1. Introduction
Biomarkers are increasingly employed in empirical studies of human populations to understand how physiological processes change with the diseases [
1,
2,
3,
4,
5]. The identification of biomarkers is strongly needed for Rett syndrome (RTT; MIM #312750), which is an X-linked neurodevelopmental disorder that affects most females [
6]. It is mainly (>90%) caused by mutations in the
methyl CpG binding protein 2 (
MECP2) gene (OMIM #300005) coding for MeCP2, which is a pleotropic protein abundantly expressed in the brain. MeCP2 is involved as an epigenetic modulator by controlling chromatin architecture and gene expression through the binding to methylated DNA [
7] and acting as a key regulator for neuronal development and function [
8,
9]. Over nine hundred
MECP2 mutations have been reported including benign and pathogenic variations [
10], with the most frequent ones (“hotspots”) comprising more than 3/4 (78%) of all the reported pathogenic mutations [
11].
Main features include a severe stereotypic intellectual disability, seizures, autonomic dysfunction, microcephaly, communication dysfunction, postural hypotonia, growth failure, and non-purpose hand use [
12]. The typical clinical picture of the disease is diagnosed as a peculiar phenotypic RTT form by a panel of key clinical elements [
13] with a median age of diagnosis at 2.7 years (interquartile range: 2–4.1 years) [
14]. The disease evolves by highlighting clinical changes, which are classified according to a sequence of disease stages. In particular, in typical RTT, a four-stage neurological regression occurs with loss of previously acquired cognitive, social, and motor skills. After 6–8 months of life with apparent normal psychomotor development, RTT children experience a phase of neurological regression during which they lose manual skills and speech and develop hand stereotypies, autistic behaviors, and walking deficiencies. Subsequently, these patients show a pseudo-stationary stage that is followed by a stage where further motor deterioration, characterized by scoliosis and worsening of the ability to walk, takes place [
6,
10].
In recent years, natural history (NH) [
15,
16], clinical severity [
17,
18], and genotype–phenotype association [
17,
19,
20,
21] have become relevant in better understanding the disease progression and developing objective measurements to be applied in trial metrics [
22].
No definitive cure for RTT is available to date [
23]. Several trials are ongoing in order to test molecules that are potentially able to improve quality of life [
24], although no objective measures to assess clinical severity at the cellular, biochemical, or molecular level are currently available.
Cumulating evidence indicates a key role of oxidative stress in the pathogenesis of RTT [
25,
26]. Isoprostanoids, a large family of compounds derived from non-enzymatic oxidation of polyunsaturated fatty acids (PUFAs) [
27,
28], have been shown to be pertinent to RTT pathological mechanisms [
26,
29,
30]. Among the isoprostanoids, neuroprostanes (NeuroPs), metabolites produced by oxidative metabolism of docosahexaenoic acid (DHA) in the neural cells, have been reported to be relevant in RTT and other conditions of neurological disease [
30,
31,
32]. Prior data strongly suggest that in vivo DHA oxidation follows preferential chemical rearrangements according to different brain diseases [
30]. Among the eight regioisomer series (4-, 7-, 10-, 11-, 13-, 14-, 17- or 20-series) generated by free radical-induced oxidation of DHA, 4(
RS)-4-F
4t-neuroprostane (4-F
4t-NeuroP) and 10(
RS)-10-F
4t-neuroprostane (10-F
4t-NeuroP) are the most investigated ones [
11,
29,
30]. In particular, plasma levels of 4-F
4t-NeuroP and 10-F
4t-NeuroP have been suggested to be biologically synthesized in vivo and related to clinical severity in different neurological diseases in order to be distinctive for different neurological conditions [
30].
The aim of the study is to assess a possible association between MECP2 mutations or RTT disease progression and plasma levels of 4-F4t-NeuroP and 10-F4t-NeuroP in typical RTT patients harboring MECP2 gene mutations.
3. Discussion
Our findings show that 4-F
4t-NeuroP or 10-F
4t-NeuroP, previously reported to be relevant to neurological diseases [
29,
30,
31], are specifically linked to RTT severity, natural history, and
MECP2 mutation type.
No cure for RTT is currently available, although a number of treatments either downstream [
24] or gene-targeted (i.e., gene therapy, gene editing, and gene re-expression) [
23] are proposed. In order to test the effects of potential treatments, the availability of objective and reliable biomarkers is of paramount relevance. The existing severity scores mainly suffer from subjectivity and lack of real-time monitoring. In recent years, biosensors based on artificial intelligence, machine learning, and internet of things have been proposed to overcome the intrinsic limitations of the subjective assessment as outcome measure in randomized clinical trials [
33] (
https://clinicaltrials.gov/ct2/show/NCT04514549, accessed date 18 April 2021).
On the other hand, reliable biomarkers should be able to guide diagnosis, prevention, severity monitoring, and follow-up, as well as assessment of treatment efficacy.
Although lipid peroxidation cannot be considered as a universal surrogate for oxidative stress evaluation [
34], consistent evidence indicates that the levels of lipid peroxidation products are valuable biomarkers in measuring oxidative stress status [
29,
35]. The amounts of 4-F
4t-NeuroP or 10-F
4t-NeuroP in brain tissue have been reported to be predictive of severity score in a murine model of Krabbe disease [
31] and symptomatic
Mecp2 stop/y mice [
30]. Moreover, in the
Mecp2−/y mouse model of RTT, significant inverse relationships between F
4-NeuroPs and brain weight were reported, thus suggesting the involvement of DHA-derived peroxidation products in the pathogenesis of microcephaly [
26], which is a key clinical feature of the RTT phenotype. Interestingly, in the present study, a significant relationship between F
4t-NeuroP (both 4-F
4t-NeuroP and 10-F
4t-NeuroP isomers) and microcephaly is observed in the examined RTT patient cohort.
Here, we investigated the role of F
4t-NeuroPs over a broad spectrum of RTT phenotypes, while previous studies investigated the role of F
4-NeuroPs in RTT [
30,
36]. The novelty of the current study implements specific isomers of F
4-NeuroPs, namely 4-F
4t-NeuroP and 10-F
4t-NeuroP, in the pathogenetic mechanisms of RTT that contribute to the presentation and evolution of the disease. The F
4t-NeuroP precursor i.e., DHA, is particularly abundant in the membranes of neurons as compared to other polyunsaturated fatty acids which are widely distributed in human tissues. DHA autoxidizes non-enzymatically and releases predominantly F
4t-NeuroPs. In addition, the free radical-induced oxidation of PUFAs has been consistently reported in RTT patients [
37,
38,
39] as well as in the brain tissue of
Mecp2 mutant experimental mice [
26]. Given that RTT is a genetic disease linked to impaired expression of a regulatory gene of genetic transcription and chromatin structure, this rare neurodevelopmental disease is considered a useful model in the comprehension of common mechanisms shared by neurodegenerative diseases. It is important to underline that although clinical severity typically worsens, RTT is considered a neurodevelopmental disorder [
40].
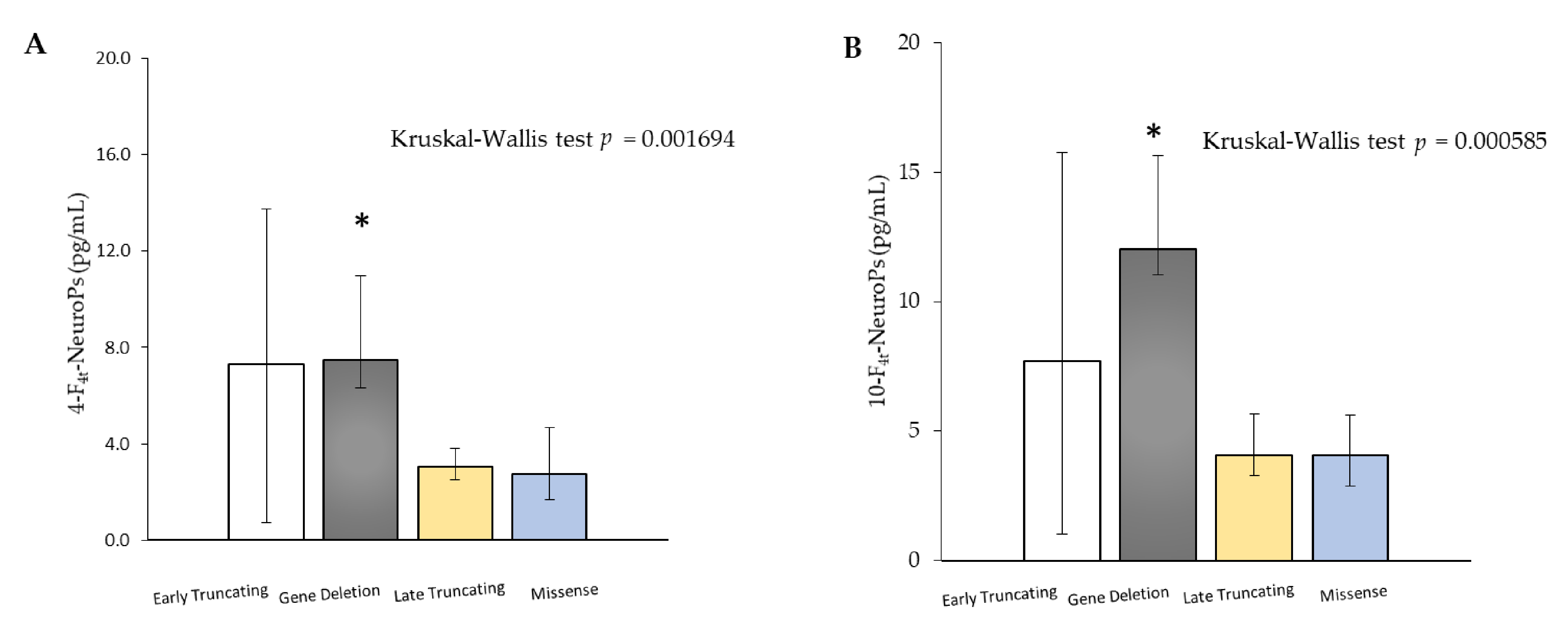
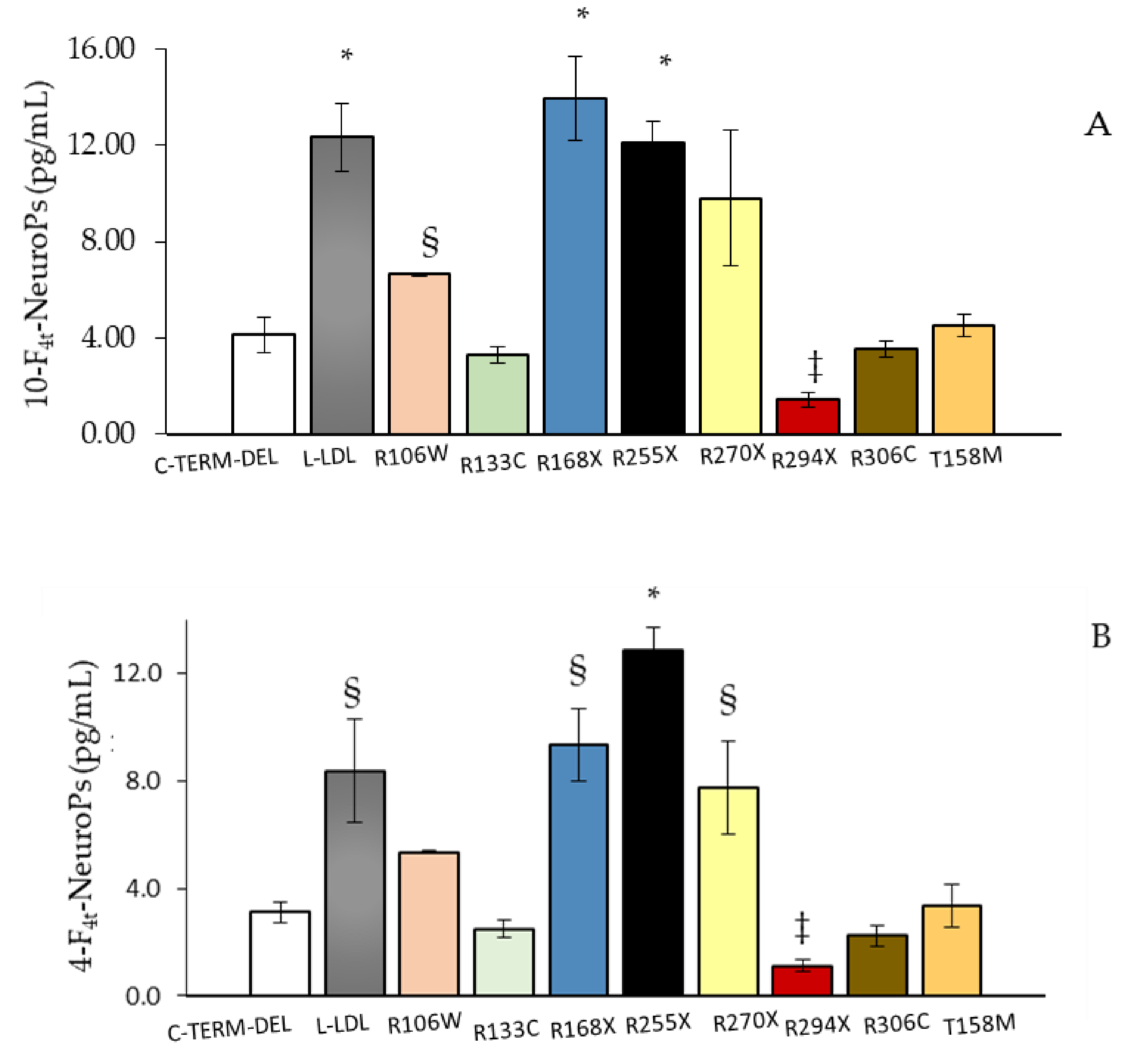
MECP2 mutation type is a strong predictor of disease severity [
12,
17,
41]. Interestingly, plasma amounts of both 4-F
4t-NeuroP and 10-F
4t-NeuroP showed different abundance as a function of
MECP2 gene mutation type. According to our results, showing higher amounts of F
4-NeuroP isomers in subjects carrying either early truncating mutations or gene deletions, a more severe disease is reported to be closely related to
MECP2 truncating mutations [
17,
20,
42,
43,
44]. Furthermore, 4-F
4t-NeuroP and 10-F
4t-NeuroP plasma levels mirrored the clinical severity known in patients carrying
MECP2 “hotspots” mutations [
17]. Therefore, F
4-NeuroP isomers could be involved in the impact of different
MECP2 mutations on ambulation, hand use, and language deficiencies [
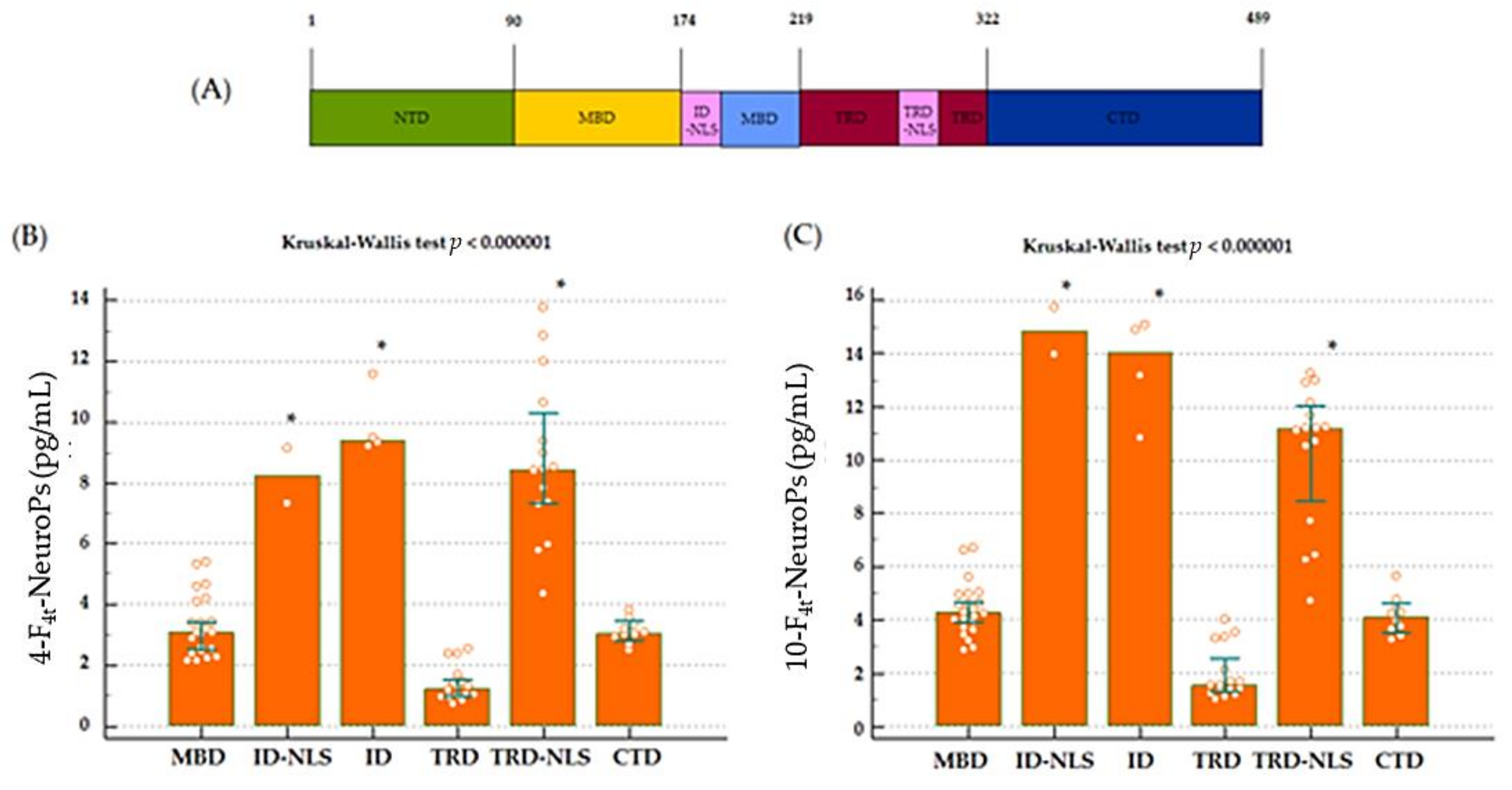
17]. Interestingly, plasma levels of both 4-F
4t-NeuroP and 10-F
4t-NeuroP are shown to be significantly different also considering the MeCP2 protein domain along which gene mutations are grouped. Moreover, mutations after the NLS MeCP2 protein domain (mutations including R294 onwards through C terminal) were found to be associated to a significant decrease in severity of motor functions [
45] and are, in our study, associated to lower plasma levels of both 4- and 10-F
4t-NeuroPs.
Since the aim here is to identify the relationship between F
4-NeuroP isomers and RTT genotype–phenotype correlation, or NH of RTT, a unique RTT population with proven
MECP2 mutations was assessed, and an intra-group comparison was performed. Likewise, a comparative study was carried out to investigate if specific mutations in
MECP2 confer different severity in RTT [
17]. Moreover, significant increase in both 4-F
4t-NeuroP and 10-F
4t-NeuroP in RTT subjects as compared to a matched control population was previously reported [
30]. A similar study, involving the RTT genotype–phenotype and, in particular NH in RTT, would be difficult to investigate in mouse RTT models given the different temporal evolution of the disease in mice compared to humans. [
26].
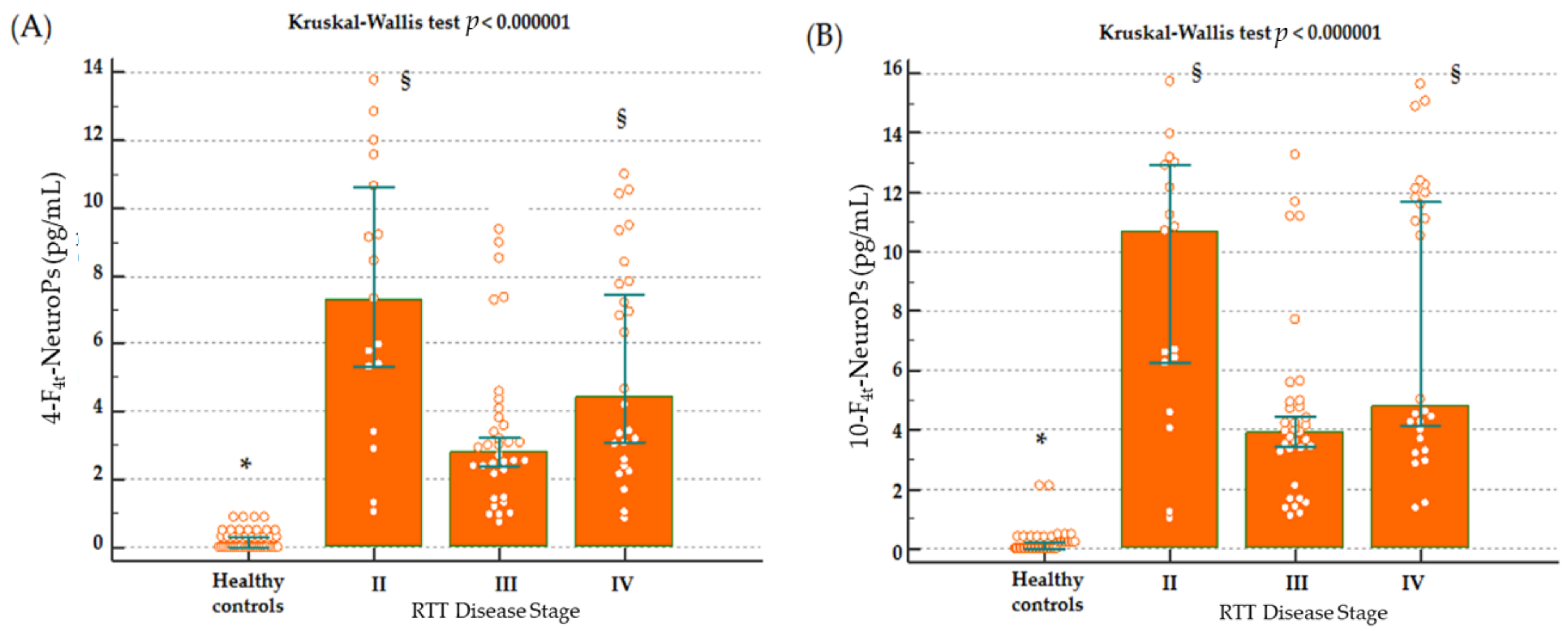
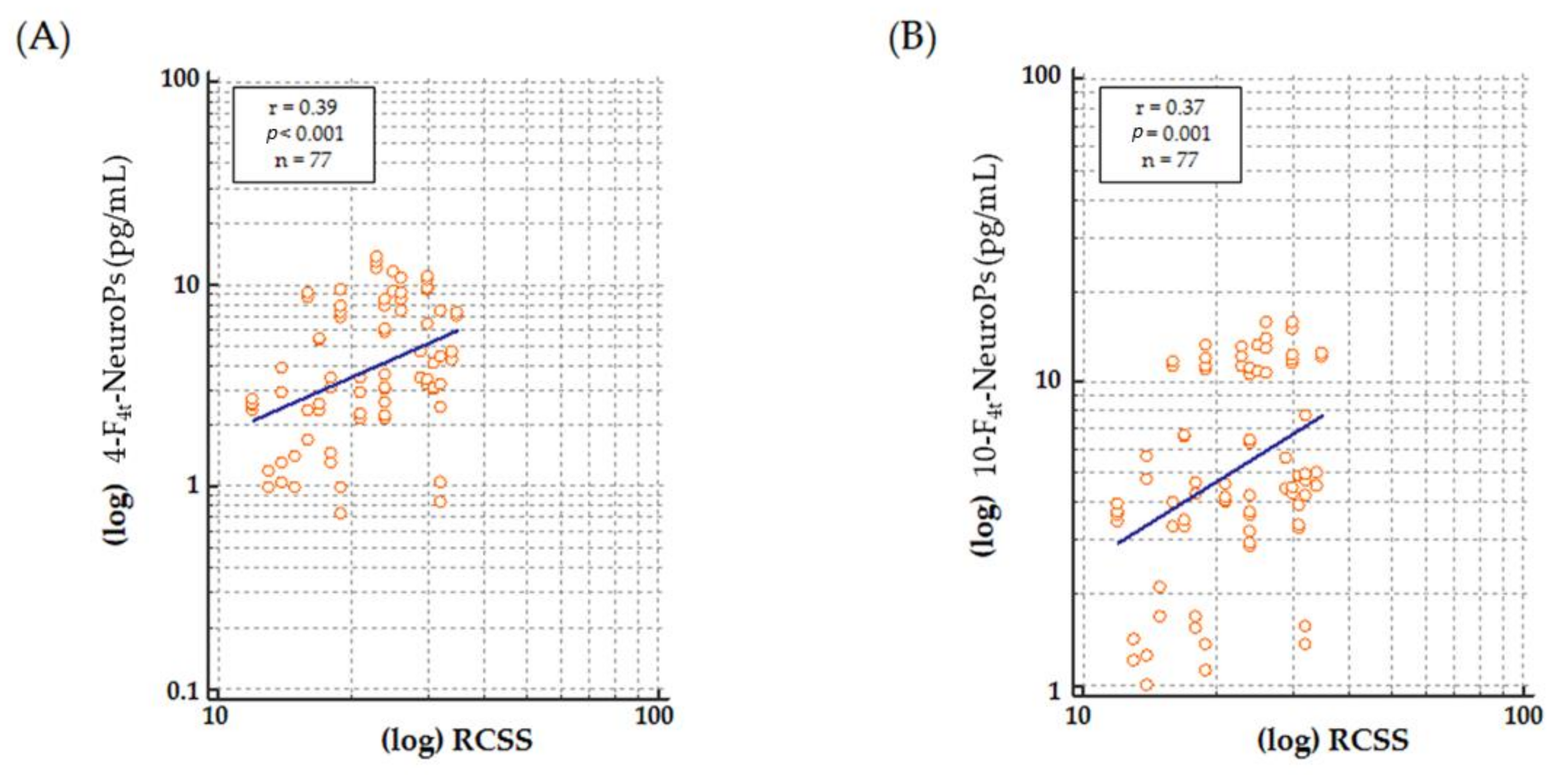
Higher levels of both 4-F
4t-NeuroP and 10-F
4t-NeuroP at stage II of the disease, is worthy of debate, as it reinforces the link between the investigated F
4-NeuroP isomers and the biological mechanisms leading to disease progression. The so defined stage II represents a period of rapid-developmental regression due to the loss of previously acquired skills such as communication and gross motor skills. Conversely, in stage III, the phenotype stabilizes, and some of the skills lost during stage II may be also be partially regained. Stage IV is the last disease stage where the late motor deterioration can last for years or decades [
10]. Thus, the time-course for 4-F
4t-NeuroP and 10-F
4t-NeuroP formation appears to be parallel to the neurological impairment resulting in the clinical presentation. One further point to be taken into account in the study is that no subjects at stage I were enrolled, since it is the early onset stagnation period, which appears between 6 and 18 months of age, and it is frequently overstepped at the time of the clinical diagnosis.
The wide clinical spectrum of RTT appears to be linked to a variety of
MECP2 gene targets [
12,
46]. Under the clinical consensus criteria [
13], key clinical features are used for the distinction between classic or typical RTT from variant or atypical forms [
43]. To date, the role of possible co-existing home mutations in influencing oxidative stress in RTT is unclear [
47], and no biochemical indicators of neurological disease progression in the natural history of RTT have been identified. More research is needed to understand the relationship between oxidized products of DHA and data coming from exome sequencing.
Although more insight is certainly needed on the mechanistic side, overall, our data indicate that 4-F4t-NeuroP and 10-F4t-NeuroP are involved in the pathophysiological mechanisms of RTT and mark the patient’s NH.
4. Materials and Methods
4.1. Subjects
A total of 77 typical RTT patients (age: range: 5.0–47.0; median 15.0; 95% C.I. for the median 13.0 to 19.0, interquartile range 10.0–23.0) with proven hotspot MECP2 gene mutation (R294X, n = 12. R133C, n = 6, R306C, n = 4, C-term deletion, n = 10, R106W, n = 2; T158M, n = 14, R270X, n = 12, R255 X, n = 3, R168X, n = 6; Large deletion, n = 8) admitted to the Child Neuropsychiatry Unit of the Azienda Ospedaliera Universitaria Senese (Siena, Italy) from Jan 2012 to June 2014 (Head: J.H.). Moreover, a total of 45 matched for gender and of comparable age (age: range: 5.00–45.0; median 15.0; 95% C.I. for the median 15.0 to 17.0, inter-quartile range 13.0–18.0) healthy control subjects were recruited. Written consent form was obtained by all the enrolled subjects or by the patient’s guardians. All sensitive clinical data were anonymized by assigning a randomly generated integer code to each RTT patients/control subjects. This study was approved by the local Ethical Committee of Siena University Hospital (Azienda Ospedaliera Universitaria Senese, Siena, Italy)and was carried out in accordance to the rules expressed in the Declaration of Helsinki Ethical Principles for Medical Research involving Human Subjects (Brazil, 2013). A structured clinical evaluation was carried out for each patient.
4.2. RTT Natural History and Clinical Severity Scoring
RTT natural history (NH) was recorded through longitudinal data, and the following key variables were considered: scoliosis, muscle tone, sitting, ambulation, hand function, and feeding.
Clinical severity was assessed by Rett Clinical Severity Score (RCSS) [
17]. RCSS is a validated RTT specific scale designed to assess the severity of key symptoms was completed by the clinicians (C.D.F. and J.H.) at the periodic medical check-ups. RCSS consists of 13 items (age of onset of regression, somatic growth, head growth, independent sitting, ambulation (independent or assisted), hand use, scoliosis, language, non-verbal communication, respiratory dysfunction, autonomic symptoms, onset of stereotypies, and seizures), providing a rating of core symptoms of RTT on a Likert scale of either 0 to 4 or 0 to 5 with a maximum total score of 58. All scores range from 0 to 4 or 0 to 5 with 0 representing the least severe and 4 or 5 representing the most severe finding.
4.3. Sample Preparation
Platelet-poor plasma samples were obtained by centrifugation (2400× g for 15 min at 4 °C) of blood aliquots collected in heparinized tubes. As an antioxidant, butylated hydroxytoluene (BHT) (90 μM prepared in absolute ethanol) was added to each plasma samples, mixed and stored at −70 °C until the time of detection and quantification of F4t-NeuroPs (4-F4t-NeuroP and 10-F4t-NeuroP) released into the circulation as unesterified F4t-NeuroPs (free F4-NeuroPs).
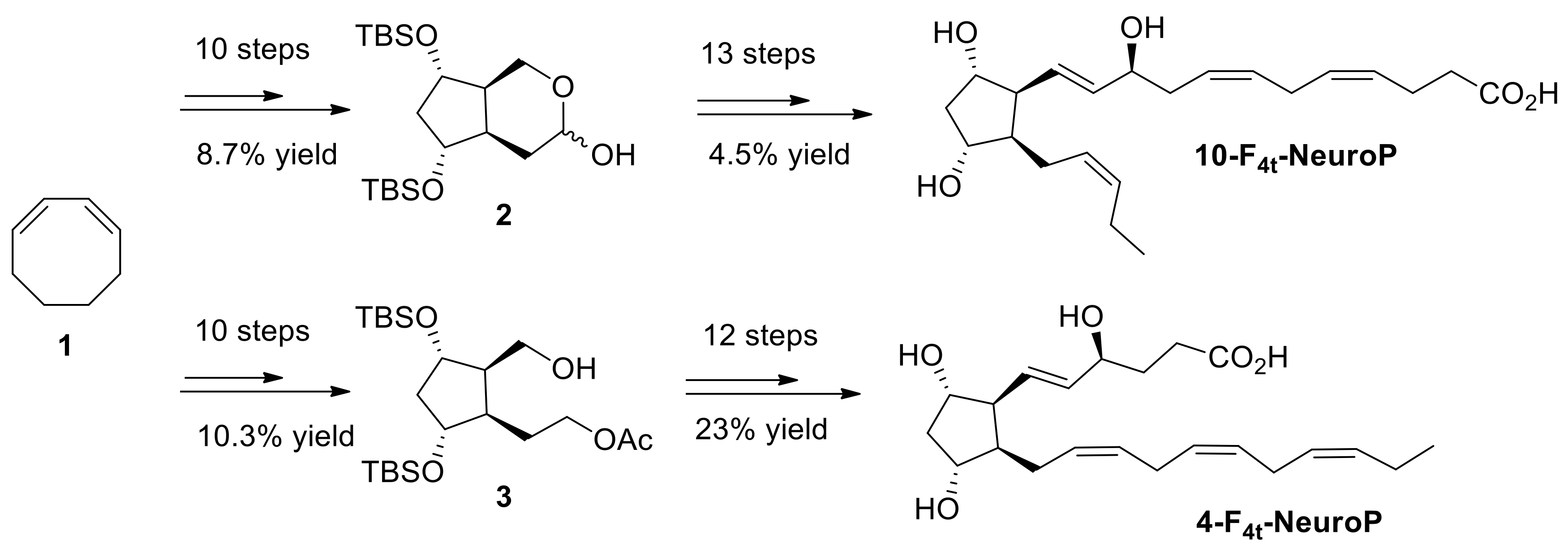
4.4. 4(RS)-F4t-NeuroP, and 10(R)-10-F4t-NeuroP and 10(S)-10-F4t-NeuroP Synthesis
Both 4- and 10-F
4t-NeuroP molecules are not commercially available for quantitative analysis in targeted lipidomics. Hence, it is in-house synthesized for the use in this study. The synthesis of the two series of 4- and 10-F
4t-NeuroPs was performed by our group in previous work [
48,
49,
50], as summarized in
Scheme 1. The two compounds were obtained in around 20–22 steps of synthesis from commercially available 1,3-cyclooctadiene 1. As an example of the synthetic work,
Scheme 1 describes the synthesis of the 4- and the 10-F
4t-NeuroP.
Following our recent strategy [
48], the two key bicyclic intermediates 2 and 3 were obtained in 10 steps and in 8.7% and 10.3% yield, respectively. The introduction of α and ω chains was performed by using regioselective protections/deprotections, oxidations, Wittig elongation, and cross metathesis coupling reactions as the main steps for the 10-F
4t-NeuroP [
50]. The final step was the saponification of the methyl esters in the presence of LiOH to obtain free acids. The 4-F
4t-NeuroP was obtained starting from intermediate 3 in 12 more steps of synthesis, giving 23% yield after optimizations, while 10-F
4t-NeuroP and its C10-epimer were obtained in 13 steps from intermediate 2 [
49].
4.5. 4-F4t-NeuroP and 10-F4t-NeuroP Measurement
Measurement of F4-NeuroPs (4-F4t-NeuroP and 10-F4t-NeuroP) was performed by a gas chromatography/negative-ion chemical ionization tandem mass spectrometry (GC/NICI-MS/MS) after sequential extraction and derivatization steps.
For purification of circulating F
4-NeuroPs, a series of clean-up procedures were carried out using known methods [
30,
51]. Briefly, the internal standard (PGF
2α-d
4, 500 pg in 50 μL ethanol) was added to each plasma sample (1 mL), together with a volume of 2 mL acidified water (pH 3), and then two sequential extractions on solid phase (SPE), the first on a C
18 cartridge and the second on an NH
2 cartridge, were performed. The C
18 cartridge (500 mg Sorbent per Cartridge, 55–105 µm Particle Size, 6cc, Waters, Milford, MA, USA) was preconditioned with methanol (5 mL) and water (5 mL), and sequentially washed after loading the sample with 10 mL water (pH 3), and 10 mL water: acetonitrile (85:15,
v/
v). Hexane: ethyl acetate: propan-2-ol (30:65:5
v/
v/
v, 5 mL) mix was used for the final eluate. Consecutively, the eluate obtained from C
18 cartridge was transferred to the NH
2 cartridge (500 mg Sorbent per Cartridge, 55–105 µm Particle Size, 6cc, Waters, Milford, MA, USA), which was preconditioned with hexane (5 mL). After loading the eluate, sequential wash with 10 mL of hexane: ethyl acetate (30:70,
v/
v), 10 mL acetonitrile: water (9:1,
v/
v), and 10 mL acetonitrile were performed, until the final elution carried out with a mix of ethyl acetate: methanol: acetic acid (10:85:5,
v/
v/
v, 5 mL). The eluate collected from the NH
2 cartridge was evaporated under nitrogen at 40 °C before carrying out the derivation step.
In the derivatization process, the carboxylic group of the F4-NeuroPs, as well as for PGF2α-d4, was converted into pentafluorobenzyl ester, while the hydroxyl group was converted to trimethylsilyl ethers. To this end, an incubation for 45 min at 40 °C was carried out in the presence of 40 μL of pentafluorobenzyl bromide (10% in acetonitrile) and 20 μL of diisopropylethylamine (10% in acetonitrile). Subsequently, the solvent was evaporated under a stream of nitrogen, and 50 μL of N,O-bis(trimethylsilyl)trifluoroacetamide with 5 μL of diisopropylethylamine (10% in acetonitrile) were added for a second incubation at 45 °C for 1 h. One more time, the samples were dried and re-suspended in 50 μL of undecane containing bis(trimethylsilyl)trifluoroacetamide (10%) for GC/NICI-MS/MS analysis (SPB 1701 GC capillary column 30 m × 0.25 mm i.d., 0.25 μm film thickness; helium as the carrier gas at 1 mL/min flow rate; methane as reagent gas, at 2.0 mL/min flow rate). The mass ions determined were the product ions at m/z 323 and m/z 303 derived from the [M-181]− precursor ions of F4-NeuroPs (m/z 593) and PGF2α-d4 (m/z 573) respectively, which corresponds to the loss of CH2C6F5 from the derivatization process. Quantification of F4-NeuroPs was performed by relating the analyte/internal standard peak area ratio (F4t-NeuroP/PGF2α-d4) to the calibration curve constructed.
4.6. Data Analysis
Differences between groups were evaluated by multiple comparisons carried out by Kruskal–Wallis test and post hoc analysis (Conover) or by one-way analysis of variance (ANOVA). The associations between variables were tested using the Spearman rank correlation at 95% confidence intervals (95% C.I.). A two-tailed p < 0.05 was considered to indicate statistical significance. Data were analyzed by using the MedCalc ver. 12.0 statistical software package (MedCalc. Software, Mariakerke, Belgium).


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}