douezan, S.; Dumond, J.; Gonzalez-Rodriguez, D.; Cuvelier, D.; Ondarcuhu, T.; Sens, P.; Dufour, S.; Murrell, M.P.; et al. Spontaneous migration of cellular aggregates from giant keratocytes to running spheroids. Proc. Natl. Acad. Sci. USA 2018, 115, 12926–12931. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Seo, C.H.; Lim, B.; Yang, J.O.; Oh, J.; Kim, M.; Lee, S.; Lee, B.; Kang, C.; Lee, S. Accurate quantification of transcriptome from RNA-Seq data by effective length normalization. Nucleic Acids Res. 2011, 39, e9. [Google Scholar] [CrossRef]
- Chae, Y.K.; Woo, J.; Kim, M.J.; Kang, S.K.; Kim, M.S.; Lee, J.; Lee, S.K.; Gong, G.; Kim, Y.H.; Soria, J.C.; et al. Expression of aquaporin 5 (AQP5) promotes tumor invasion in human non small cell lung cancer. PLoS ONE 2008, 3, e2162. [Google Scholar] [CrossRef]
- Flodby, P.; Li, C.; Liu, Y.; Wang, H.; Rieger, M.E.; Minoo, P.; Crandall, E.D.; Ann, D.K.; Borok, Z.; Zhou, B. Cell-specific expression of aquaporin-5 (Aqp5) in alveolar epithelium is directed by GATA6/Sp1 via histone acetylation. Sci. Rep. 2017, 7, 3473. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and cell migration. Pflug. Arch. 2008, 456, 693–700. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef]
- Leithner, A.; Eichner, A.; Muller, J.; Reversat, A.; Brown, M.; Schwarz, J.; Merrin, J.; de Gorter, D.J.; Schur, F.; Bayerl, J.; et al. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat. Cell Biol. 2016, 18, 1253–1259. [Google Scholar] [CrossRef]
- Antonello, Z.A.; Reiff, T.; Ballesta-Illan, E.; Dominguez, M. Robust intestinal homeostasis relies on cellular plasticity in enteroblasts mediated by miR-8-Escargot switch. EMBO J. 2015, 34, 2025–2041. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Zhang, M.; Heallen, T.; Leach, J.; Tao, G.; Xiao, Y.; Bai, Y.; Li, W.; Willerson, J.T.; Martin, J.F. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci. Signal 2015, 8, ra41. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Description | 1 Fold Change(log2) |
|---|---|---|
| AQP3 | Aquaporin 3 (Gill blood group) | 62.82 |
| TPPP3 | Tubulin polymerization-promoting protein family member 3 | 24.03 |
| S100P | S100 calcium binding protein P | 14.16 |
| FGL1 | Fibrinogen-like 1 | 13.25 |
| SNORA17 | Small nucleolar RNA, H/ACA box 17 | 12.62 |
| SPDEF | SAM pointed domain containing ETS transcription factor | 8.35 |
| AGR2 | Anterior gradient 2 | 8.11 |
| LXN | Latexin | 7.43 |
| PDK4 | Pyruvate dehydrogenase kinase, isozyme 4 | 7.20 |
| CP | Ceruloplasmin (ferroxidase) | 6.94 |
| MARCH4 | Membrane-associated ring finger (C3HC4) 4, E3 ubiquitin protein ligase | −6.93 |
| RPL21 | Ribosomal protein L21 | −7.08 |
| FOSL1 | Fos-related antigen 1 isoform 2 | −7.22 |
| LINC00707 | Long intergenic non-protein coding RNA 707 | −7.49 |
| CYR61 | Cysteine-rich, angiogenic inducer, 61 | −8.17 |
| ANKRD1 | Ankyrin repeat domain 1 (cardiac muscle) | −8.26 |
| MIR663A | MicroRNA 663a | −8.46 |
| IL32 | Interleukin 32 | −9.88 |
| IL11 | Interleukin 11 | −10.85 |
| EDN2 | Endothelin 2 | −12.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, S.; Choe, C.; Roh, S. AQP3 Increases Intercellular Cohesion in NSCLC A549 Cell Spheroids through Exploratory Cell Protrusions. Int. J. Mol. Sci. 2021, 22, 4287. https://doi.org/10.3390/ijms22084287
Min S, Choe C, Roh S. AQP3 Increases Intercellular Cohesion in NSCLC A549 Cell Spheroids through Exploratory Cell Protrusions. International Journal of Molecular Sciences. 2021; 22(8):4287. https://doi.org/10.3390/ijms22084287
Chicago/Turabian StyleMin, Sol, Chungyoul Choe, and Sangho Roh. 2021. "AQP3 Increases Intercellular Cohesion in NSCLC A549 Cell Spheroids through Exploratory Cell Protrusions" International Journal of Molecular Sciences 22, no. 8: 4287. https://doi.org/10.3390/ijms22084287
APA StyleMin, S., Choe, C., & Roh, S. (2021). AQP3 Increases Intercellular Cohesion in NSCLC A549 Cell Spheroids through Exploratory Cell Protrusions. International Journal of Molecular Sciences, 22(8), 4287. https://doi.org/10.3390/ijms22084287