
Mitocryptide-2: Identification of Its Minimum Structure for Specific Activation of FPR2–Possible Receptor Switching from FPR2 to FPR1 by Its Physiological C-terminal Cleavages


Abstract
1. Introduction
2. Results
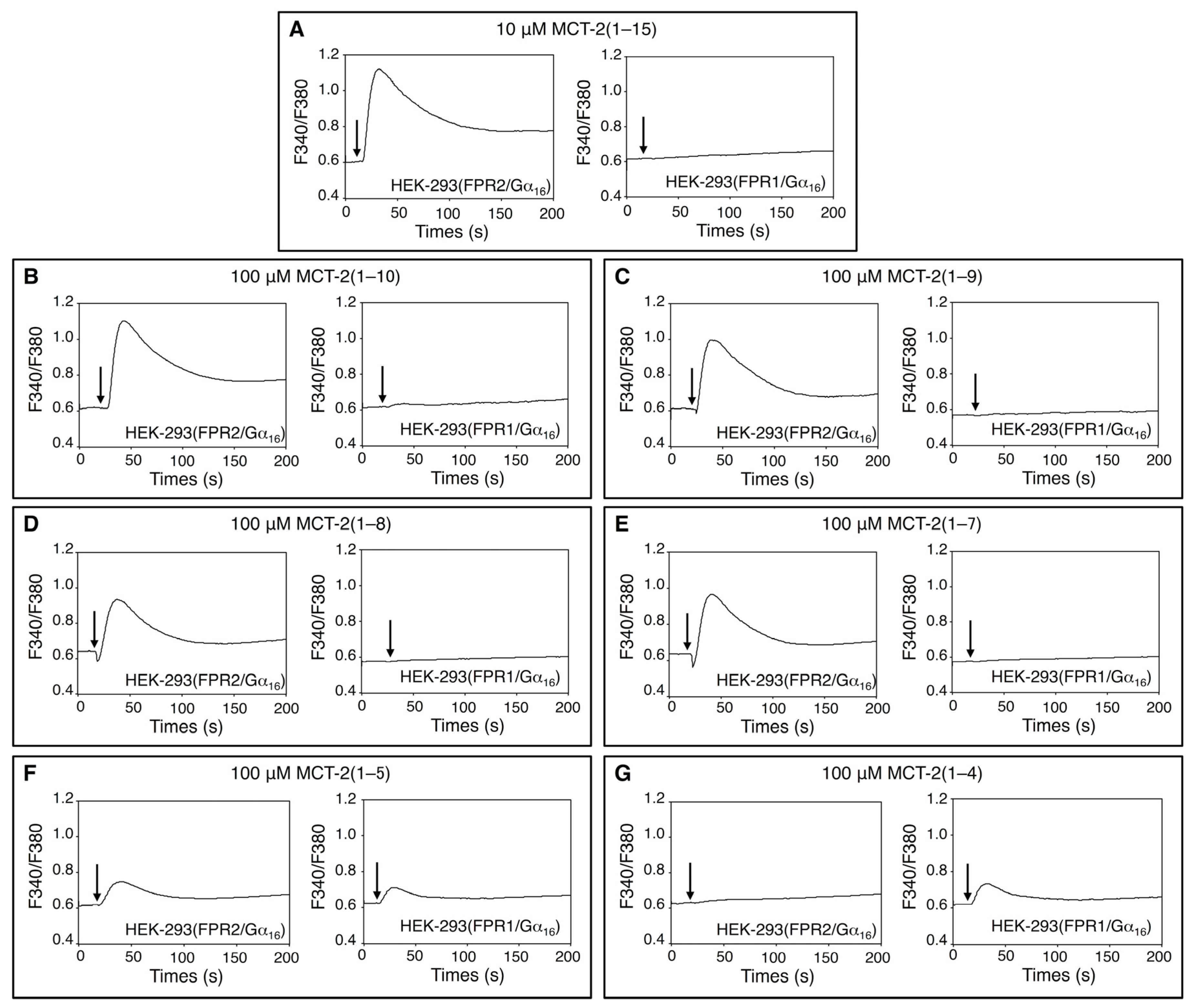
2.1. Effects of MCT-2(1–15) and Its Derivatives on [Ca2+]i in HEK-293 Cells Stably Expressing FPR1 or FPR2
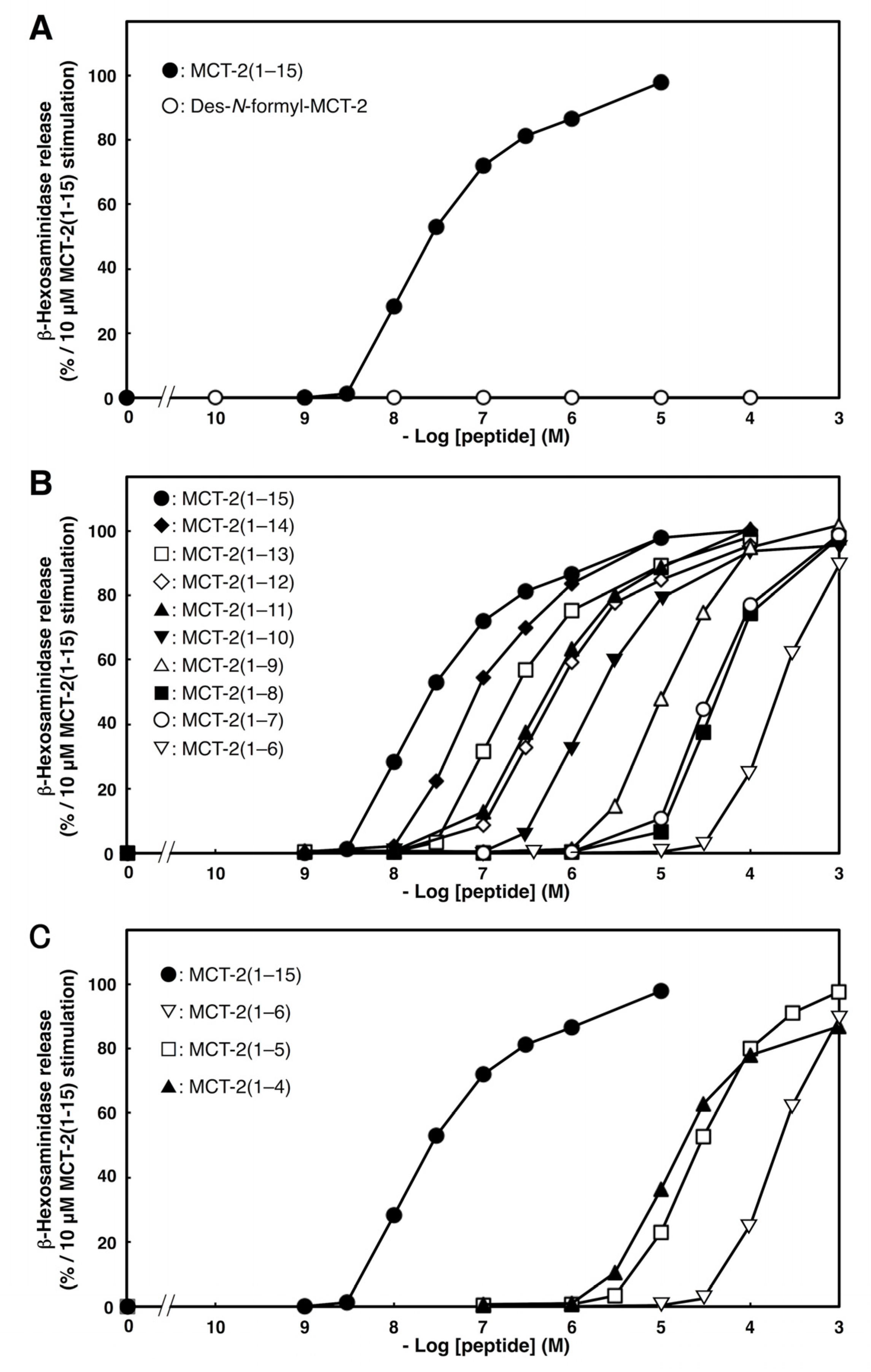
2.2. Effects of C- or N-Terminal Truncations of MCT-2(1–15) on β-Hexosaminidase Release from Neutrophilic/Granulocytic Differentiated HL-60 Cells
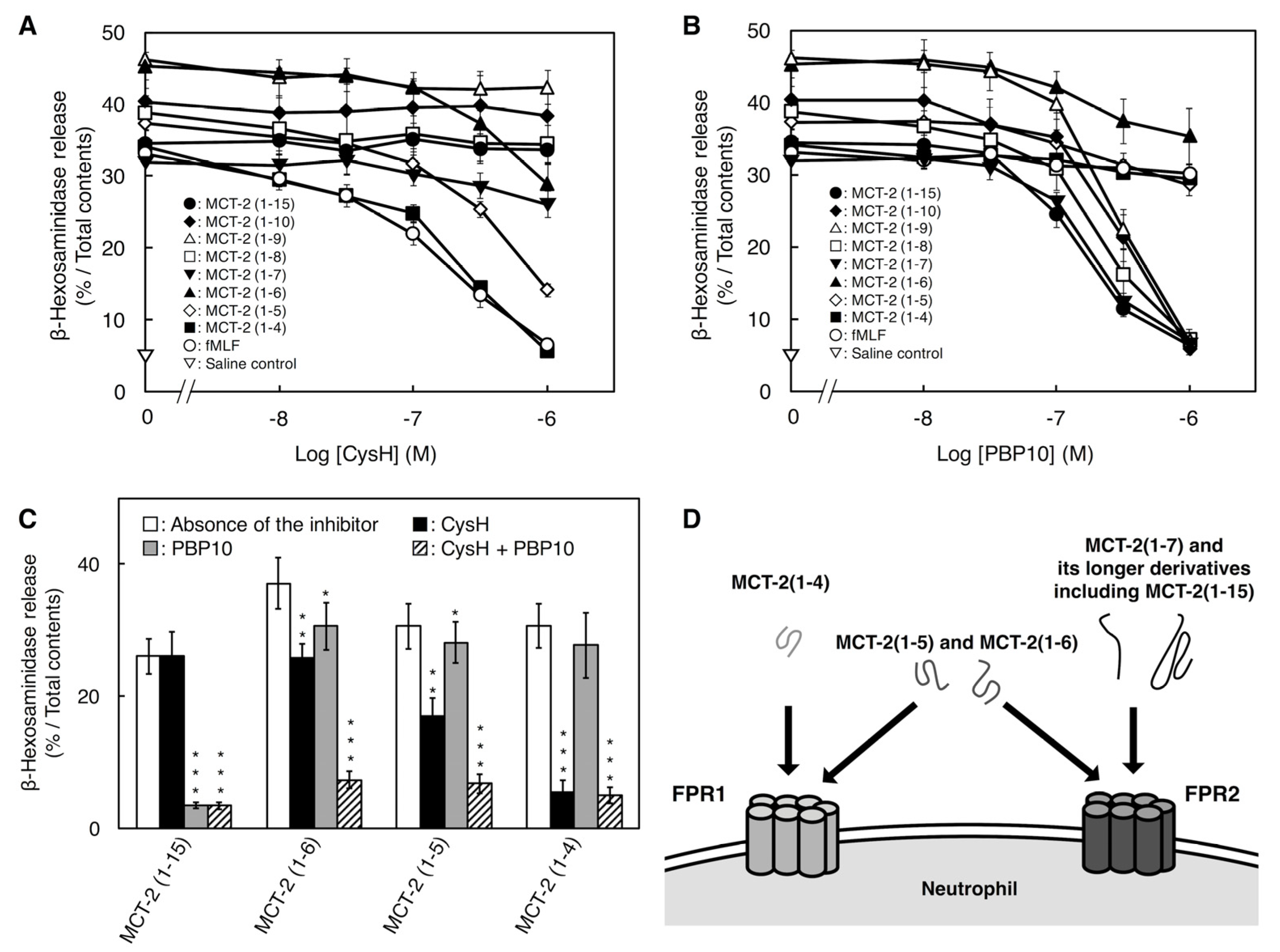
2.3. Involvement of FPR1 and FPR2 in β-Hexosaminidase Release Stimulated by MCT-2(1–15) and Its Derivatives
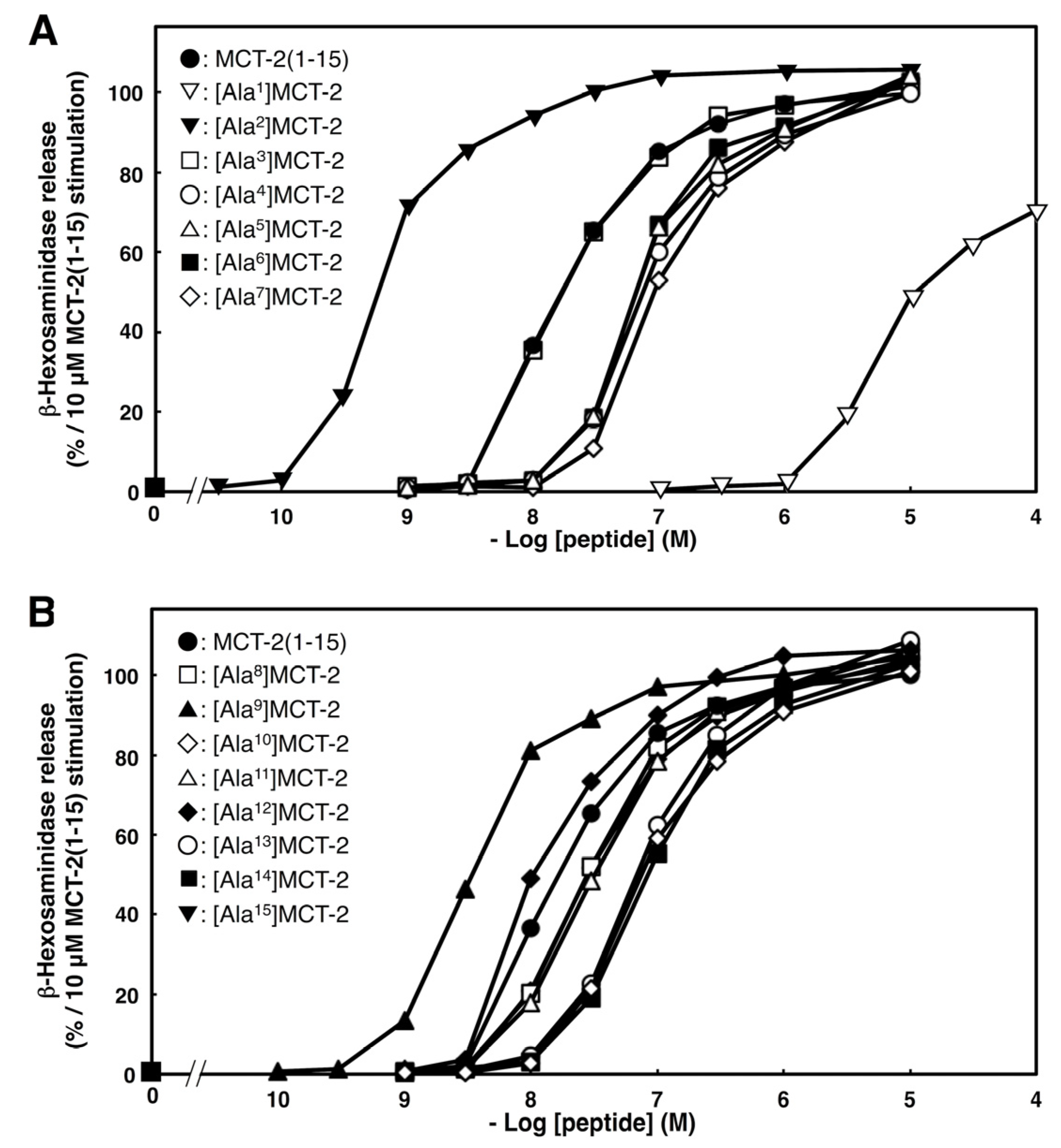
2.4. Effects of Substituting Ala for Each Amino Acid Residue of MCT-2(1–15) on β-Hexosaminidase Release by Differentiated HL-60 Cells
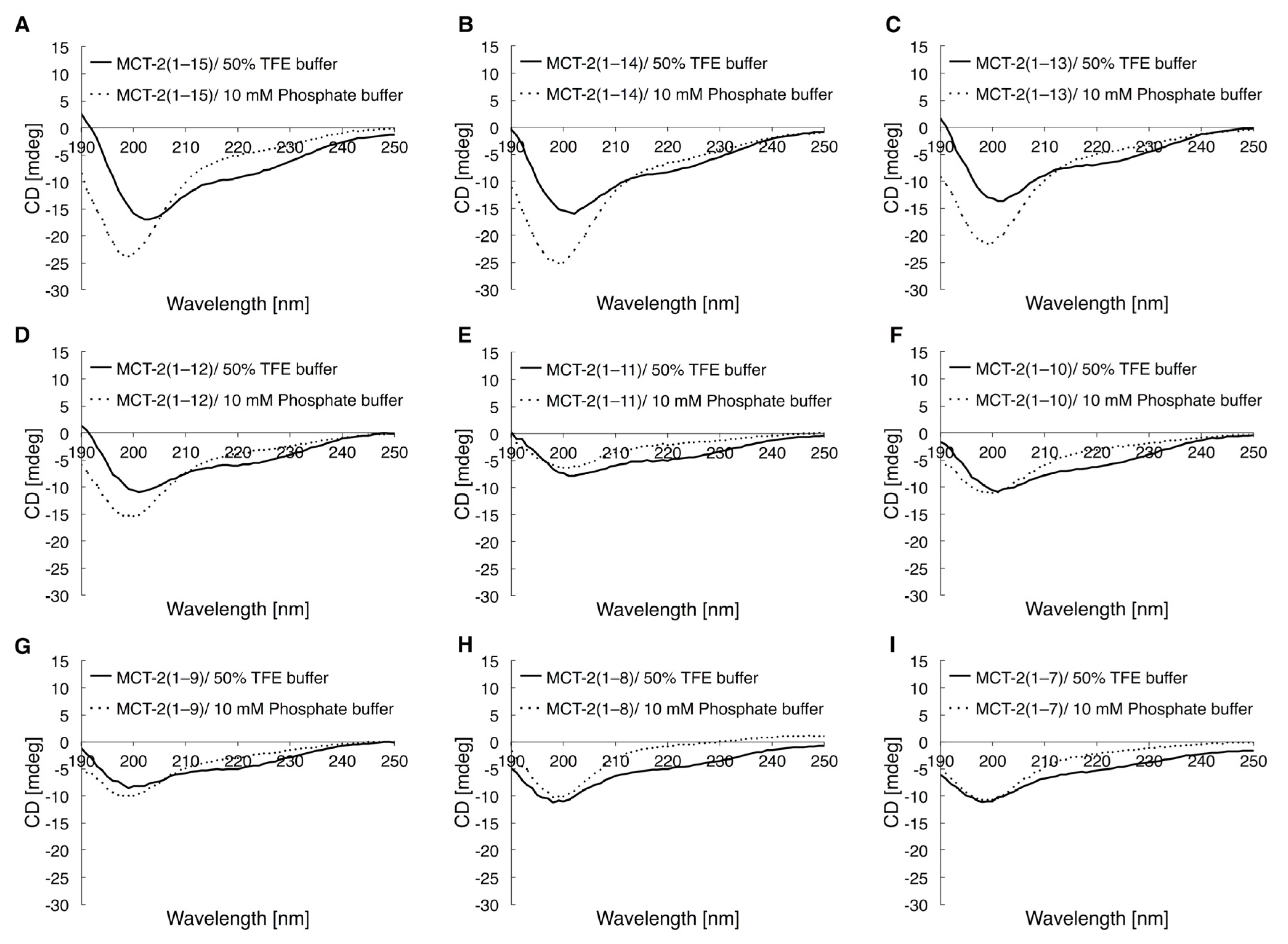
2.5. Circular Dichroic Spectra of MCT-2(1–15) and Its Derivatives
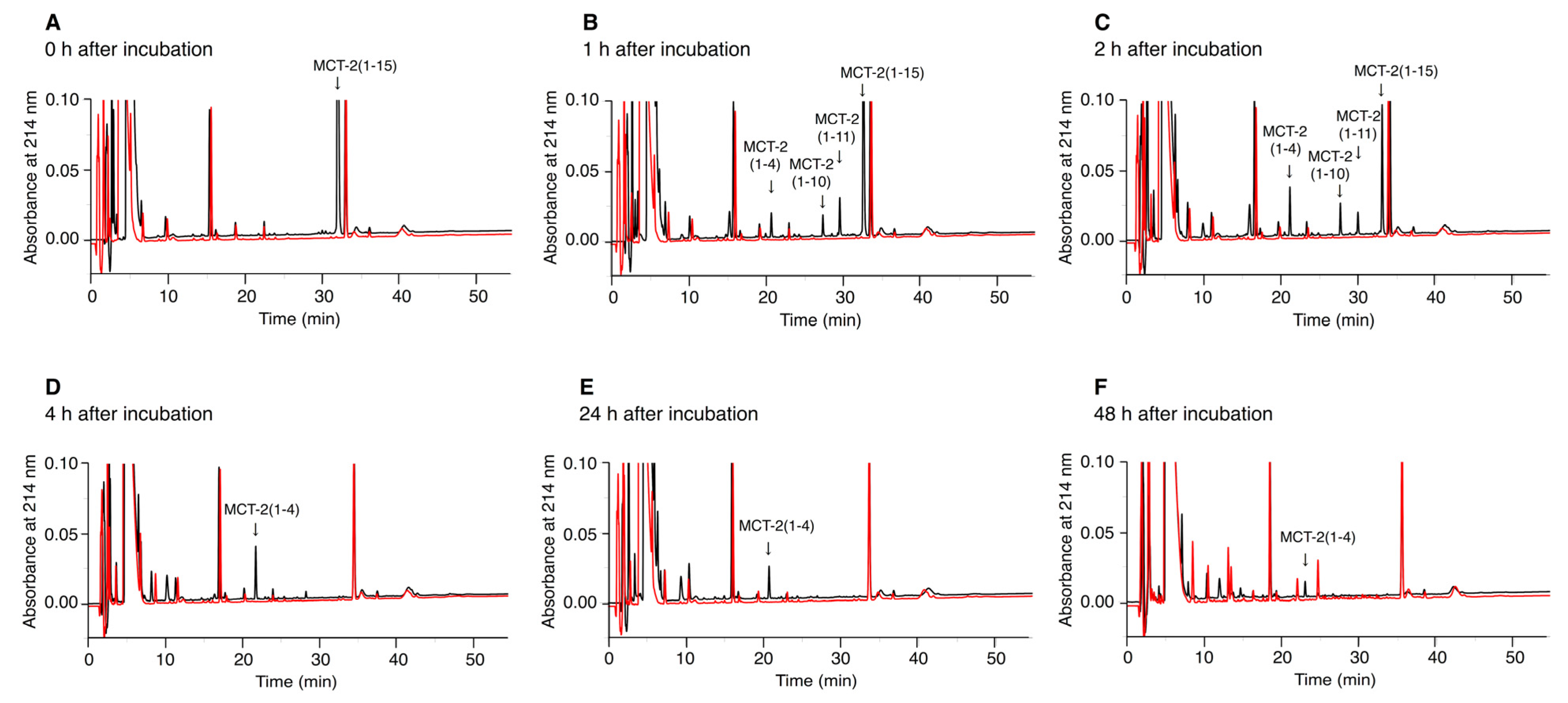
2.6. Time-Dependent Alterations of the Molecular Forms of MCT-2(1–15) in Serum
3. Discussion
3.1. Recognition Mechanisms of MCT-2(1–15) and Its Derivatives by Formyl-Peptide Receptors
3.2. Secondary Structures of MCT-2(1–15) and Its Derivatives for the Interaction with FPR2
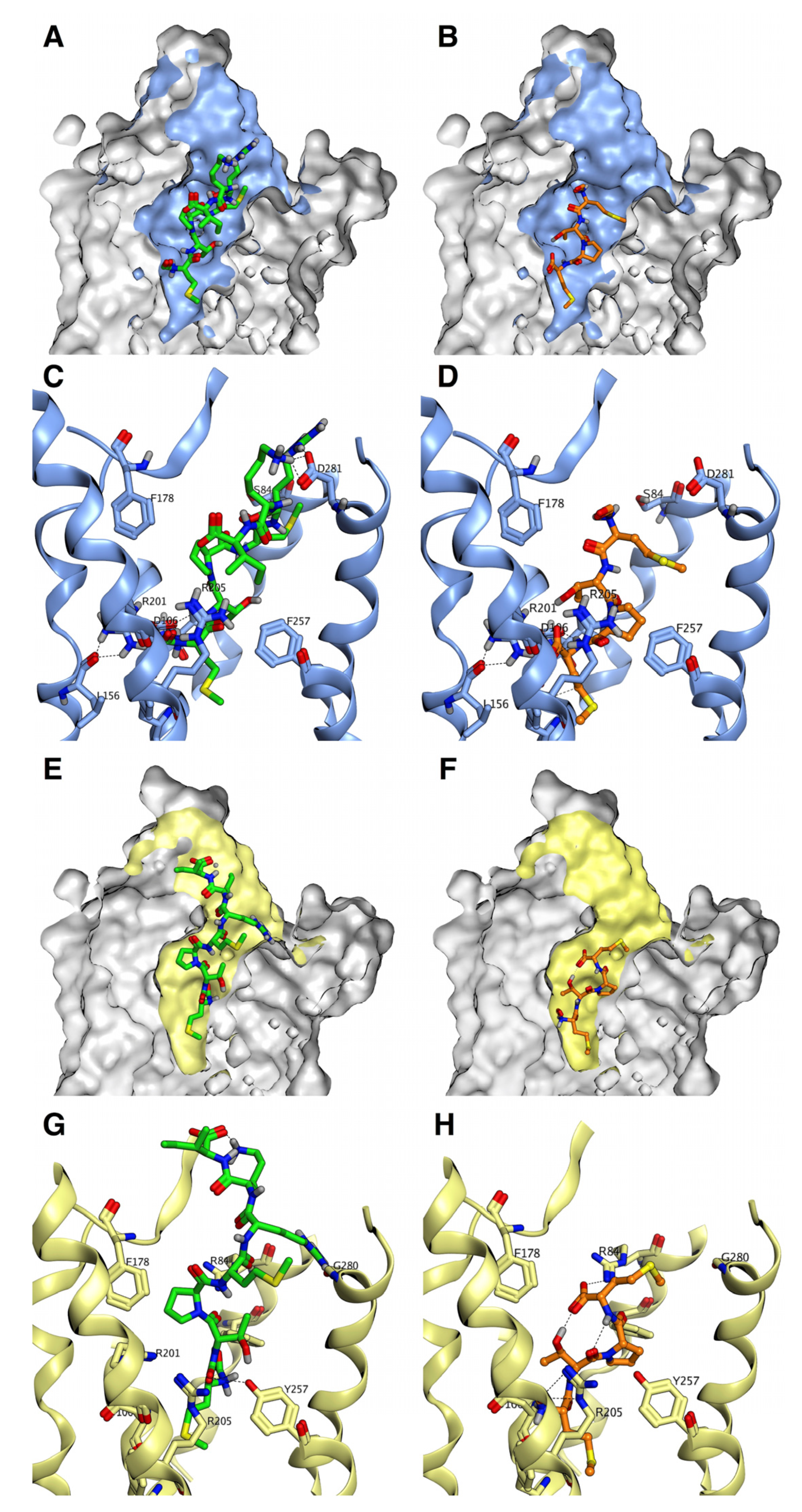
3.3. Binding Characteristics of MCT-2(1–15) and Its Derivatives to FPR1 and FPR2
3.4. Alteration of the Molecular Forms of MCT-2(1–15) in the Bloodstream
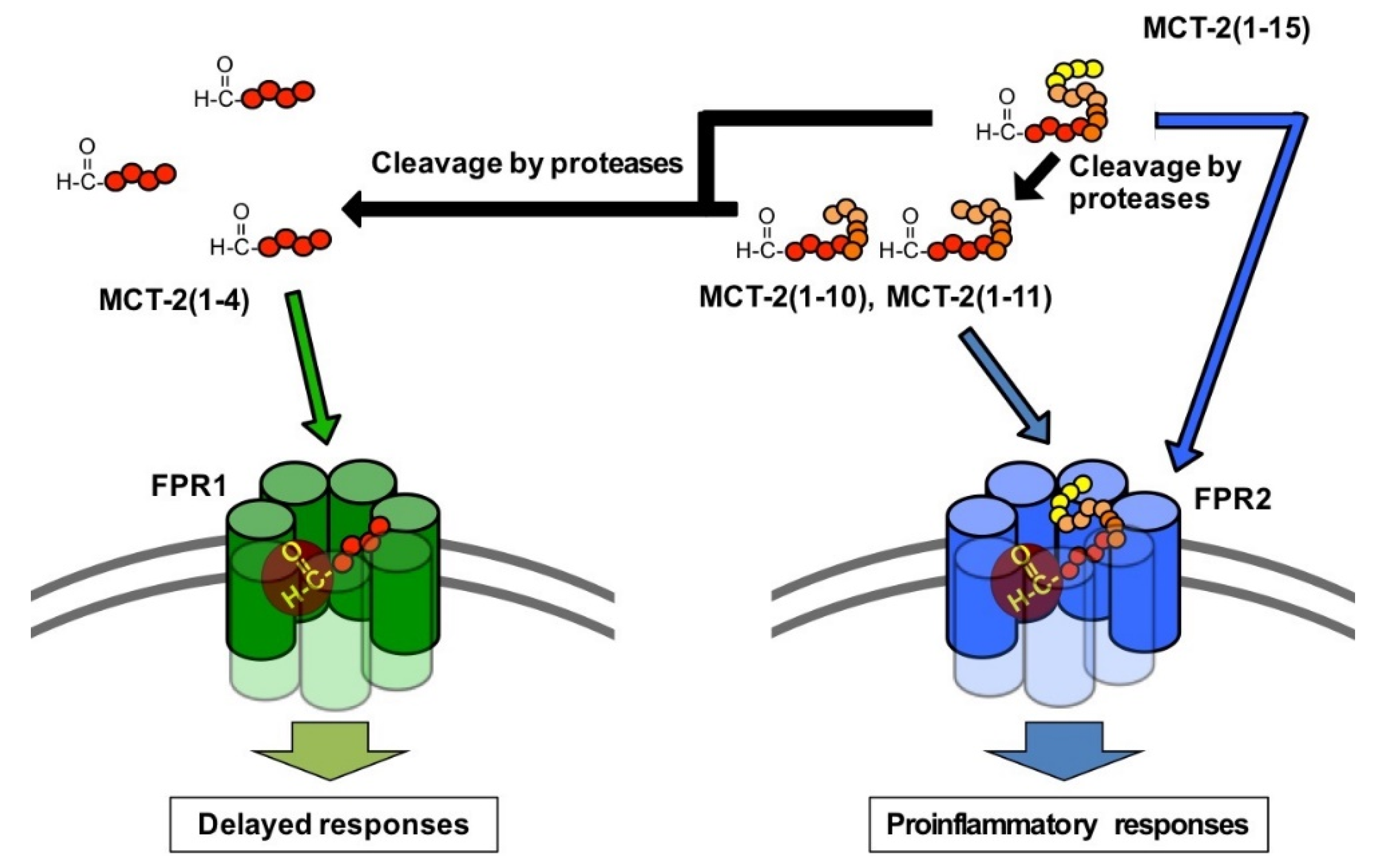
3.5. Possible Physiological Roles of the Receptor Preference Shift of MCT-2(1–15) from FPR2 to FPR1
4. Materials and Methods
4.1. Peptides
4.2. Preparation of Cells
4.3. Establishment of HEK-293 Cells Stably Expressing FPR1 or FPR2
4.4. Measurement of [Ca2+]i
4.5. Assay of β-Hexosaminidase Release
4.6. Measurement of Circular Dichroic Spectra
4.7. Analysis of Time-Dependent Alterations of MCT-2 Molecular Forms in Serum
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| [Ca2+]i | Concentration of intracellular free Ca2+ |
| CD | Circular dichroic |
| CysH | Cyclosporin H |
| EM | Electron microscopy |
| fMLF | Formyl-Met-Leu-Phe |
| FPR1 | Formyl peptide receptor 1 |
| FPR2 | Formyl peptide receptor 2 |
| HBHS | HEPES-buffered Hank’s solution |
| MCT-2 | Mitocryptide-2 |
| MS | Mass spectrometry |
| mtDAMPs | Mitochondrial damage-associated molecular patterns |
| RP | Reverse phase |
| TFE | 2,2,2-trifluoroethanol |
References
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Lay, K. Molecular mechanisms of leukocyte recruitment in the inflammatory process. Cardiovasc. Res. 1996, 32, 733–742. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, E.; Corcoran, B.A.; Wahl, S.M. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl. Acad. Sci. USA 1975, 72, 1059–1062. [Google Scholar] [CrossRef]
- Becker, E.L. The relationship of the chemotactic behavior of the complement-derived factors, C3a, C5a, and C567, and a bacterial chemotactic factor to their ability to activate the proesterase 1 of rabbit polymorphonuclear leukocytes. J. Exp. Med. 1972, 135, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.N.; Henson, P.M.; Otani, A.; Hugli, T.E. Chemotactic response to human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under stimulated in vivo conditions. J. Immunol. 1978, 120, 109–115. [Google Scholar]
- Marasco, W.A.; Phan, S.H.; Krutzsch, H.; Showell, H.J.; Feltner, D.E.; Nairn, R.; Becker, E.L.; Ward, P.A. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol. Chem. 1984, 259, 5430–5439. [Google Scholar] [CrossRef]
- Walz, A.; Peveri, P.; Aschauer, H.; Baggiolini, M. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem. Biophys. Res. Commun. 1987, 149, 755–761. [Google Scholar] [CrossRef]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef]
- Ueki, N.; Someya, K.; Matsuo, Y.; Wakamatsu, K.; Mukai, H. Cryptides: Functional cryptic peptides hidden in protein structures. Biopolymers (Pep. Sci.) 2007, 88, 190–198. [Google Scholar] [CrossRef]
- Mukai, H.; Hokari, Y.; Seki, T.; Takao, T.; Kubota, M.; Matsuo, Y.; Tsukagoshi, H.; Kato, M.; Kimura, H.; Shimonishi, Y.; et al. Discovery of mitocryptide-1, a neutrophil-activating cryptide from healthy porcine heart. J. Biol. Chem. 2008, 283, 30596–30605. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Seki, T.; Nakano, H.; Hokari, Y.; Takao, T.; Kawanami, M.; Tsukagoshi, H.; Kimura, H.; Kiso, Y.; Shimonishi, Y.; et al. Mitocryptide-2: Purification, identification, and characterization of a novel cryptide that activates neutrophils. J. Immunol. 2009, 182, 5072–5080. [Google Scholar] [CrossRef] [PubMed]
- Hokari, Y.; Seki, T.; Nakano, H.; Matsuo, Y.; Fukamizu, A.; Munekata, E.; Kiso, Y.; Mukai, H. Isolation and identification of novel neutrophil-activating cryptides hidden in mitochondrial cytochrome c. Prot. Pept. Lett. 2012, 19, 680–687. [Google Scholar] [CrossRef]
- Marutani, T.; Hattori, T.; Tsutsumi, K.; Koike, Y.; Harada, A.; Noguchi, K.; Kiso, Y.; Mukai, H. Mitochondrial protein-derived cryptides: Are endogenous N-formylated peptides including mitocryptide-2 components of mitochondrial damage-associated molecular patterns? Biopolymers (Pep. Sci.) 2016, 106, 580–587. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.M.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Hauser, C.J.; Sursal, T.; Rodriguez, E.K.; Appleton, P.T.; Zhang, Q.; Itagaki, K. Mitochondrial DAMPs from femoral reamings activate neutrophils via formyl peptide receptors and P44/42 MAP kinase. J. Orthop. Trauma 2010, 24, 534–538. [Google Scholar] [CrossRef]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef]
- Hu, Q.; Wood, C.R.; Cimen, S.; Venkatachalam, A.B.; Alwayn, I.P.J. Mitochondrial damage-associated molecular patterns (MTDs) are released during hepatic ischemia reperfusion and induce inflammatory responses. PLoS ONE 2015, 10, e0140105. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J.N. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kyung, S.Y.; Rogers, A.J.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: Derivation and validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [PubMed]
- Unuma, K.; Aki, T.; Matsuda, S.; Funakoshi, T.; Yoshida, K.; Uemura, K. Elimination and active extrusion of liver mitochondrial proteins during lipopolysaccharide administration in rat. Hepatol. Res. 2013, 43, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Deng, S.; Zhao, S.; Ai, Y.; Zhang, L.; Pan, P.; Su, X.; Tan, H.; Wu, D. Intra-Peritoneal Administration of mitochondrial DNA provokes acute lung injury and systemic inflammation via toll-like receptor 9. Int. J. Mol. Sci. 2016, 17, 1425. [Google Scholar] [CrossRef]
- Deng, S.Y.; Zhang, L.M.; Ai, Y.H.; Pan, P.H.; Zhao, S.P.; Su, X.L.; Wu, D.D.; Tan, H.Y.; Zhang, L.N.; Tsung, A. Role of interferon regulatory factor-1 in lipopolysaccharide-induced mitochondrial damage and oxidative stress responses in macrophages. Int. J. Mol. Med. 2017, 40, 1261–1269. [Google Scholar] [CrossRef]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 2017, 26, 508. [Google Scholar] [CrossRef]
- Hazeldine, J.; Hampson, P.; Opoku, F.A.; Foster, M.; Lord, J.M. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 2015, 46, 975–984. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Zhang, B.; Asadi, S.; Weng, Z.; Sismanopoulos, N.; Theoharides, T.C. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE 2012, 7, e49767. [Google Scholar] [CrossRef]
- Walker, J.E.; Carroll, J.; Altman, M.C.; Fearnley, I.M. Mass spectrometric characterization of the thirteen subunits of bovine respiratory complexes that are encoded in mitochondrial DNA. Methods Enzymol. 2009, 456, 111–131. [Google Scholar]
- Ott, M.; Amunts, A.; Brown, A. Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef]
- Boulay, F.; Tardif, M.; Brouchon, L.; Vignais, P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem. Biophys. Res. Commun. 1990, 168, 1103–1109. [Google Scholar] [CrossRef]
- Quehenberger, O.; Prossnitz, E.R.; Cavanagh, S.L.; Cochrane, C.G.; Ye, R.D. Multiple domains of the N-formyl peptide receptor are required for high-affinity ligand binding. Construction and analysis of chimeric N-formyl peptide receptors. J. Biol. Chem. 1993, 268, 18167–18175. [Google Scholar] [CrossRef]
- Murphy, P.M. The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 1994, 12, 593–633. [Google Scholar] [CrossRef]
- Seki, T.; Fukamizu, A.; Kiso, Y.; Mukai, H. Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem. Biophys. Res. Commun. 2011, 404, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Gabl, M.; Holdfeldt, A.; Winther, M.; Forsman, H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016, 114, 22–39. [Google Scholar] [CrossRef]
- Weiß, E.; Kretschmer, D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Goulopoulou, S.; Webb, R.C. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H768–H777. [Google Scholar] [CrossRef]
- Babbin, B.A.; Jesaitis, A.J.; Ivanov, A.I.; Kelly, D.; Laukoetter, M.; Nava, P.; Parkos, C.A.; Nusrat, A. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 2007, 179, 8112–8121. [Google Scholar] [CrossRef]
- Zhang, X.G.; Hui, Y.N.; Huang, X.F.; Du, H.J.; Zhou, J.; Ma, J.X. Activation of formyl peptide receptor-1 enhances restitution of human retinal pigment epithelial cell monolayer under electric fields. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Julian, M.W.; Bao, S.; McCullers, M.K.; Lai, J.P.; Knoell, D.L.; Crouser, E.D. Formyl peptide receptor ligands promote wound closure in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 264–269. [Google Scholar] [CrossRef]
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Le, Y.; Gao, J.L.; Zhao, J.; Wang, J.M.; Wang, A. Formylpeptide receptors mediate rapid neutrophil mobilization to accelerate wound healing. PLoS ONE 2014, 9, e90613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Yang, T.; Su, Z.; Fang, D.; Wang, Y.; Fang, J.; Hou, X.; Le, Y.; Chen, K.; et al. Formylpeptide receptor 1 mediates the tumorigenicity of human hepatocellular carcinoma cells. Oncoimmunology 2015, 5, e1078055. [Google Scholar] [CrossRef] [PubMed]
- Lind, S.; Gabl, M.; Holdfeldt, A.; Mårtensson, J.; Sundqvist, M.; Nishino, K.; Dahlgren, C.; Mukai, H.; Forsman, H. Identification of residues critical for FPR2 activation by the cryptic peptide mitocryptide-2 originating from the mitochondrial DNA-encoded cytochrome b. J. Immunol. 2019, 202, 2710–2719. [Google Scholar] [CrossRef]
- Gabl, M.; Sundqvist, M.; Holdfeldt, A.; Lind, S.; Mårtensson, J.; Christenson, K.; Marutani, T.; Dahlgren, C.; Mukai, H.; Forsman, H. Mitocryptides from human mitochondrial DNA-encoded proteins activate neutrophil formyl peptide receptors: Receptor preference and signaling properties. J. Immunol. 2018, 200, 3269–3282. [Google Scholar] [CrossRef]
- Offermanns, S.; Simon, M.I. Gα15 and Gα16 couple a wide variety of receptors to phospholipase C. J. Biol. Chem. 1995, 270, 15175–15180. [Google Scholar] [CrossRef]
- Williams, L.T.; Snyderman, R.; Pike, M.C.; Lefkowitz, R.J. Specific receptor sites for chemotactic peptides on human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 1204–1208. [Google Scholar] [CrossRef]
- Niedel, J.; Kahane, I.; Lachman, L.; Cuatrecasas, P. A subpopulation of cultured human promyelocytic leukemia cells (HL-60) displays the formyl peptide chemotactic receptor. Proc. Natl. Acad. Sci. USA 1980, 77, 1000–1004. [Google Scholar] [CrossRef]
- Chaplinski, T.J.; Niedel, J.E. Cyclic nucleotide-induced maturation of human promyelocytic leukemia cells. J. Clin. Investig. 1982, 70, 953–964. [Google Scholar] [CrossRef]
- Krautwurst, D.; Seifert, R.; Hescheler, J.; Schultz, G. Formyl peptides and ATP stimulate Ca2+ and Na+ inward currents through non-selective cation channels via G-proteins in dibutyryl cyclic AMP-differentiated HL-60 cells. Involvement of Ca2+ and Na+ in the activation of β-glucuronidase release and superoxide production. Biochem. J. 1992, 288, 1025–1035. [Google Scholar]
- Gysin, B.; Schwyzer, R. Hydrophobic and electrostatic interactions between adrenocorticotropin-(1-24)-tetracosapeptide and lipid vesicles. amphiphilic primary structures. Biochemistry 1984, 23, 1811–1818. [Google Scholar] [CrossRef]
- Sargent, D.F.; Schwyzer, R. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc. Natl. Acad. Sci. USA 1986, 83, 5774–5778. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, H.; Edward Zhou, X.; Verma, K.R.; de Waal, P.W.; Jang, W.; Xu, T.H.; Wang, L.; Meng, X.; Zhao, G.; et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat. Commun. 2020, 11, 885. [Google Scholar] [CrossRef]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef]
- Kim, S.D.; Kim, Y.K.; Lee, H.Y.; Kim, Y.S.; Jeon, S.G.; Baek, S.H.; Song, D.K.; Ryu, S.H.; Bae, Y.S. The agonists of formyl peptide receptors prevent development of severe sepsis after microbial infection. J. Immunol. 2010, 185, 4302–4310. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Kwon, S.; Lee, S.K.; Kook, M.; Lee, H.Y.; Song, K.D.; Lee, H.K.; Baek, S.H.; Park, C.B.; Bae, Y.S. The immune-stimulating peptide WKYMVm has therapeutic effects against ulcerative colitis. Exp. Mol. Med. 2013, 45, e40. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Park, W.S.; Ahn, S.Y.; Sung, D.K.; Sung, S.I.; Kim, J.H.; Chang, Y.S. WKYMVm hexapeptide, a strong formyl peptide receptor 2 agonist, attenuates hyperoxia-induced lung injuries in newborn mice. Sci. Rep. 2019, 9, 6815. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Mukai, H.; Kawai, K.; Suzuki, Y.; Yamashita, K.; Munekata, E. Stimulation of dog gastropancreatic hormone release by neuromedin B and its analogues. Am. J. Physiol. 1987, 252, E765–E771. [Google Scholar] [CrossRef]
- Mukai, H.; Munekata, E.; Higashijima, T. G protein antagonist. A novel hydrophobic peptide competes with receptor for G protein binding. J. Biol. Chem. 1992, 267, 16237–16243. [Google Scholar] [CrossRef]
- Mukai, H.; Kikuchi, M.; Suzuki, Y.; Munekata, E. A mastoparan analog without lytic effects and its stimulatory mechanisms in mast cells. Biochem. Biophys. Res. Commun. 2007, 362, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Wakamatsu, K.; Mukai, H. Mastoparan as a G protein-activator. In Methods and Tools in Biosciences and Medicine: Animal Toxins; Birkhauser: Basel, Switzerland, 2000; pp. 116–126. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | EC50 (nM) | Maximum Effect (%) a | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |||||
| MCT-2(1–15) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | I | N | 20.0 ± 3.39 | 100 |
| MCT-2(1–14) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | I | 96.7 ± 17.6 | 102 | |
| MCT-2(1–13) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | 186 ± 33.6 | 100 ± 3 | ||
| MCT-2(1–12) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | 633 ± 153 | 98 ± 3 | |||
| MCT-2(1–11) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | 547 ± 93.9 | 103 ± 3 | ||||
| MCT-2(1–10) | formyl | - | M | T | P | M | R | K | I | N | P | L | 1800 ± 173 | 98 ± 3 | |||||
| MCT-2(1–9) | formyl | - | M | T | P | M | R | K | I | N | P | 10,760 ± 850 | 104 ± 3 | ||||||
| MCT-2(1–8) | formyl | - | M | T | P | M | R | K | I | N | 41,333 ± 2517 | 100 ± 2 | |||||||
| MCT-2(1–7) | formyl | - | M | T | P | M | R | K | I | 35,500 ± 500 | 101 ± 2 | ||||||||
| MCT-2(1–6) | formyl | - | M | T | P | M | R | K | 202,667 ± 2309 | 90 ± 2 ** | |||||||||
| MCT-2(1–5) | formyl | - | M | T | P | M | R | 26,000 ± 1581 | 104 ± 2 | ||||||||||
| MCT-2(1–4) | formyl | - | M | T | P | M | 18,000 ± 1323 | 88 ± 2 *** | |||||||||||
| Des-N-formyl MCT-2 | M | T | P | M | R | K | I | N | P | L | M | K | L | I | N | >1,000,000 | 0 | ||
| Peptide | Sequence | EC50 (nM) | Maximum Effect (%) a | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |||||
| MCT-2(1–15) | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | I | N | 20.0 ± 3.39 | 100 |
| [Ala1]MCT-2 | formyl | - | A | T | P | M | R | K | I | N | P | L | M | K | L | I | N | >10,000 | 70 ± 2 *** |
| [Ala2]MCT-2 | formyl | - | M | A | P | M | R | K | I | N | P | L | M | K | L | I | N | 0.61 ± 0.07 | 106 ± 2 |
| [Ala3]MCT-2 | formyl | - | M | T | A | M | R | K | I | N | P | L | M | K | L | I | N | 16.3 ± 0.29 | 102 ± 1 |
| [Ala4]MCT-2 | formyl | - | M | T | P | A | R | K | I | N | P | L | M | K | L | I | N | 68.0 ± 2.65 | 100 ± 2 |
| [Ala5]MCT-2 | formyl | - | M | T | P | M | A | K | I | N | P | L | M | K | L | I | N | 61.3 ± 0.58 | 104 ± 1 |
| [Ala6]MCT-2 | formyl | - | M | T | P | M | R | A | I | N | P | L | M | K | L | I | N | 64.0 ± 3.00 | 103 ± 2 |
| [Ala7]MCT-2 | formyl | - | M | T | P | M | R | K | A | N | P | L | M | K | L | I | N | 94.7 ± 3.21 | 104 ± 2 |
| [Ala8]MCT-2 | formyl | - | M | T | P | M | R | K | I | A | P | L | M | K | L | I | N | 31.0 ± 3.97 | 106 ± 1 |
| [Ala9]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | A | L | M | K | L | I | N | 3.02 ± 1.21 | 105 ± 1 |
| [Ala10]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | A | M | K | L | I | N | 73.0 ± 13.3 | 100 ± 2 |
| [Ala11]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | L | A | K | L | I | N | 36.7 ± 11.0 | 102 ± 1 |
| [Ala12]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | L | M | A | L | I | N | 12.3 ± 0.29 | 106 ± 3 |
| [Ala13]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | A | I | N | 83.7 ± 3.54 | 107 ± 3 |
| [Ala14]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | A | N | 76.7 ± 11.9 | 103 ± 3 |
| [Ala15]MCT-2 | formyl | - | M | T | P | M | R | K | I | N | P | L | M | K | L | I | A | 29.3 ± 1.49 | 104 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marutani, T.; Nishino, K.; Miyaji, T.; Kamada, K.; Ohura, K.; Kiso, Y.; Mukai, H. Mitocryptide-2: Identification of Its Minimum Structure for Specific Activation of FPR2–Possible Receptor Switching from FPR2 to FPR1 by Its Physiological C-terminal Cleavages. Int. J. Mol. Sci. 2021, 22, 4084. https://doi.org/10.3390/ijms22084084
Marutani T, Nishino K, Miyaji T, Kamada K, Ohura K, Kiso Y, Mukai H. Mitocryptide-2: Identification of Its Minimum Structure for Specific Activation of FPR2–Possible Receptor Switching from FPR2 to FPR1 by Its Physiological C-terminal Cleavages. International Journal of Molecular Sciences. 2021; 22(8):4084. https://doi.org/10.3390/ijms22084084
Chicago/Turabian StyleMarutani, Takayuki, Kodai Nishino, Tomoyuki Miyaji, Keisuke Kamada, Koji Ohura, Yoshiaki Kiso, and Hidehito Mukai. 2021. "Mitocryptide-2: Identification of Its Minimum Structure for Specific Activation of FPR2–Possible Receptor Switching from FPR2 to FPR1 by Its Physiological C-terminal Cleavages" International Journal of Molecular Sciences 22, no. 8: 4084. https://doi.org/10.3390/ijms22084084
APA StyleMarutani, T., Nishino, K., Miyaji, T., Kamada, K., Ohura, K., Kiso, Y., & Mukai, H. (2021). Mitocryptide-2: Identification of Its Minimum Structure for Specific Activation of FPR2–Possible Receptor Switching from FPR2 to FPR1 by Its Physiological C-terminal Cleavages. International Journal of Molecular Sciences, 22(8), 4084. https://doi.org/10.3390/ijms22084084