
Role of Electrostatic Interactions in Calcitonin Prefibrillar Oligomer-Induced Amyloid Neurotoxicity and Protective Effect of Neuraminidase

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Effects of Treatment with NAA
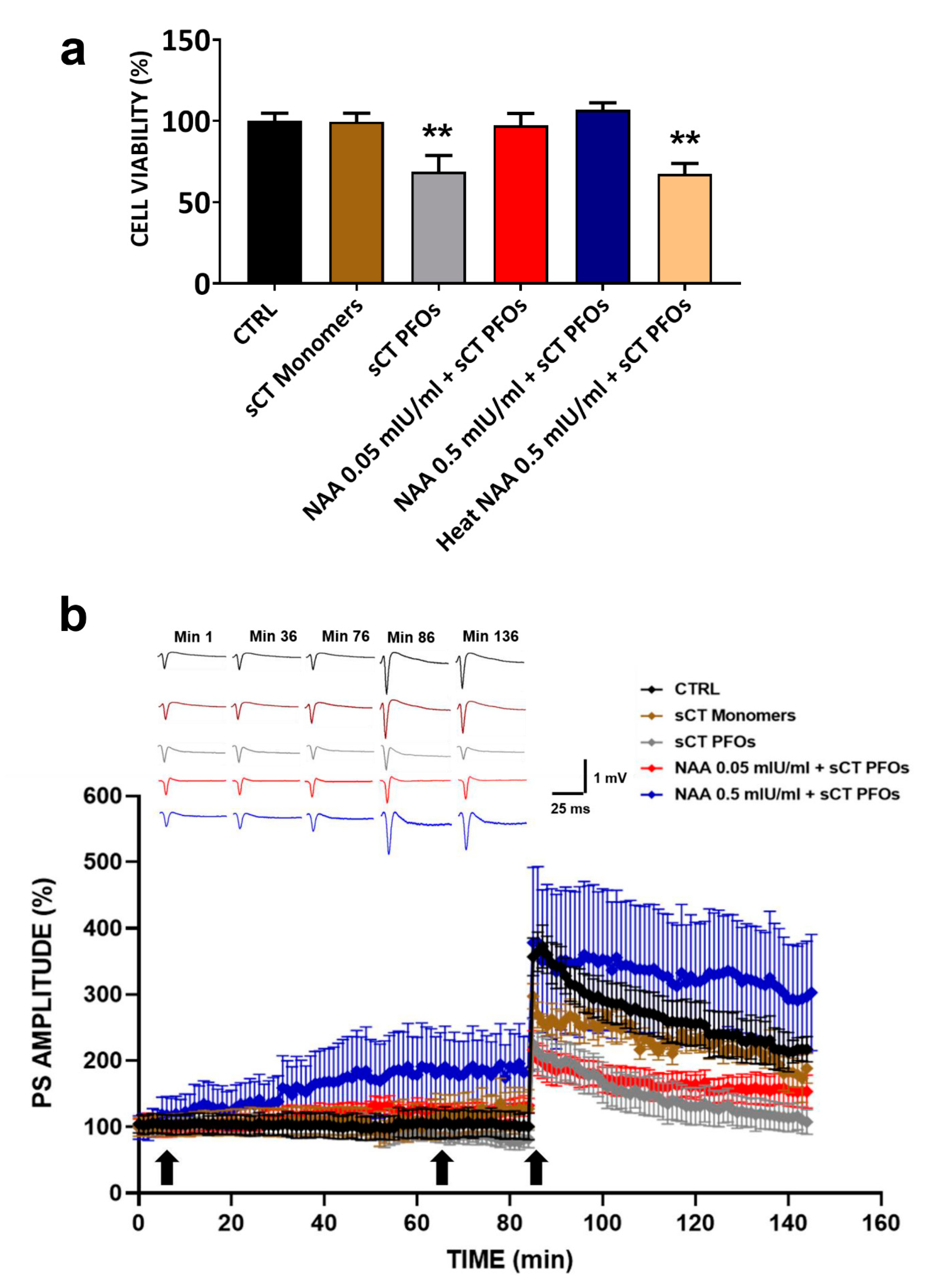
2.2. Effects of the Combined Treatment with NAA and sCT PFOs
3. Discussion
3.1. Effects of Treatment with Different NAA Concentrations on Cell Viability and Synaptic Transmission
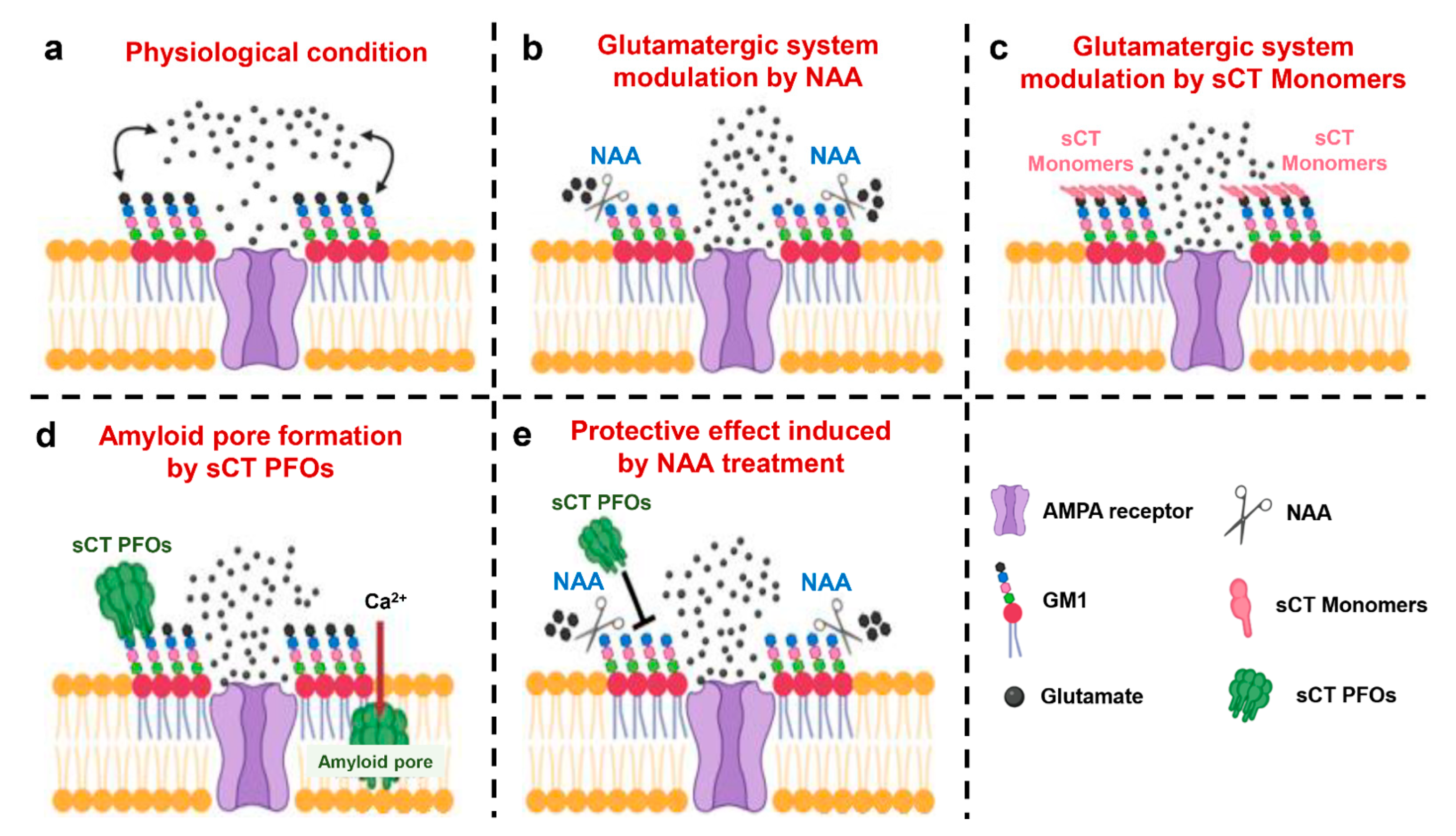
3.2. NAA Protection Effect from Amyloid Neurotoxicity Induced by sCT PFOs
4. Materials and Methods
4.1. Animals
4.2. Cell Cultures
4.3. sCT Sample Preparation by Size Exclusion Chromatography (SEC)
4.4. NAA Solution Preparation
4.5. Assessment of Cell Viability by MTS Assay
4.6. Extracellular Recordings in Mouse Hippocampal Slices
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Macchia, G.; Paradisi, S.; Frank, C.; Camerini, S.; Chistolini, P.; Gaudiano, M.C.; Petrucci, T.C.; Malchiodi-Albedi, F. Native metastable prefibrillar oligomers are the most neurotoxic species among amyloid aggregates. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1622–1629. [Google Scholar] [CrossRef]
- Malchiodi-Albedi, F.; Contrusciere, V.; Raggi, C.; Fecchi, K.; Rainaldi, G.; Paradisi, S.; Matteucci, A.; Santini, M.T.; Sargiacomo, M.; Frank, C.; et al. Lipid raft disruption protects mature neurons against amyloid oligomer toxicity. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 406–415. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Gaudiano, M.C.; Malchiodi-Albedi, F. The Slowly Aggregating Salmon Calcitonin: A Useful Tool for the Study of the Amyloid Oligomers Structure and Activity. Int. J. Mol. Sci. 2011, 12, 9277–9295. [Google Scholar] [CrossRef]
- Vieira, M.N.N.; Forny-Germano, L.; Saraiva, L.M.; Sebollela, A.; Martinez, A.M.B.; Houzel, J.-C.; De Felice, F.G.; Ferreira, S.T. Soluble oligomers from a non-disease related protein mimic Aβ-induced tau hyperphosphorylation and neurodegeneration. J. Neurochem. 2007, 103, 736–748. [Google Scholar] [CrossRef]
- Gancar, M.; Kurin, E.; Bednarikova, Z.; Marek, J.; Mucaji, P.; Nagy, M.; Gazova, Z. Amyloid Aggregation of Insulin: An Interaction Study of Green Tea Constituents. Sci. Rep. 2020, 10, 9115. [Google Scholar] [CrossRef]
- Taguchi, H.; Kawai-Noma, S. Amyloid oligomers: Diffuse oligomer-based transmission of yeast prions. FEBS J. 2010, 277, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Bucciantini, M.; Capanni, C.; Taddei, N.; Dobson, C.M.; Stefani, M. Solution conditions can promote formation of either amyloid protofilaments or mature fibrils from the HypF N-terminal domain. Protein Sci. 2008, 10, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Diociaiuti, M.; Polzi, L.Z.; Valvo, L.; Malchiodi-Albedi, F.; Bombelli, C.; Gaudiano, M.C. Calcitonin Forms Oligomeric Pore-Like Structures in Lipid Membranes. Biophys. J. 2006, 91, 2275–2281. [Google Scholar] [CrossRef]
- Belfiore, M.; Cariati, I.; Matteucci, A.; Gaddini, L.; Macchia, G.; Fioravanti, R.; Frank, C.; Tancredi, V.; D’Arcangelo, G.; Diociaiuti, M. Calcitonin native prefibrillar oligomers but not monomers induce membrane damage that triggers NMDA-mediated Ca2+-influx, LTP impairment and neurotoxicity. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Schubert, D.; Behl, C.; Lesley, R.; Brack, A.; Dargusch, R.; Sagara, Y.; Kimura, H. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schubert, D. Cytotoxic Amyloid Peptides Inhibit Cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Reduction by Enhancing MTT Formazan Exocytosis. J. Neurochem. 1997, 69, 2285–2293. [Google Scholar] [CrossRef]
- Schengrund, C.-L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Aureli, M.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Lipid Rafts in Neurodegeneration and Neuroprotection. Mol. Neurobiol. 2014, 50, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Malchiodi-Albedi, F.; Paradisi, S.; Matteucci, A.; Frank, C.; Diociaiuti, M. Amyloid Oligomer Neurotoxicity, Calcium Dysregulation, and Lipid Rafts. Int. J. Alzheimer’s Dis. 2011, 2011, 906964. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef]
- Bucciantini, M.; Rigacci, S.; Stefani, M. Amyloid aggregation: Role of biological membranes and the aggregate–membrane system. J. Phys. Chem. Lett. 2014, 5, 517–527. [Google Scholar] [CrossRef]
- Perissinotto, F.; Rondelli, V.; Parisse, P.; Tormena, N.; Zunino, A.; Almásy, L.; Merkel, D.; Bottyán, L.; Sajti, S.; Casalis, L. GM1 Ganglioside role in the interaction of Alpha-synuclein with lipid membranes: Morphology and structure. Biophys. Chem. 2019, 255, 106272. [Google Scholar] [CrossRef]
- Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J. 2015, 32, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Diociaiuti, M.; Bombelli, C.; Zanetti-Polzi, L.; Belfiore, M.; Fioravanti, R.; Macchia, G.; Giordani, C. The Interaction between Amyloid Prefibrillar Oligomers of Salmon Calcitonin and a Lipid-Raft Model: Molecular Mechanisms Leading to Membrane Damage, Ca2+-Influx and Neurotoxicity. Biomolecules 2019, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ Oligomers Are Rapidly Sequestered from Brain ISF In Vivo and Bind GM1 Ganglioside on Cellular Membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Malenka, R.C. Long-Term Potentiation—A Decade of Progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef]
- Aureli, M.; Mauri, L.; Ciampa, M.G.; Prinetti, A.; Toffano, G.; Secchieri, C.; Sonnino, S. GM1 Ganglioside: Past Studies and Future Potential. Mol. Neurobiol. 2016, 53, 1824–1842. [Google Scholar] [CrossRef]
- Hoffman, K.B.; Kessler, M.; Lynch, G. Sialic acid residues indirectly modulate the binding properties of AMPA-type glutamate receptors. Brain Res. 1997, 753, 309–314. [Google Scholar] [CrossRef]
- Wieraszko, A.; Seifert, W. The role of monosialoganglioside GM1 in the synaptic plasticity: In vitro study on rat hippocampal slices. Brain Res. 1985, 345, 159–164. [Google Scholar] [CrossRef]
- Wang, S.S.; Rymer, D.L.; Good, T.A. Reduction in cholesterol and sialic acid content protects cells from the toxic effects of β-amyloid peptides. J. Biol. Chem. 2001, 276, 42027–42034. [Google Scholar] [CrossRef]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F.; et al. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J. 2011, 26, 818–831. [Google Scholar] [CrossRef]
- Oropesa-Nuñez, R.; Keshavan, S.; Dante, S.; Diaspro, A.; Mannini, B.; Capitini, C.; Cecchi, C.; Stefani, M.; Chiti, F.; Canale, C. Toxic HypF-N Oligomers Selectively Bind the Plasma Membrane to Impair Cell Adhesion Capability. Biophys. J. 2018, 114, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 2017, 549, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Louhimies, S. Directive 86/609/EEC on the Protection of Animals Used for Experimental and Other Scientific Purposes. Altern. Lab. Anim. 2002, 30, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Cariati, I.; Scimeca, M.; Pallone, G.; Bonanno, E.; Tancredi, V.; D’Arcangelo, G.; Frank, C. Effects of short-term aerobic exercise in a mouse model of Niemann-Pick type C disease on synaptic and muscle plasticity. Ann. Ist. Super. Sanita 2019, 55, 330–337. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| TIME (min) | CTRL (PS% Amplitude) | NAA 0.005 mIU/mL (PS% Amplitude) | NAA 0.05 mIU/mL (PS% Amplitude) | NAA 0.5 mIU/mL (PS% Amplitude) | NAA 10 mIU/mL (PS% Amplitude) | SIGNIFICANCE between Groups at Different Times |
|---|---|---|---|---|---|---|
| 5 | 99.4 ± 14.1 | 103.3 ± 11.0 | 112.8 ± 24.0 | 103.6 ± 18.4 | 109.8 ± 27.5 | no significance |
| 35 | 101.5 ± 16.9 | 94.0 ± 3.4 | 125.7 ± 28.1 | 146.1 ± 27.2 | 179.4 ± 52.1 | CTRL vs. NAA 10 mIU/mL, *** p < 0.001 |
| 65 | 95.6 ± 17.5 | 100.5 ± 12.4 | 124.2 ± 22.8 | 171.9 ± 35.9 | 233.5 ± 62.6 | CTRL vs. NAA 0.5 mIU/mL, *** p < 0.001; CTRL vs. NAA 10 mIU/mL, **** p < 0.0001 |
| TIME (min) | CTRL (PS% Amplitude) | NAA 0.05 mIU/mL (PS% Amplitude) | NAA 0.5 mIU/mL (PS% Amplitude) | SIGNIFICANCE between Groups at Different Times |
|---|---|---|---|---|
| 5 | 100.5 ± 17.4 | 114.7 ± 20.9 | 106.9 ± 21.0 | no significance |
| 60 | 100.8 ± 20.9 | 130.7 ± 21.2 | 159.0 ± 33.5 | CTRL vs. NAA 0.5 mIU/mL, ** p < 0.01 |
| 66 | 354.0 ± 39.3 | 316.3 ± 55.4 | 354.1 ± 69.1 | no significance |
| 125 | 224.3 ± 26.5 | 231.2 ± 36.3 | 307.4 ± 68.5 | CTRL vs. NAA 0.5 mIU/mL, ** p < 0.01 |
| TIME (min) | CTRL (PS% Amplitude) | sCT Monomers (PS% Amplitude) | sCT PFOs (PS% Amplitude) | NAA 0.05 mIU/mL + sCT PFOs (PS% Amplitude) | NAA 0.5 mIU/mL + sCT PFOs (PS% Amplitude) | SIGNIFICANCE between Groups at Different Times |
|---|---|---|---|---|---|---|
| 1 | 104.7 ± 13.9 | 100.3 ± 14.4 | 99.9 ± 9.7 | 99.6 ± 12.9 | 99.3 ± 18.0 | no significance |
| 36 | 102.0 ± 18.7 | 108.6 ± 21.7 | 105.7 ± 15.8 | 117.8 ± 12.9 | 156.2 ± 44.6 | no significance |
| 76 | 104.2 ± 18.6 | 128.7 ± 25.8 | 87.2 ± 16.9 | 124.4 ± 11.3 | 184.5 ± 52.8 | CTRL vs. NAA 0.5 mIU/mL + sCT PFOs, *** p < 0.001 |
| 86 | 356.8 ± 28.6 | 297.3 ± 19.1 | 207.5 ± 38.6 | 216.1 ± 29.1 | 378.3 ± 113.8 | CTRL vs. sCT PFOs and NAA 0.05 mIU/mL + sCT PFOs, **** p < 0.0001 |
| 136 | 226.4 ± 32.7 | 201.0 ± 12.7 | 105.5 ± 20.1 | 157.6 ± 22.9 | 313.9 ± 79.8 | CTRL vs. sCT PFOs, **** p < 0.0001; CTRL vs. NAA 0.05 mIU/mL + sCT PFOs, ** p < 0.01; CTRL vs. NAA 0.5 mIU/mL + sCT PFOs, *** p < 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cariati, I.; Bonanni, R.; Marini, M.; Rinaldi, A.M.; Zarrilli, B.; Tancredi, V.; Frank, C.; D’Arcangelo, G.; Diociaiuti, M. Role of Electrostatic Interactions in Calcitonin Prefibrillar Oligomer-Induced Amyloid Neurotoxicity and Protective Effect of Neuraminidase. Int. J. Mol. Sci. 2021, 22, 3947. https://doi.org/10.3390/ijms22083947
Cariati I, Bonanni R, Marini M, Rinaldi AM, Zarrilli B, Tancredi V, Frank C, D’Arcangelo G, Diociaiuti M. Role of Electrostatic Interactions in Calcitonin Prefibrillar Oligomer-Induced Amyloid Neurotoxicity and Protective Effect of Neuraminidase. International Journal of Molecular Sciences. 2021; 22(8):3947. https://doi.org/10.3390/ijms22083947
Chicago/Turabian StyleCariati, Ida, Roberto Bonanni, Mario Marini, Anna Maria Rinaldi, Beatrice Zarrilli, Virginia Tancredi, Claudio Frank, Giovanna D’Arcangelo, and Marco Diociaiuti. 2021. "Role of Electrostatic Interactions in Calcitonin Prefibrillar Oligomer-Induced Amyloid Neurotoxicity and Protective Effect of Neuraminidase" International Journal of Molecular Sciences 22, no. 8: 3947. https://doi.org/10.3390/ijms22083947
APA StyleCariati, I., Bonanni, R., Marini, M., Rinaldi, A. M., Zarrilli, B., Tancredi, V., Frank, C., D’Arcangelo, G., & Diociaiuti, M. (2021). Role of Electrostatic Interactions in Calcitonin Prefibrillar Oligomer-Induced Amyloid Neurotoxicity and Protective Effect of Neuraminidase. International Journal of Molecular Sciences, 22(8), 3947. https://doi.org/10.3390/ijms22083947