
Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation


Abstract
1. Introduction
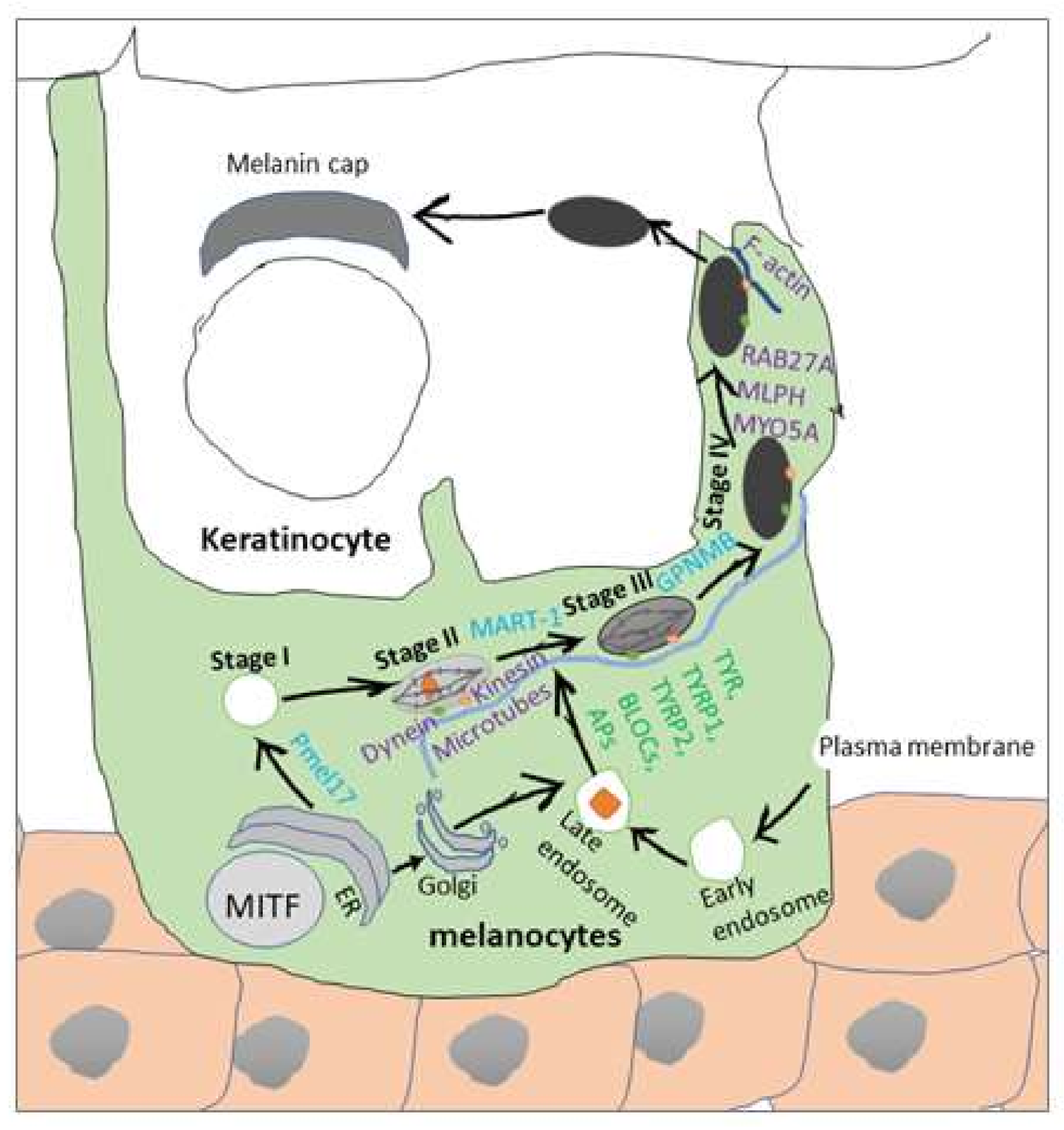
2. Melanogenesis Process
3. Melanogenesis Regulation
Involvement of Signaling Pathways in Melanogenesis Regulation
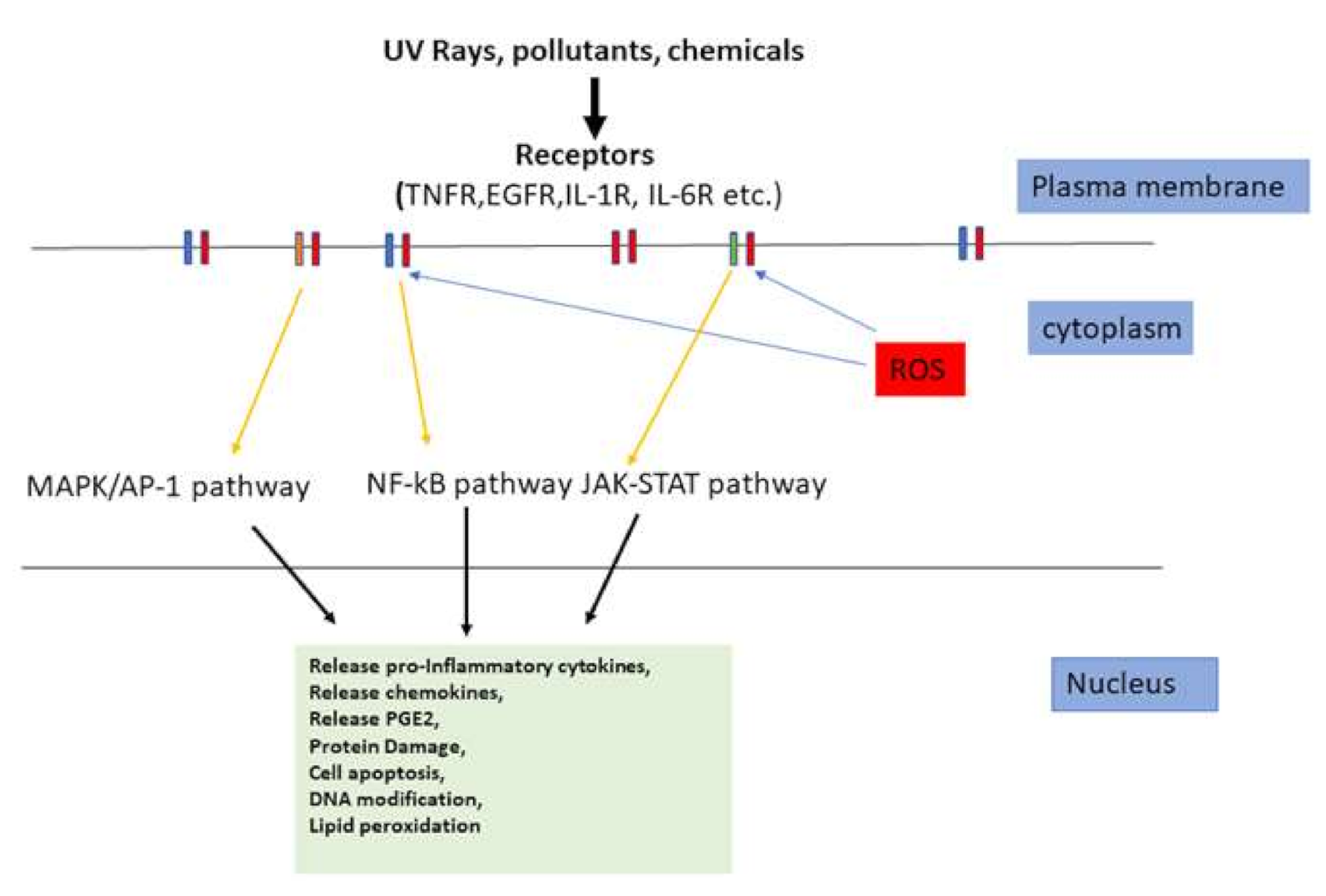
4. Skin Inflammation
5. Inflammatory Cytokines
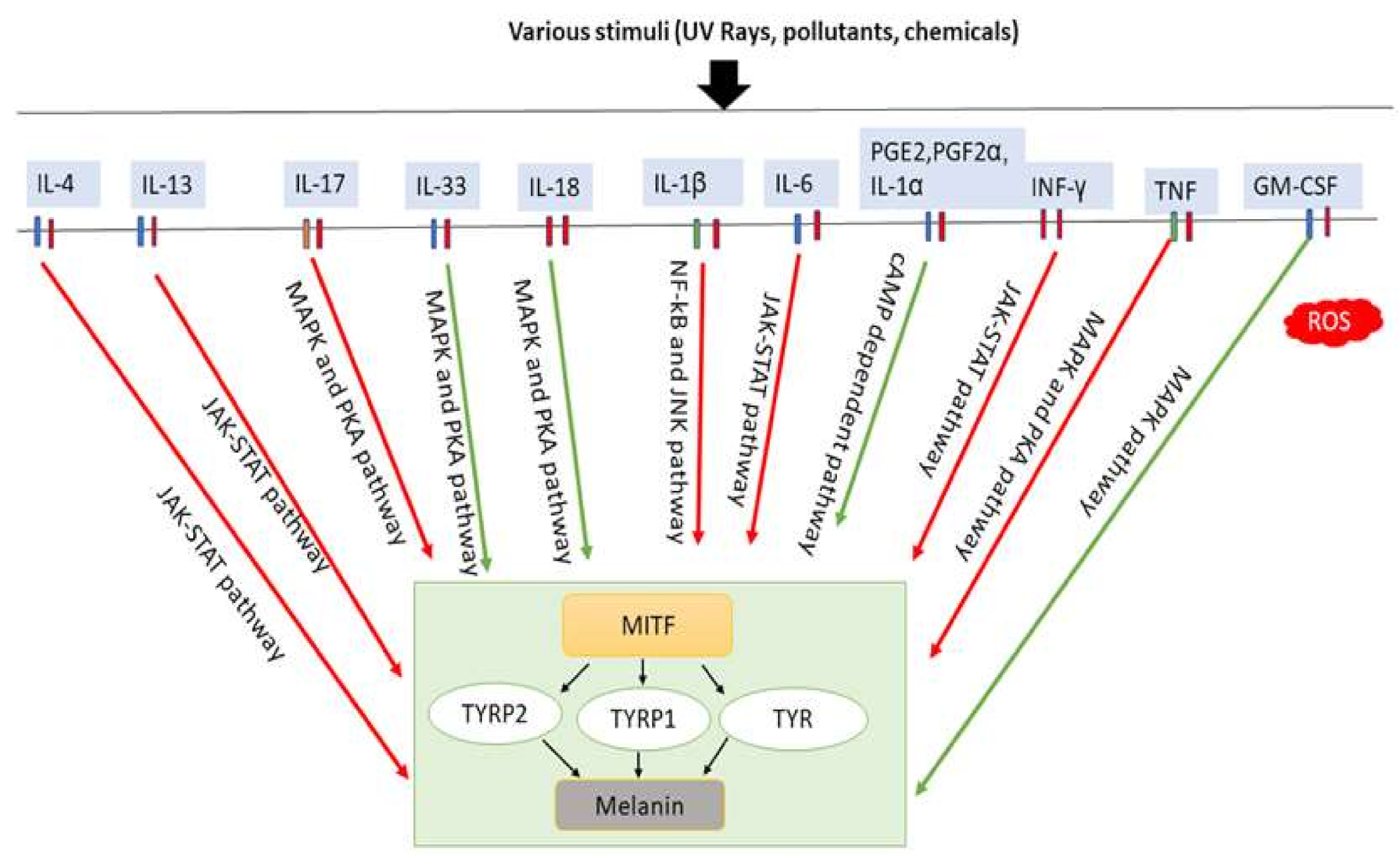
6. Inflammatory Cytokines Induce Melanogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokines | Main Source | Cells Used for Experiment | Impact on Melanogenesis | Mechanisms of Melanogenesis | Refs. |
|---|---|---|---|---|---|
| IL-4 | Th-cells | Melanocytes | Inhibition | JAK2-STAT6 signaling pathway is used for downregulating the expression of MITF, TYRP1, TYRP2 | [16] |
| IL-13 | Th2 | γδ T-cell | Inhibition | Suppress tyrosinase and DCT through the JAK2–STAT6 | [107] |
| IL-17 | Epithelial cells, Endothelial cells, Fibroblasts | Melanocytes, Primary pooled human keratinocytes | Inhibition | TNF and IL-17 simultaneously inhibit melanin formation through PKA and MAPK signaling pathways | [93] |
| IL-33 | Keratinocytes, Fibroblasts | Keratinocytes | Promotion | Activate p38 and PKA pathways and thus promote MITF, TYR, TYRP1, TYRP2 expression | [111,112] |
| IL-18 | Monocytes/Macrophages, Keratinocytes | Melanocytes | Promotion | Increase the activity of tyrosinase and upregulate the expression Of TYRP1 and TYRP2 | [113] |
| IL-1α | Langerhans cells, Melanocytes, keratinocytes | Primary melanocytes and swine skin | Promotion | Melanin deposition is increased when IL-1α is combined with KGF | [114] |
| IL-1β | Monocytes/Macrophages, Keratinocytes | Melanoma cell lines (LB2259-MEL and CP50-MEL) | Inhibition | Through the NF-κB and JNK pathways, it downregulates MITF-M expression | [115] |
| IL-6 | Macrophages, T-cells, Adipocyte | Melanocytes | Inhibition | Tyrosinase activity is declined | [15] |
| PGE2 and PGF2α | Fibroblasts, Keratinocytes | Keratinocytes | Promotion | Activate cAMP-dependent pathway and stimulate melanocyte dendrite formation | [116] |
| INF-γ | T-cells, NK cells, NKT cells | B16F10 | Inhibition | Block melanosomal maturation and upregulate STAT1 phosphorylation | [117,118] |
| TNF | Monocytes, Macrophages, Keratinocytes, Dendritic cells, Th1, Th17 and Th22 | Melanocytes, Primary pooled human keratinocytes | Promotion | Stimulate PKA and MAPK signaling pathways with combination of IL17, and thus inhibit melanin formation | [93] |
| GM-CSF | Macrophages, Keratinocytes and Th cells | Melanocytes | Promotion | Stimulate MAPK pathway and promote melanocyte proliferation, melanin and synthesis | [119] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| PMEL17 | Melanocytes lineage-specific antigen gp100 |
| MART-1 | Melanoma antigen recognized by T cell-1 |
| GPNMB | Glycoprotein nonmetastatic melanoma protein b |
| TYR | Tyrosinase |
| TYRP1 | Tyrosinase-related protein-1 |
| TYRP2 | Tyrosinase-related protein-2 |
| BLOCs | Biogenesis of lysosome-related organelles complexes |
| AP-1 | Activator protein 1 |
| MLPH | Melanophilin |
| MYO5A | Myosin-Va |
| RAB27A | Ras-related protein Rab27A |
| MITF | Microphthalmia-associated transcription factor |
| DAMPs | Damage-associated molecular patterns |
| PAMPs | Pathogen-associated molecular patterns |
| PRRs | Pattern-recognition receptors |
| TLRs | Toll-like receptors; |
| NLRs | NOD-like receptors |
| RLRs | Retinoic acid-inducible gene-I-like receptors |
| MyD88 | Myeloid differentiation primary response protein-88 |
| TAM | Tyro3, Axl, and Mer receptors |
| NF-κβ | Nuclear factor kappa-B |
| IRF | Interferon regulatory factor |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| IKK | IκB kinase |
| JAK-STAT | Janus kinase-signal transducer and activator of transcription |
| MAPKs | Mitogen-activated protein kinases |
| MKK | MAPK kinase |
| MKKK | MAPK kinase kinase |
| Erk1/2 | Etxracellular signal-regulated kinase ½ |
| JNK | c-Jun N-terminal kinases |
| UVR | Ultraviolet radiation |
| TNFR | Tumor necrosis factor receptor |
| EGFR | Epidermal growth factor receptor |
| PGE2 | Prostaglandin E2 |
| PGF2α | Prostaglandin F2α |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| IFN-γ | Interferon-γ |
| PKA | Protein kinase A |
| NK cell | Natural killer cell |
| NKT cell | Natural killer T cell |
| Th | T helper cell |
| cAMP | Cyclic adenosine monophosphate |
| PAX3 | Paired box protein Pax-3 |
| SOX10 | Sex-determining region Y-box 10 |
| KIT | Stem cell growth factor receptor (CD117) |
| SCF | Stem cell growth factor |
| EDN | Endothelin |
| NGF | Nerve growth factor |
| HGF | Hepatocyte growth factor |
| α-MSH | α-Melanocyte-stimulating hormone |
| ACTH | Adrenocorticotropic hormone |
| MLPH | melanophilin |
| CREB | cAMP responsive-element binding protein |
| SIK | Salt-inducible kinase |
| CRTC | CREB-regulated transcription coactivator |
| MITF-M | Melanocytes-specific MITF isoform |
References
- Chuong, C.M.; Nickoloff, B.J.; Elias, P.M.; Goldsmith, L.A.; Macher, E.; Maderson, P.A.; Sundberg, J.P.; Tagami, H.; Plonka, P.M.; Thestrup-Pederson, K.; et al. What is the ‘true’ function of skin? Exp. Dermatol. 2002, 11, 159–187. [Google Scholar] [CrossRef]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef]
- Bonaventure, J.; Domingues, M.J.; Larue, L. Cellular and molecular mechanisms controlling the migration of melanocytes and melanoma cells. Pigment. Cell Melanoma Res. 2013, 26, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Jan Borovansky, P.A.R. Melanins and Melanosomes Biosynthesis, Biogenesis, Physiological, and Pathological Functions; Jan Borovansky, P.A.R., Ed.; Wiley-Blackwell: Weinheim, Germany, 2011; p. 429. [Google Scholar]
- Lei, T.C.; Virador, V.; Yasumoto, K.; Vieira, W.D.; Toyofuku, K.; Hearing, V.J. Stimulation of melanoblast pigmentation by 8-methoxypsoralen:the involvement of microphthalmia-associated transcription factor, the protein kinase a signal pathway, and proteasome-mediated degradation. J. Investig. Dermatol. 2002, 119, 1341–1349. [Google Scholar] [CrossRef]
- Sviderskaya, E.V.; Hill, S.P.; Balachandar, D.; Barsh, G.S.; Bennett, D.C. Agouti signaling protein and other factors modulating differentiation and proliferation of immortal melanoblasts. Dev. Dyn. 2001, 221, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Tolleson, W.H. Human melanocyte biology, toxicology, and pathology. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2005, 23, 105–161. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Martin, S.F. Contact dermatitis: From pathomechanisms to immunotoxicology. Exp. Dermatol. 2012, 21, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 2011, 11, 505–518. [Google Scholar] [CrossRef]
- Behrends, U.; Peter, R.U.; Hintermeier-Knabe, R.; Eissner, G.; Holler, E.; Bornkamm, G.W.; Caughman, S.W.; Degitz, K. Ionizing radiation induces human intercellular adhesion molecule-1 in vitro. J. Investig. Dermatol. 1994, 103, 726–730. [Google Scholar] [CrossRef]
- Fuchs, J.; Kern, H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: A clinical study using solar simulated radiation. Free Radic. Biol. Med. 1998, 25, 1006–1012. [Google Scholar] [CrossRef]
- Bäsler, K.; Brandner, J.M. Tight junctions in skin inflammation. Pflugers Arch. 2017, 469, 3–14. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.; Kassem, L.M.; Nordlund, J.J. Interleukins 1 alpha and 6 and tumor necrosis factor-alpha are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J. Investig. Dermatol. 1991, 96, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Choi, H.; Han, J.; Jin, S.H.; Park, J.Y.; Shin, D.W.; Lee, T.R.; Kim, K.; Lee, A.Y.; Noh, M. IL-4 inhibits the melanogenesis of normal human melanocytes through the JAK2-STAT6 signaling pathway. J. Investig. Dermatol. 2013, 133, 528–536. [Google Scholar] [CrossRef]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell Mol. Life Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Fistarol, S.K.; Itin, P.H. Disorders of pigmentation. J. Dtsch. Dermatol. Ges. 2010, 8, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Tsatmali, M.; Ancans, J.; Thody, A.J. Melanocyte function and its control by melanocortin peptides. J. Histochem. Cytochem. 2002, 50, 125–133. [Google Scholar] [CrossRef]
- Costin, G.E.; Hearing, V.J. Human skin pigmentation: Melanocytes modulate skin color in response to stress. FASEB J. 2007, 21, 976–994. [Google Scholar] [CrossRef]
- Dessinioti, C.; Stratigos, A.J.; Rigopoulos, D.; Katsambas, A.D. A review of genetic disorders of hypopigmentation: Lessons learned from the biology of melanocytes. Exp. Dermatol. 2009, 18, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G. Melanocyte Activation Mechanisms and Rational Therapeutic Treatments of Solar Lentigos. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G.; Ishida, K. Inhibitors of intracellular signaling pathways that lead to stimulated epidermal pigmentation: Perspective of anti-pigmenting agents. Int. J. Mol. Sci. 2014, 15, 8293–8315. [Google Scholar] [CrossRef]
- Seiberg, M. Keratinocyte-melanocyte interactions during melanosome transfer. Pigment. Cell Res. 2001, 14, 236–242. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis--pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Yaar, M.; Byers, H.R.; Goukassian, D.; Fine, R.E.; Gonsalves, J.; Gilchrest, B.A. Kinesin participates in melanosomal movement along melanocyte dendrites. J. Investig. Dermatol. 2000, 114, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Byers, H.R.; Yaar, M.; Eller, M.S.; Jalbert, N.L.; Gilchrest, B.A. Role of cytoplasmic dynein in melanosome transport in human melanocytes. J. Investig. Dermatol. 2000, 114, 990–997. [Google Scholar] [CrossRef]
- Schiaffino, M.V. Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 2010, 42, 1094–1104. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199. [Google Scholar] [CrossRef]
- Hoashi, T.; Watabe, H.; Muller, J.; Yamaguchi, Y.; Vieira, W.D.; Hearing, V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005, 280, 14006–14016. [Google Scholar] [CrossRef]
- Hoashi, T.; Sato, S.; Yamaguchi, Y.; Passeron, T.; Tamaki, K.; Hearing, V.J. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J. 2010, 24, 1616–1629. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, W.; Zhu, C.; Yuan, X.; Li, D.; Gu, W.; Ma, H.; Xie, X.; Gao, T. Silencing of GPNMB by siRNA inhibits the formation of melanosomes in melanocytes in a MITF-independent fashion. PLoS ONE 2012, 7, e42955. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis--controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404. [Google Scholar] [CrossRef]
- Park, H.Y.; Perez, J.M.; Laursen, R.; Hara, M.; Gilchrest, B.A. Protein kinase C-beta activates tyrosinase by phosphorylating serine residues in its cytoplasmic domain. J. Biol. Chem. 1999, 274, 16470–16478. [Google Scholar] [CrossRef]
- Kondo, T.; Hearing, V.J. Update on the regulation of mammalian melanocyte function and skin pigmentation. Expert Rev. Dermatol. 2011, 6, 97–108. [Google Scholar] [CrossRef]
- Kuroda, T.S.; Ariga, H.; Fukuda, M. The actin-binding domain of Slac2-a/melanophilin is required for melanosome distribution in melanocytes. Mol. Cell Biol. 2003, 23, 5245–5255. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, K.; Naeyaert, J.M.; Lambert, J. The quest for the mechanism of melanin transfer. Traffic 2006, 7, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Fisher, D.E. Lighting a path to pigmentation: Mechanisms of MITF induction by UV. Pigment. Cell Melanoma Res. 2010, 23, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y. Biology of melanocytes. In Fitzpatrick’s Dermatology in General Medicine; McGraw-Hill: New York, NY, USA, 2008; pp. 591–608. [Google Scholar]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Mujahid, N.; Liang, Y.; Murakami, R.; Choi, H.G.; Dobry, A.S.; Wang, J.; Suita, Y.; Weng, Q.Y.; Allouche, J.; Kemeny, L.V.; et al. A UV-Independent Topical Small-Molecule Approach for Melanin Production in Human Skin. Cell Rep. 2017, 19, 2177–2184. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Brenner, M.; Hearing, V.J. The regulation of skin pigmentation. J. Biol. Chem. 2007, 282, 27557–27561. [Google Scholar] [CrossRef]
- Bennett, D.C.; Lamoreux, M.L. The color loci of mice—a genetic century. Pigment. Cell Res. 2003, 16, 333–344. [Google Scholar] [CrossRef]
- Yuan, X.H.; Jin, Z.H. Paracrine regulation of melanogenesis. Br. J. Dermatol. 2018, 178, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A.; Pittelkow, M.R.; Schallreuter, K.U. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J. Investig. Dermatol. 2006, 126, 1948–1965. [Google Scholar] [CrossRef]
- Besmer, P.; Murphy, J.E.; George, P.C.; Qiu, F.H.; Bergold, P.J.; Lederman, L.; Snyder, H.W., Jr.; Brodeur, D.; Zuckerman, E.E.; Hardy, W.D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 1986, 320, 415–421. [Google Scholar] [CrossRef]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. Embo J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396, 370–373. [Google Scholar] [CrossRef]
- Takeda, K.; Yasumoto, K.; Takada, R.; Takada, S.; Watanabe, K.; Udono, T.; Saito, H.; Takahashi, K.; Shibahara, S. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 2000, 275, 14013–14016. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.J.; Erickson, C.A.; Takada, S.; Burrus, L.W. Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev. Biol. 2001, 233, 22–37. [Google Scholar] [CrossRef]
- Dunn, K.J.; Brady, M.; Ochsenbauer-Jambor, C.; Snyder, S.; Incao, A.; Pavan, W.J. WNT1 and WNT3a promote expansion of melanocytes through distinct modes of action. Pigment. Cell Res. 2005, 18, 167–180. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000, 14, 158–162. [Google Scholar] [PubMed]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef]
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 2002, 158, 1079–1087. [Google Scholar] [CrossRef]
- Benson, H.A.E.; Grice, J.E.; Mohammed, Y.; Namjoshi, S.; Roberts, M.S. Topical and Transdermal Drug Delivery: From Simple Potions to Smart Technologies. Curr. Drug Deliv. 2019, 16, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R. Skin antioxidants: Their role in aging and in oxidative stress—New approaches for their evaluation. Biomed Pharm. 1999, 53, 181–192. [Google Scholar] [CrossRef]
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. p53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 5), S322–S328. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Cavaillon, J.M. Stress molecules in sepsis and systemic inflammatory response syndrome. FEBS Lett. 2007, 581, 3723–3733. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Hendrayani, S.F.; Al-Harbi, B.; Al-Ansari, M.M.; Silva, G.; Aboussekhra, A. The inflammatory/cancer-related IL-6/STAT3/NF-κB positive feedback loop includes AUF1 and maintains the active state of breast myofibroblasts. Oncotarget 2016, 7, 41974–41985. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Henríquez-Olguín, C.; Altamirano, F.; Valladares, D.; López, J.R.; Allen, P.D.; Jaimovich, E. Altered ROS production, NF-κB activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. Biochim. Biophys. Acta 2015, 1852, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.L.; Labasi, J.M.; Zhu, Y.; Tang, X.; McClure, K.; Gabel, C.A.; Athar, M.; Bickers, D.R. Role of p38 MAPK in UVB-induced inflammatory responses in the skin of SKH-1 hairless mice. J. Investig. Dermatol. 2005, 124, 1318–1325. [Google Scholar] [CrossRef]
- Wang, F.; Lee, E.; Lowes, M.A.; Haider, A.S.; Fuentes-Duculan, J.; Abello, M.V.; Chamian, F.; Cardinale, I.; Krueger, J.G. Prominent production of IL-20 by CD68+/CD11c+ myeloid-derived cells in psoriasis: Gene regulation and cellular effects. J. Investig. Dermatol. 2006, 126, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef]
- Dhar, A.; Young, M.R.; Colburn, N.H. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: The JB6 model is predictive. Mol. Cell Biochem. 2002, 234–235, 185–193. [Google Scholar] [CrossRef]
- Endoh, I.; Di Girolamo, N.; Hampartzoumian, T.; Cameron, B.; Geczy, C.L.; Tedla, N. Ultraviolet B irradiation selectively increases the production of interleukin-8 in human cord blood-derived mast cells. Clin. Exp. Immunol. 2007, 148, 161–167. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J. Investig. Dermatol. 2008, 128, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Beak, S.M.; Lee, Y.S.; Kim, J.A. NADPH oxidase and cyclooxygenase mediate the ultraviolet B-induced generation of reactive oxygen species and activation of nuclear factor-kappaB in HaCaT human keratinocytes. Biochimie 2004, 86, 425–429. [Google Scholar] [CrossRef]
- Glady, A.; Tanaka, M.; Moniaga, C.S.; Yasui, M.; Hara-Chikuma, M. Involvement of NADPH oxidase 1 in UVB-induced cell signaling and cytotoxicity in human keratinocytes. Biochem. Biophys. Rep. 2018, 14, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shao, Y.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J. Cell Biochem. 2009, 107, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Wu, J.X.; Fan, Y.; Adamson, E.D. UV activates growth factor receptors via reactive oxygen intermediates. J. Cell Biol. 1996, 133, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Tober, K.L.; Thomas-Ahner, J.M.; Kusewitt, D.F.; Oberyszyn, T.M. Effects of UVB on E prostanoid receptor expression in murine skin. J. Investig. Dermatol. 2007, 127, 214–221. [Google Scholar] [CrossRef]
- Soontrapa, K.; Honda, T.; Sakata, D.; Yao, C.; Hirata, T.; Hori, S.; Matsuoka, T.; Kita, Y.; Shimizu, T.; Kabashima, K.; et al. Prostaglandin E2-prostaglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 6668–6673. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch. Dermatol. Res. 2010, 302, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.S.; Wang, Z.Q.; Voorhees, J.; Fisher, G. EGF receptor crosstalks with cytokine receptors leading to the activation of c-Jun kinase in response to UV irradiation in human keratinocytes. Cell Signal. 2001, 13, 139–144. [Google Scholar] [CrossRef]
- Blanton, R.A.; Kupper, T.S.; McDougall, J.K.; Dower, S. Regulation of interleukin 1 and its receptor in human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 1273–1277. [Google Scholar] [CrossRef]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Alvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar] [CrossRef]
- Peus, D.; Meves, A.; Vasa, R.A.; Beyerle, A.; O’Brien, T.; Pittelkow, M.R. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic. Biol. Med. 1999, 27, 1197–1202. [Google Scholar] [CrossRef]
- Madson, J.G.; Hansen, L.A. Multiple mechanisms of Erbb2 action after ultraviolet irradiation of the skin. Mol. Carcinog. 2007, 46, 624–628. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Finkel, T. Signal. transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An. inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.F.; Akalu, Y.T.; Suarez-Farinas, M.; Gonzalez, J.; Mitsui, H.; Lowes, M.A.; Orlow, S.J.; Manga, P.; Krueger, J.G. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: Potential relevance to psoriasis. J. Investig. Dermatol. 2013, 133, 2741–2752. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef]
- Reiner, S.L.; Seder, R.A. Dealing from the evolutionary pawnshop: How lymphocytes make decisions. Immunity 1999, 11, 1–10. [Google Scholar] [CrossRef][Green Version]
- Barata, L.T.; Ying, S.; Meng, Q.; Barkans, J.; Rajakulasingam, K.; Durham, S.R.; Kay, A.B. IL-4- and IL-5-positive T lymphocytes, eosinophils, and mast cells in allergen-induced late-phase cutaneous reactions in atopic subjects. J. Allergy Clin. Immunol. 1998, 101, 222–230. [Google Scholar] [CrossRef]
- Min, B.; Prout, M.; Hu-Li, J.; Zhu, J.; Jankovic, D.; Morgan, E.S.; Urban, J.F., Jr.; Dvorak, A.M.; Finkelman, F.D.; LeGros, G.; et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 2004, 200, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Singh, J.; Rani, R.; Gokhale, R.S.; Sharma, V.K.; Marfatia, Y.S.; Begum, R. Interleukin-4 genetic variants correlate with its transcript and protein levels in patients with vitiligo. Br. J. Dermatol. 2012, 167, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Salgame, P.; Abrams, J.S.; Clayberger, C.; Goldstein, H.; Convit, J.; Modlin, R.L.; Bloom, B.R. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991, 254, 279–282. [Google Scholar] [CrossRef]
- Basak, P.Y.; Adiloglu, A.K.; Ceyhan, A.M.; Tas, T.; Akkaya, V.B. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J. Am. Acad. Dermatol. 2009, 60, 256–260. [Google Scholar] [CrossRef]
- Nouri-Koupaee, A.; Mansouri, P.; Jahanbini, H.; Sanati, M.H.; Jadali, Z. Differential expression of mRNA for T-bet and GATA-3 transcription factors in peripheral blood mononuclear cells of patients with vitiligo. Clin. Exp. Dermatol. 2015, 40, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Rael, E.L.; Lockey, R.F. Interleukin-13 signaling and its role in asthma. World Allergy Organ J. 2011, 4, 54–64. [Google Scholar] [CrossRef]
- Nishimura, Y.; Nitto, T.; Inoue, T.; Node, K. IL-13 attenuates vascular tube formation via JAK2-STAT6 pathway. Circ. J. 2008, 72, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Renauld, J.C. New insights into the role of cytokines in asthma. J. Clin. Pathol. 2001, 54, 577–589. [Google Scholar] [CrossRef]
- Kim, K.; Han, J.; Lee, T.R.; Shin, D.W.; Chang, H.; Cho, A.R.; Choi, S.J.; Jo, S.J.; Kwon, O. Comparative Analysis of Human Epidermal and Peripheral Blood γδ T Cell Cytokine Profiles. Ann. Dermatol. 2014, 26, 308–313. [Google Scholar] [CrossRef]
- Han, J.; Lee, E.; Kim, E.; Yeom, M.H.; Kwon, O.; Yoon, T.H.; Lee, T.R.; Kim, K. Role of epidermal γδ T-cell-derived interleukin 13 in the skin-whitening effect of Ginsenoside F1. Exp. Dermatol. 2014, 23, 860–862. [Google Scholar] [CrossRef]
- Speeckaert, R.; Lambert, J.; Grine, L.; Van Gele, M.; De Schepper, S.; van Geel, N. The many faces of interleukin-17 in inflammatory skin diseases. Br. J. Dermatol. 2016, 175, 892–901. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Komine, M.; Tsuda, H.; Tominaga, S.; Ohtsuki, M. Ultraviolet B irradiation induces the expression of IL-33 mRNA and protein in normal human epidermal keratinocytes. J. Dermatol. Sci. 2012, 65, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Song, J.; Ping, F.; Shang, J. Enhancement of the p38 MAPK and PKA signaling pathways is associated with the pro-melanogenic activity of Interleukin 33 in primary melanocytes. J. Dermatol. Sci. 2014, 73, 110–116. [Google Scholar] [CrossRef]
- Zhou, J.; Shang, J.; Song, J.; Ping, F. Interleukin-18 augments growth ability of primary human melanocytes by PTEN inactivation through the AKT/NF-κB pathway. Int. J. Biochem. Cell Biol. 2013, 45, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Hu, Y.; Li, W.H.; Eisinger, M.; Seiberg, M.; Lin, C.B. The role of keratinocyte growth factor in melanogenesis: A possible mechanism for the initiation of solar lentigines. Exp. Dermatol. 2010, 19, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Kholmanskikh, O.; van Baren, N.; Brasseur, F.; Ottaviani, S.; Vanacker, J.; Arts, N.; van der Bruggen, P.; Coulie, P.; De Plaen, E. Interleukins 1alpha and 1beta secreted by some melanoma cell lines strongly reduce expression of MITF-M and melanocyte differentiation antigens. Int. J. Cancer 2010, 127, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Leopardi, S.; Printup, S.; Malhi, N.; Seiberg, M.; Lapoint, R. Proteinase-activated receptor-2 stimulates prostaglandin production in keratinocytes: Analysis of prostaglandin receptors on human melanocytes and effects of PGE2 and PGF2alpha on melanocyte dendricity. J. Investig. Dermatol. 2004, 122, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.T.; Ganju, P.; Singh, A.; Vijayan, V.; Kirty, K.; Yadav, S.; Puntambekar, S.; Bajaj, S.; Dani, P.P.; Kar, H.K.; et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ling, J.; Wang, Y.; Shang, J.; Ping, F. Cross-talk between interferon-gamma and interleukin-18 in melanogenesis. J. Photochem. Photobiol. B 2016, 163, 133–143. [Google Scholar] [CrossRef]
- Videira, I.F.; Moura, D.F.; Magina, S. Mechanisms regulating melanogenesis. An. Bras. Dermatol. 2013, 88, 76–83. [Google Scholar] [CrossRef]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Byrne, S.N.; Beaugie, C.; O’Sullivan, C.; Leighton, S.; Halliday, G.M. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am. J. Pathol. 2011, 179, 211–222. [Google Scholar] [CrossRef]
- Ali, S.; Huber, M.; Kollewe, C.; Bischoff, S.C.; Falk, W.; Martin, M.U. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18660–18665. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Moulin, D.; Donzé, O.; Talabot-Ayer, D.; Mézin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef] [PubMed]
- Pushparaj, P.N.; Tay, H.K.; H’Ng, S.C.; Pitman, N.; Xu, D.; McKenzie, A.; Liew, F.Y.; Melendez, A.J. The cytokine interleukin-33 mediates anaphylactic shock. Proc. Natl. Acad. Sci. USA 2009, 106, 9773–9778. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Oboki, K.; Kajiwara, N.; Morii, E.; Aozasa, K.; Flavell, R.A.; Okumura, K.; Saito, H.; Nakae, S. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J. Immunol. 2009, 183, 7890–7897. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hueber, A.J.; Alves-Filho, J.C.; Asquith, D.L.; Michels, C.; Millar, N.L.; Reilly, J.H.; Graham, G.J.; Liew, F.Y.; Miller, A.M.; McInnes, I.B. IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur. J. Immunol. 2011, 41, 2229–2237. [Google Scholar] [CrossRef]
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K.; et al. An.IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J. Immunol. 2008, 181, 5981–5989. [Google Scholar] [CrossRef]
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054. [Google Scholar] [CrossRef]
- Hsu, C.L.; Chhiba, K.D.; Krier-Burris, R.; Hosakoppal, S.; Berdnikovs, S.; Miller, M.L.; Bryce, P.J. Allergic inflammation is initiated by IL-33-dependent crosstalk between mast cells and basophils. PLoS ONE 2020, 15, e0226701. [Google Scholar] [CrossRef] [PubMed]
- Cevikbas, F.; Steinhoff, M. IL-33: A novel danger signal system in atopic dermatitis. J. Investig. Dermatol. 2012, 132, 1326–1329. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1. Rev. Infect. Dis. 1984, 6, 51–95. [Google Scholar] [CrossRef]
- Kampschmidt, R.F. The numerous postulated biological manifestations of interleukin-1. J. Leukoc. Biol. 1984, 36, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Kupper, T.S. Immune and inflammatory processes in cutaneous tissues. Mechanisms and speculations. J. Clin. Investig. 1990, 86, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef]
- Sims, J.E.; March, C.J.; Cosman, D.; Widmer, M.B.; MacDonald, H.R.; McMahan, C.J.; Grubin, C.E.; Wignall, J.M.; Jackson, J.L.; Call, S.M.; et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 1988, 241, 585–589. [Google Scholar] [CrossRef]
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K.; et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647. [Google Scholar] [CrossRef]
- Groves, R.W.; Sherman, L.; Mizutani, H.; Dower, S.K.; Kupper, T.S. Detection of interleukin-1 receptors in human epidermis. Induction of the type II receptor after organ culture and in psoriasis. Am. J. Pathol. 1994, 145, 1048–1056. [Google Scholar]
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 2002, 1592, 265–280. [Google Scholar] [CrossRef]
- Tang, A.; Gilchrest, B.A. Regulation of keratinocyte growth factor gene expression in human skin fibroblasts. J. Dermatol. Sci. 1996, 11, 41–50. [Google Scholar] [CrossRef]
- Groves, R.W.; Ross, E.; Barker, J.N.; Ross, J.S.; Camp, R.D.; MacDonald, D.M. Effect of in vivo interleukin-1 on adhesion molecule expression in normal human skin. J. Investig. Dermatol. 1992, 98, 384–387. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kondo, S.; Sauder, D.N.; Kono, T.; Galley, K.A.; McKenzie, R.C. Differential modulation of interleukin-1 alpha (IL-1 alpha) and interleukin-1 beta (IL-1 beta) in human epidermal keratinocytes by UVB. Exp. Dermatol. 1994, 3, 29–39. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Pentland, A.P.; Mahoney, M.; Jacobs, S.C.; Holtzman, M.J. Enhanced prostaglandin synthesis after ultraviolet injury is mediated by endogenous histamine stimulation. A mechanism for irradiation erythema. J. Clin. Investig. 1990, 86, 566–574. [Google Scholar] [CrossRef]
- Pentland, A.P.; Mahoney, M.G. Keratinocyte prostaglandin synthesis is enhanced by IL-1. J. Investig. Dermatol. 1990, 94, 43–46. [Google Scholar] [CrossRef]
- Fosslien, E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann. Clin. Lab. Sci. 2000, 30, 3–21. [Google Scholar]
- Pentland, A.P.; Needleman, P. Modulation of keratinocyte proliferation in vitro by endogenous prostaglandin synthesis. J. Clin. Investig. 1986, 77, 246–251. [Google Scholar] [CrossRef]
- Scott, G.; Jacobs, S.; Leopardi, S.; Anthony, F.A.; Learn, D.; Malaviya, R.; Pentland, A. Effects of PGF2alpha on human melanocytes and regulation of the FP receptor by ultraviolet radiation. Exp. Cell Res. 2005, 304, 407–416. [Google Scholar] [CrossRef]
- Holtzman, M.J.; Zhang, V.; Hussain, H.; Roswit, W.T.; Wilson, J.D. Prostaglandin H synthase and lipoxygenase gene families in the epithelial cell barrier. Ann. N. Y. Acad. Sci. 1994, 744, 58–77. [Google Scholar] [CrossRef]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Carnaud, C.; Lee, D.; Donnars, O.; Park, S.H.; Beavis, A.; Koezuka, Y.; Bendelac, A. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999, 163, 4647–4650. [Google Scholar]
- Frucht, D.M.; Fukao, T.; Bogdan, C.; Schindler, H.; O’Shea, J.J.; Koyasu, S. IFN-gamma production by antigen-presenting cells: Mechanisms emerge. Trends Immunol. 2001, 22, 556–560. [Google Scholar] [CrossRef]
- Flaishon, L.; Hershkoviz, R.; Lantner, F.; Lider, O.; Alon, R.; Levo, Y.; Flavell, R.A.; Shachar, I. Autocrine secretion of interferon gamma negatively regulates homing of immature B cells. J. Exp. Med. 2000, 192, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell 2010, 6, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Malek, Z.; Scott, M.C.; Suzuki, I.; Tada, A.; Im, S.; Lamoreux, L.; Ito, S.; Barsh, G.; Hearing, V.J. The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation. Pigment. Cell Res. 2000, 13 (Suppl. 8) (Suppl. 8), 156–162. [Google Scholar] [CrossRef]
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J. Investig. Dermatol. 2012, 132, 1869–1876. [Google Scholar] [CrossRef]
- Gregg, R.K.; Nichols, L.; Chen, Y.; Lu, B.; Engelhard, V.H. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J. Immunol. 2010, 184, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, Y.; Sun, Y.; Shi, W.; Yang, J.; Zhu, L.; Li, M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Derm. Venereol. 2015, 95, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Chu, C.Q.; Eedy, D.J.; Feldmann, M.; Brennan, F.M.; Breathnach, S.M. Localization of tumour necrosis factor-alpha (TNF-alpha) and its receptors in normal and psoriatic skin: Epidermal cells express the 55-kD but not the 75-kD TNF receptor. Clin. Exp. Immunol. 1993, 94, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-γ and tumor necrosis factor-α in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Kim, D.S.; Kim, H.J.; Seo, K.I.; Kim, K.H.; Chung, J.H.; Eun, H.C.; Jung, H.C. Growth related secretion and production of GM-CSF by epithelial cell line. J. Dermatol. Sci. 2001, 25, 53–58. [Google Scholar] [CrossRef]
- Wu, X.G.; Hong, W.S.; Xu, A. GM-CSF: A possible prognostic serum biomarker of vitiligo patients’ considered for transplantation treatment with cultured autologous melanocytes: A pilot study. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1409–1411. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.R.; Ansary, T.M.; Komine, M.; Ohtsuki, M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. Int. J. Mol. Sci. 2021, 22, 3970. https://doi.org/10.3390/ijms22083970
Hossain MR, Ansary TM, Komine M, Ohtsuki M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. International Journal of Molecular Sciences. 2021; 22(8):3970. https://doi.org/10.3390/ijms22083970
Chicago/Turabian StyleHossain, Md Razib, Tuba M. Ansary, Mayumi Komine, and Mamitaro Ohtsuki. 2021. "Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation" International Journal of Molecular Sciences 22, no. 8: 3970. https://doi.org/10.3390/ijms22083970
APA StyleHossain, M. R., Ansary, T. M., Komine, M., & Ohtsuki, M. (2021). Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. International Journal of Molecular Sciences, 22(8), 3970. https://doi.org/10.3390/ijms22083970