
Impact of Sodium–Glucose Co-Transporter 2 Inhibitors on Cardiac Protection


Abstract
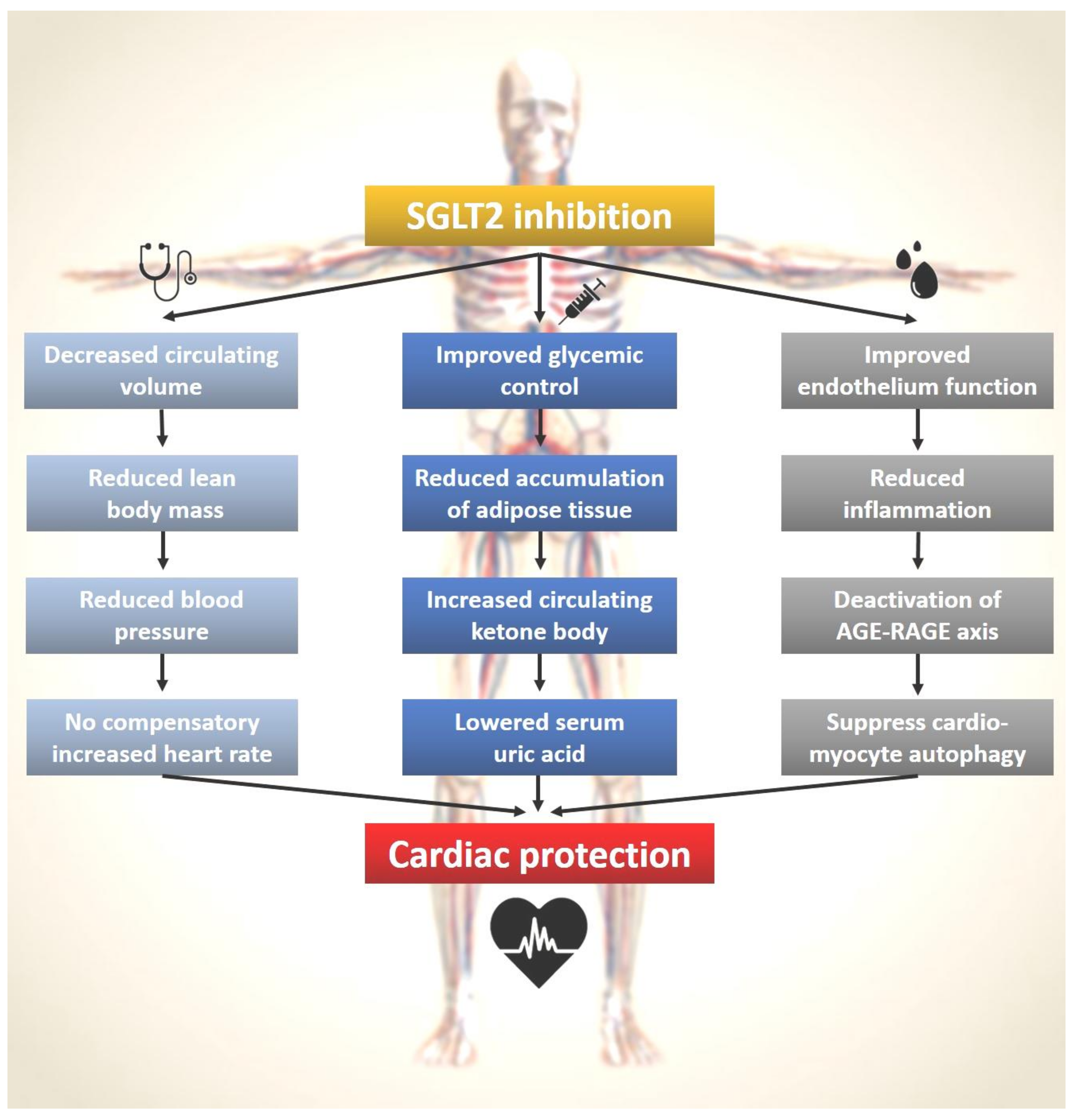
:1. Introduction
2. Clinical Trials
3. Physiological Perspectives on Cardiac Protections
3.1. Glycemia
3.2. Circulating Volume
3.3. Body Mass
3.4. Blood Pressure
3.5. Heart Rate
3.6. Adipose Tissue
3.7. Ketone Bodies
3.8. Uric Acid
3.9. Endothelial and Vascular Functions
3.10. Inflammation
3.11. AGE-Mediated Effects
3.12. Autophagy
3.13. Crosstalk between Autophagy and Innate Immunity
4. Adverse Effects of SGLT2 Inhibitors
5. Summary
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawshani, A.; Rawshani, A.; Franzén, S.; Sattar, N.; Eliasson, B.; Svensson, A.M.; Zethelius, B.; Miftaraj, M.; McGuire, D.K.; Rosengren, A.; et al. Risk factors, mortality, and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2018, 379, 633–644. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Zelniker, T.A.; Braunwald, E. Clinical Benefit of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; MattheusDevins, T.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z.I. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Barsotti, E.; Clerico, A.; Muscelli, E. Renal handling of ketones in response to sodium glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care 2017, 40, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Ridderstråle, M.; Andersen, K.R.; Zeller, C.; Kim, G.; Woerle, H.J.; Broedl, U.C.; EMPA-REG H2H-SU trial investigators. Comparison of empagliflozin and glimperidie as add-on to metformin in patients with type 2 diabetes: A 104 week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014, 2, 691–700. [Google Scholar] [CrossRef]
- Del Prato, S.; Nauck, M.; Durán-Garcia, S.; Maffei, L.; Rohwedder, K.; Theuerkauf, A.; Parikh, S. Long-term glycaemic response and tolerability of dapagliflozin versus a sulphonylurea as add-on therapy to metformin in patients with type 2 diabetes: 4-year data. Diabetes Obes. Metab. 2015, 17, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Van Baar, M.J.B.; van Ruiten, C.C.; Muskiet, M.H.A.; Bloemendaal, L.; Ijzerman, R.G.; van Raalte, D.H. SGLT2 Inhibitors in Combination Therapy: From Mechanisms to Clinical Considerations in Type 2 Diabetes Management. Diabetes Care 2018, 41, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, C.C.; Gansevoort, R.T. Sodium-glucose cotransporter 2 inhibitors: Extending the indication to non-diabetic kidney disease? Nephrol. Dial. Transplant. 2020, 35 (Suppl. S1), i33–i42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambers Heersprink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [Green Version]
- Berl, T.; Quinttnat-Pelletier, F.; Verbails, J.; Schrier, R.W.; Bichet Dg Ouyang, J.; Czerwiec, F.S.; Saltwater Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J. Am. Soc. Nephrol. 2010, 21, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaux, G. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Semin. Nephrol. 2009, 29, 239–256. [Google Scholar] [CrossRef]
- Ohara, K.; Masuda, T.; Morinari, M.; Okada, M.; Miki, A.; Nakagawa, S.; Murakami, T.; Oka, K.; Asakura, M.; Miyazawa, Y.; et al. The extracellular volume status predicts body fluid response to SGLT2 inhibitor dapagliflozin in diabetic kidney disease. Diabetol. Metab. Syndr. 2020, 12, 37. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Norton, L.; DeFronzo, R.A. Efficacy and safety of SGLT2 inhibitors in the treatment of type 2 diabetes mellitus. Curr. Diab. Rep. 2012, 12, 230–238. [Google Scholar] [CrossRef]
- Nishimura, R.; Osonoi, T.; Kanada, S.; Jinnouchi, H.; Sugio, K.; Omiya, H.; Ubukata, M.; Sakai, S.; Samukawa, Y. Effects of luseogliflozin, a sodium-glucose co-transporter 2 inhibitor, on 24-h glucose variability assessed by continuous glucose monitoring in Japanese patients with type 2 diabetes mellitus: A randomized, double-blind, placebo-controlled, crossover study. Diabetes Obes. Metab. 2015, 17, 800–804. [Google Scholar]
- Kohan, D.E.; Fioretto, P.; Tang, W.; List, J.F. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014, 85, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology 2016, 157, 1029–1042. [Google Scholar] [CrossRef]
- Storgaard, H.; Gluud, L.L.; Bennett, C.; Grøndahl, M.F.; Christensen, M.B.; Knop, F.K.; Vilsbøll, T. Benefits and harms of sodium-glucose co-transporter 2 inhibitors in patients with type 2 diabetes: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0166125. [Google Scholar] [CrossRef]
- Henry, R.R.; Murray, A.V.; Marmolejo, M.H.; Hennicken, D.; Ptaszynska, A.; List, J.F. Dapagliflozin, metformin XR, or both: Initial pharmacotherapy for type 2 diabetes, a randomised controlled trial. Int. J. Clin. Pract. 2012, 66, 446–456. [Google Scholar] [CrossRef]
- Yoshida, A.; Kusakabe, H.; Takamura, T.; Kaku, K.; Suganami, H. The SGLT2 Inhibitor Tofogliflozin Reduces Weight in People with Type 2 Diabetes due to Fluid Loss Initially and to Lipolysis in Late. Diabetes 2018, 67 (Suppl. S1). [Google Scholar] [CrossRef]
- Cefalu, W.T.; Leiter, L.A.; Yoon, K.H.; Arias, P.; Niskanen, L.; Xie, J.; Balis, D.A.; Canovatchel, W.; Meininger, G. Efficacy and safety of canagliflozin versus glimperide in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 2013, 382, 941–950. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R.V.; Bakris, G.L. Blood pressure effects of sodium-glucose co-transport 2 (SGLT2) inhibitors. J. Am. Soc. Hypertens. 2014, 8, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Pharmacodynamics, efficacy and safety of sodium-glucose co-transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs 2015, 75, 33–59. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Kawasoe, S.; Maruguchi, Y.; Kajiya, S.; Uenomachi, H.; Miyata, M.; Kawasoe, M.; Kubozono, T.; Ohishi, M. Mechanism of the blood pressure-lowering effect of sodium-glucose cotransporter 2 inhibitors in obese patients with type 2 diabetes. BMC Pharm. Toxicol. 2017, 18, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, K.D.; Cherney, D. Renal angiotensinogen and sodium-glucose cotransporter-2 inhibition: Insights from experimental diabetic kidney disease. Am. J. Nephrol. 2019, 49, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia. 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M. Hemodynamic effects of sodium-glucose cotransporter 2 inhibitors. J. Clin. Med. Res. 2017, 9, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Matsubayashi, Y.; Nojima, T.; Yoshida, A.; Suganami, H.; Yamada, T.; Fujihara, K.; Tanaka, S.; Kaku, K.; Sone, H. Influence of SGLT2 Inhibitor on Resting Heart Rate (RHR) and Factors Related to Its Changes. Diabetes 2018, 67 (Suppl. S1). [Google Scholar] [CrossRef]
- Wan, N.; Rahman, A.; Hitomi, H.; Nishiyama, A. The Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Sympathetic Nervous Activity. Front. Endocrinol. 2018, 9, 421. [Google Scholar] [CrossRef] [Green Version]
- Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ. Tech. Rep. Ser. 2000, 894, 1–253.
- Pan, H.; Guo, J.; Su, Z. Advances in understanding the interrelations between leptin resistance and obesity. Physiol. Behav. 2014, 130, 157–169. [Google Scholar] [CrossRef]
- Packer, M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis. Diabetes Obes. Metab. 2018, 20, 1361–1366. [Google Scholar] [CrossRef]
- Ansaldo, A.M.; Montecucco, F.; Sahebkar, A.; Dallegri, F.; Carbone, F. Epicardial adipose tissue and cardiovascular diseases. Int. J. Cardiol. 2019, 278, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Gra-Menendez, S. Effects of Dapagliflozin on Epicardial Fat Thickness in Patients with Type 2 Diabetes and Obesity. Obesity 2020, 28, 1068–1074. [Google Scholar] [CrossRef]
- Prattichizzo, F.; de Nigris, V.; Micheloni, S.; Sala, L.L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Staels, B. Cardiovascular Protection by Sodium Glucose Cotransporter 2 Inhibitors: Potential Mechanisms. Am. J. Med. 2017, 130, S30–S39. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. Cardiovascular protection in the EMPA-REG OUTCOME trial: A ‘thrifty substrate’ hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes: Novel Translational Insights into the Heart Failure Benefits of SGLT2 Inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Perkins, B.A.; Cherney, D.Z.I. Uric acid as a biomarker and a therapeutic target in diabetes. Can. J. Diabetes 2015, 39, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; MazzaliIs, M. There a Pathogenetic Role for Uric Acid in Hypertension and Cardiovascular and Renal Disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Trujillo, A.; Vijapurkar, U.; Damaraju, C.V.; Meininger, G. Effect of canagliflozin on serum uric acid in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2015, 17, 426–429. [Google Scholar] [CrossRef] [Green Version]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamaia, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Doblado, M.; Moley, K.H. Facilitative glucose transporter 9, a unique hexose and urate transporter. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E831–E835. [Google Scholar] [CrossRef] [Green Version]
- Mcgill, J.B. The SGLT2 inhibitor empagliflozin for the treatment of type 2 diabetes mellitus: A bench to bedside review. J. Diab. Ther. 2014, 5, 43–63. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, S.; Jinnouchi, H.; Kurinami, N.; Hieshima, K.; Yoshida, A.; Jinnouchi, K.; Nishimura, H.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; et al. The SGLT2 Inhibitor Dapagliflozin Significantly Improves the Peripheral Microvascular Endothelial Function in Patients with Uncontrolled Type 2 Diabetes Mellitus. Intern. Med. 2018, 57, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of Empagliflozin on Endothelial Function in Patients with Type 2 Diabetes and Cardiovascular Disease: Results from the Multicenter, Randomized, Placebo-Controlled, Double-Blind EMBLEM Trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef] [Green Version]
- Striepe, K.; Jumar, A.; Ott, C.; Karg, M.V.; Schneider, M.P.; Kannenkeril, D.; Schmieder, R.E. Effects of the selective sodium-glucose cotransporter 2 inhibitor empagliflozin on vascular function and central hemodynamics in patients with type 2 diabetes mellitus. Circulation 2017, 136, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.M. Diabetes and Cardiovascular Disease: Emerging Therapeutic Approaches. Arter. Thromb. Vasc. Biol. 2019, 39, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim KMChoi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; Kim, Y.B.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE–/– mice fed a Western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Kolhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Park, S.; Lakatta, E.G. RAGE signaling in inflammation and arterial aging. Front. Biosci. 2009, 14, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Kosmopoulos, M.; Drekolias, D.; Zavras, P.D.; Piperi, C.; Papavassiliou, A.G. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinßius, E.; Agdauletova, S.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics. 2021, 11, 4502–4515. [Google Scholar] [CrossRef]
- Rahadian, A.; Fukuda, D.; Salim, H.M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Sata, M. Canagliflozin prevents diabetes-induced vascular dysfunction in ApoE-deficient mice. J. Atheroscler. Thromb. 2020, 27, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular Mechanisms of Autophagy in the Cardiovascular System. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Xu, Y.; Wang, D.; Chen, F.; Tu, Z.; Qian, J.; Xu Sh Xu, Y.; Hwa, J.; Li, J.; Shang, H.; et al. Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell 2021, 1–24. [Google Scholar] [CrossRef]
- Clancy, C.E.; Chen-Izu, Y.; Bers, D.M.; Belardinelli, L.; Boyden, P.A.; Csernoch, L.; Despa, S.; Fermini, B.; Hool, L.C.; Izu, L.; et al. Deranged sodium to sudden death. J. Physiol. 2015, 593, 1331–1345. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Chen, C.C.; Lin, M.H.; Su, H.T.; Ho, M.Y.; Yeh, J.K.; Tsai, M.L.; Hsieh, I.C.; Wen, M.S. TLR9 Binding to Beclin 1 and Mitochondrial SIRT3 by a Sodium-Glucose Co-Transporter 2 Inhibitor Protects the Heart from Doxorubicin Toxicity. Biology 2020, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, P.T.; Wang, X.; Zhao, Y.; Meacham, C.E.; Zou, Z.; Bordieanu, B.; Johanns, M.; Vertommen, D.; Wijshake, T.; et al. TLR9 and Beclin 1 Crosstalk Regulates Muscle AMPK Activation in Exercise. Nature 2020, 578, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Rabin-Court, A.; Song, J.D.; Cardone, R.L.; Wang, Y.; Kibbey, R.G.; Shulman, G.I. Dehydration and insulinopenia are necessary and sufficient for euglycemic ketoacidosis in SGLT2 inhibitor-treated rats. Nat. Commun. 2019, 10, 548. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.; Mahmoud, A.N.; Elgendy, I.Y.; Abuzaid, A.; Barakat, A.F.; Elgendy, A.Y.; Al-Ani, M.; Mentias, A.; Nairooz, R.; Bavry, A.A.; et al. Cardiovascular outcomes with sodium–glucose cotransporter-2 inhibitors in patients with type II diabetes mellitus: A meta-analysis of placebo-controlled randomized trials. Int. J. Cardiol. 2017, 227, 352–358. [Google Scholar] [CrossRef]
- Chen, T.H.; Li, Y.R.; Chen, S.W.; Lin, Y.S.; Sun, C.C.; Chen, D.Y.; Mao, C.T.; Wu, M.; Chang, C.H.; Chu, P.H.; et al. Sodium-glucose cotransporter 2 inhibitors versus metformin as first-line therapy in patients with type 2 diabetes mellitus: A multi-institution database study. Cardiovasc. Diabetol. 2020, 19, 189. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| General Population | Special Population * | |||||
|---|---|---|---|---|---|---|
| Clinical Trials | EMPA-REG OUTCOME [5] | CANVAS Program [6] | DECLAR-TIMI 58 [7] | CREDENCE [8] (patients with CKD) * | DAPA-HF [9] (patients with HF) * | EMPEROR-Reduced [10] (patients with HF) * |
| Intervention | Empagliflozin 10 or 25 mg qd vs. placebo | Canagliflozin 100 or 300 mg qd vs. placebo | Dapagliflozin 10 mg qd vs. placebo | Canagliflozin 100 mg qd vs. placebo | Dapagliflozin 10 mg qd vs. placebo | Empagliflozin 10 mg qd vs. placebo |
| Population | 7020 T2DM patients with 99.2% established CVD | 10,142 T2DM patients with 65.6% established CVD and 34.4% CV risk factors | 17,160 T2DM patients with 40.6% established CVD and 59.4% CV risk factors | 4401 T2DM patients with 50.4% established CVD and 49.6% CV risk factors | 4744 patients with 42% T2DM and 58% without T2DM | 3730 patients with 50% T2DM and 50% without T2DM |
| Follow-up | 3.1 years | 3.6 years | 4.2 years | 2.6 years | 1.5 years | 1.3 years |
| Baseline A1c | 7–10% | 7–10.5% | 6.5–12.0% | 6.5–12.0% | No restriction | No restriction |
| eGFR | ≥30 | ≥30 | ≥60 | 30–89 | ≥30 | ≥20 |
| Primary outcomes | 3P MACE, HR 0.86, 95% CI 0.74–0.99 | 3P MACE, HR 0.86, 95% CI 0.75–0.97 | 3P MACE, HR 0.93 95% CI 0.84–1.03; CV death or HHF, HR 0.83, 95% CI 0.73–0.95 | New ESRD or 2x creatinine level or renal or CV death, HR 0.66, 95% CI 0.53–0.81 | HHF, CV death, urgent hospital visit, and IV therapy for HF, HR 0.74, 95% CI 0.65–0.85 | Adjudicated CV death or HHF, HR 0.75, 95% CI 0.65–0.86 |
| CV death | HR 0.62, 95% CI 0.49–0.77 | HR 0.87, 95% CI 0.72–1.06 | HR 0.98, 95% CI 0.82–1.17 | HR 0.78, 95% CI 0.61–1.00 | HR 0.82, 95% CI 0.69–0.98 | HR 0.92, 95% CI 0.75–1.12 |
| All-cause mortality | HR 0.68, 95% CI 0.57–0.82 | HR 0.87, 95% CI 0.74–1.01 | HR 0.93, 95% CI 0.82–1.04 | HR 0.83, 95% CI 0.68–1.02 | HR 0.83, 95% CI 0.71–0.97 | HR 0.92, 95% CI 0.77–1.10 |
| MI | HR 1.18, 95% CI 0.70–1.09 | HR 1.18, 95% CI 0.73–1.09 | HR 1.18, 95% CI 0.77–1.01 | |||
| HHF | HR 0.65, 95% CI 0.50–0.85 | HR 0.65, 95% CI 0.52–0.87 | HR 0.65, 95% CI 0.61–0.88 | HR 0.61, 95% CI 0.47–0.80 | HR 0.70, 95% 0.59–0.83 | HR 0.69, 95% CI 0.59–0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, V.C.-C.; Li, Y.-R.; Wang, C.-Y. Impact of Sodium–Glucose Co-Transporter 2 Inhibitors on Cardiac Protection. Int. J. Mol. Sci. 2021, 22, 7170. https://doi.org/10.3390/ijms22137170
Wu VC-C, Li Y-R, Wang C-Y. Impact of Sodium–Glucose Co-Transporter 2 Inhibitors on Cardiac Protection. International Journal of Molecular Sciences. 2021; 22(13):7170. https://doi.org/10.3390/ijms22137170
Chicago/Turabian StyleWu, Victor Chien-Chia, Yan-Rong Li, and Chao-Yung Wang. 2021. "Impact of Sodium–Glucose Co-Transporter 2 Inhibitors on Cardiac Protection" International Journal of Molecular Sciences 22, no. 13: 7170. https://doi.org/10.3390/ijms22137170
APA StyleWu, V. C.-C., Li, Y.-R., & Wang, C.-Y. (2021). Impact of Sodium–Glucose Co-Transporter 2 Inhibitors on Cardiac Protection. International Journal of Molecular Sciences, 22(13), 7170. https://doi.org/10.3390/ijms22137170