
Modeling Neoplastic Growth in Renal Cell Carcinoma and Polycystic Kidney Disease



Abstract
1. Introduction
2. Neoplastic Character and Vasculature
3. Molecular Pathways and Genes Implicated in RCC
4. Cell Metabolism
5. Oxygen in ADPKD
6. Ion Channel Signaling
7. Primary Cilia
8. Non-Coding RNAs
8.1. MicroRNAs
8.2. Long Non-Coding RNAs
9. Drosophila Modeling for PKD
10. Pharmacological Strategies
10.1. Smac Treatment
10.2. Melatonin Treatment
11. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yao, Y.; Dai, W. Genomic instability and cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [PubMed]
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Znaor, A.; Lortet-Tieulent, J.; Laversanne, M.; Jemal, A.; Bray, F. International variations and trends in renal cell carcinoma incidence and mortality. Eur. Urol. 2015, 67, 519–530. [Google Scholar] [CrossRef]
- Ljunberg, B.; Campbell, S.C.; Choi, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Amiji, M.M.; Ramesh, R. Diagnostic and Therapeutic Applications of Exosomes in Cancer, 1st ed.; Elsevier: London, UK, 2018. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of renal cell carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO classification of tumours of the urinary system and male genital organs—Part A: Renal, penile, and testicular tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Haake, S.M.; Rathmell, W.K. Renal cancer subtypes: Should we be lumping or splitting for therapeutic decision making? Cancer 2017, 123, 200–209. [Google Scholar] [CrossRef]
- Wolf, M.M.; Rathmell, W.K.; Beckermann, K.E. Modeling clear cell renal cell carcinoma and therapeutic implications. Oncogene 2020, 39, 3413–3426. [Google Scholar] [CrossRef]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef]
- Kovacs, G.; Akhtar, M.; Beckwith, B.J.; Bugert, P.; Cooper, C.S.; Delahunt, B.; Eble, J.N.; Fleming, S.; Ljungberg, B.; Medeiros, L.J.; et al. The Heidelberg classification of renal cell tumours. J. Pathol. 1997, 183, 131–133. [Google Scholar] [CrossRef]
- Linehan, W.M.; Walther, M.M.; Zbar, B. The genetic basis of cancer of the kidney. J. Urol. 2003, 170, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Manley, B.J.; Hakimi, A.A. Molecular profiling of renal cell carcinoma: Building a bridge towards clinical impact. Curr. Opin. Urol. 2016, 26, 383–387. [Google Scholar] [CrossRef]
- Zekri, J.; Ahmed, N.; Coleman, R.E.; Hancock, B.W. The skeletal metastatic complications of renal cell carcinoma. Int. J. Oncol. 2001, 19, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Janzen, N.K.; Perry, K.T.; Schulam, P.G. Laparoscopic radical nephrectomy and minimally invasive surgery for kidney cancer. Kidney Cancer 2003, 99–117. [Google Scholar] [CrossRef]
- Lam, J.S.; Klatte, T.; Kim, H.L.; Patard, J.J.; Breda, A.; Zisman, A.; Pantuck, A.J.; Figlin, R.A. Prognostic factors and selection for clinical studies of patients with kidney cancer. Crit. Rev. Oncol. Hematol. 2008, 65, 235–262. [Google Scholar] [CrossRef]
- Frees, S.; Breuksch, I.; Haber, T.; Bauer, H.K.; Chavez-Munoz, C.; Raven, P.; Moskalev, I.; Costa, N.D.; Tan, Z.; Daugaard, M.; et al. Calcium-sensing receptor (CaSR) promotes development of bone metastasis in renal cell carcinoma. Oncotarget 2018, 9, 15766–15779. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.I.; Tsodikov, A.; Kunju, L.P.; Chinnaiyan, A.M.; Palapattu, G.S.; Morgan, T.M.; Alva, A.; Tomlins, S.; Wu, A.; Montgomery, J.S.; et al. Papillary renal cell carcinoma revisited: A comprehensive histomorphologic study with outcome correlations. Hum. Pathol. 2014, 45, 1139–1146. [Google Scholar] [CrossRef]
- Durinck, S.; Stawiski, E.W.; Pavia-Jimenez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar] [PubMed]
- Shuch, B.; Amin, A.; Armstrong, A.J.; Eble, J.N.; Ficarra, V.; Lopez-Beltran, A.; Martignoni, G.; Rini, B.I.; Kutikov, A. Understanding pathologic variants of renal cell carcinoma: Distilling therapeutic opportunities from biologic complexity. Eur. Urol. 2015, 67, 85–97. [Google Scholar] [CrossRef]
- Sakamoto, H.; Yamasaki, T.; Sumiyoshi, T.; Utsunomiya, N.; Takeda, M.; Kamba, T.; Nakamura, E.; Ogawa, O. A family case with germline TSC1 and mtDNA mutations developing bilateral eosinophilic chromophobe renal cell carcinomas without other typical phenotype of tuberous sclerosis. J. Clin. Pathol. 2018, 71, 936–943. [Google Scholar] [CrossRef]
- Wang, S.; Yu, Z.H.; Chai, K.Q. Identification of CFTR as a novel key gene in chromophobe renal cell carcinoma through bioinformatics analysis. Oncol. Lett. 2019, 18, 1767–1774. [Google Scholar] [CrossRef]
- Chowdhury, A.; Chakraborty, D.; Bhattacharya, P.; Dey, R. Multilocular cystic renal cell carcinoma a diagnostic dilemma: A case report in a 30-year-old woman. Urol. Ann. 2013, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, W.S.; Chung, J. SETD2, GIGYF2, FGFR3, BCR, KMT2C, and TSC2 as candidate genes for differentiating multilocular cystic renal neoplasm of low malignant potential from clear cell renal cell carcinoma with cystic change. Investig. Clin. Urol. 2019, 60, 148. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Papanicolau-Sengos, A.; Chintala, S.; Wei, L.; Liu, B.; Hu, Q.; Miles, K.M.; Conroy, J.M.; Glenn, S.T.; Costantini, M.; et al. Collecting duct carcinoma of the kidney is associated with CDKN2A deletion and SLC family gene up-regulation. Oncotarget 2016, 7, 29901–29915. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Neumann, H.P.; Richard, S. Von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef]
- Breslow, N.E.; Beckwith, J.B. Epidemiological features of Wilms’ tumor: Results of the National Wilms’ Tumor Study. J. Natl. Cancer Inst. 1982, 68, 429–436. [Google Scholar] [CrossRef]
- Weksberg, R.; Brzezinski, J. Identifying new Wilms tumour predisposition genes. Lancet Child Adolesc. Health 2019, 3, 285–287. [Google Scholar] [CrossRef]
- Gladell, P.; Paner, M.D. Clear Cell Renal Carcinoma: Fuhrman Nuclear Grade. Available online: https://www.auanet.org/education/auauniversity/education-products-and-resources/pathology-for-urologists/kidney/renal-cell-carcinomas/clear-cell-renal-cell-carcinoma-fuhrman-nuclear-grade (accessed on 25 February 2021).
- Williamson, S.R. Grading. Available online: https://www.pathologyoutlines.com/topic/kidneytumormalignantnucleargrading.html (accessed on 21 January 2021).
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Sparapani, S.; Millet-Boureima, C.; Oliver, J.; Mu, K.; Hadavi, P.; Kalostian, T.; Ali, N.; Avelar, C.M.; Bardies, M.; Barrow, B.; et al. The biology of vasopressin. Biomedicines 2021, 9, 89. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.C.M.; Harris, P.C. Molecular pathogenesis of ADPKD: The polycystin complex gets complex. Kidney Int. 2005, 67, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Zbar, B. Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int. 1997, 51, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Bonsib, S.M. Renal cystic diseases and renal neoplasms: A mini-review. Clin. J. Am. Soc. Nephrol. 2009, 4, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jin, B.; Xu, L.; Fu, G.; Meng, H.; Liu, B.; Li, J.; Xia, D. Cystic renal cell carcinoma: A report of 67 cases including 4 cases with concurrent renal cell carcinoma. BMC Urol. 2014, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, B.; Song, N.; Hua, L.; Wang, Z.; Gu, M.; Yin, C. Diagnosis and treatment of cystic renal cell carcinoma. World J. Surg. Oncol. 2013, 11, 158. [Google Scholar] [CrossRef]
- Ishikawa, I.; Saito, Y.; Onouchi, A.; Kitada, H.; Suzuki, S.; Kurihara, S.; Yuri, T.; Shinoda, A. Development of acquired cystic disease and adenocarcinoma of the kidney in glomerulonephritic chronic hemodialysis patients. Clin. Nephrol. 1980, 14, 1–6. [Google Scholar]
- Grantham, J. Polycystic kidney disease: Neoplasia in disguise. Am. J. Kidney Dis. 1990, 15, 110–116. [Google Scholar] [CrossRef]
- de Werra, C.; Donzelli, I.; Perone, M.; Micco, R.D.; Orabona, G. Multifocal and multicentric tumors. In Multiple Primary Malignancies. Updates in Surgery; Springer: Milano, Italy, 2009; pp. 129–142. [Google Scholar]
- Nargund, A.M.; Pham, C.G.; Dong, Y.; Wang, P.I.; Osmangeyoglu, H.U.; Xie, Y.; Aras, O.; Han, S.; Oyama, T.; Takeda, S.; et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep. 2017, 18, 2893–2906. [Google Scholar] [CrossRef]
- Drusian, L.; Boletta, A. MTORC1-driven accumulation of the oncometabolite fumarate as a potential critical step in renal cancer progression. Mol. Cell. Oncol. 2019, 6, 1537709. [Google Scholar] [CrossRef]
- Shao, X.; Somlo, S.; Igarashi, P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 2002, 13, 1837–1846. [Google Scholar] [CrossRef]
- Mandriota, S.J.; Turner, K.J.; Davies, D.R.; Murray, P.G.; Morgan, N.V.; Sowter, H.M.; Wykoff, C.C.; Maher, E.R.; Harris, A.L.; Ratcliffe, P.J.; et al. HIF activation identifies early lesions in VHL kidneys: Evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002, 1, 459–468. [Google Scholar] [CrossRef]
- Perretta-Tejedor, N.; Jafree, D.J.; Long, D.A. Endothelial-epithelial communication in polycystic kidney disease: Role of vascular endothelial growth factor signalling. Cell. Signal. 2020, 72, 109624. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, P353–P364. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef]
- Huang, J.L.; Woolf, A.S.; Long, D.A. Angiogenesis and autosomal dominant polycystic kidney disease. Pediatr. Nephrol. 2013, 28, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Bello-Reuss, E.; Holubec, K.; Rajaraman, S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney. Int. 2001, 60, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Popov, V.; Walocha, J.A.; Wen, J.; Bello-Reuss, E. Evidence of angiogenesis and microvascular regression in autosomal-dominant polycystic kidney disease kidneys: A corrosion cast study. Kidney Int. 2006, 70, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Woolf, A.S.; Kolatsi-Joannou, M.; Baluk, P.; Sandford, R.N.; Peters, D.J.M.; McDonald, D.M.; Price, K.L.; Winyard, P.J.D.; Long, D.A. Vascular endothelial growth factor C for polycystic kidney diseases. J. Am. Soc. Nephrol. 2016, 27, 69–77. [Google Scholar] [CrossRef]
- Ogunlade, O.; Connell, J.J.; Huang, J.L.; Zhang, E.; Lythgoe, M.F.; Long, D.A.; Beard, P. In vivo three-dimensional photoacoustic imaging of the renal vasculature in preclinical rodent models. Am. J. Physiol. Renal Physiol. 2018, 314, F1145–F1153. [Google Scholar] [CrossRef] [PubMed]
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Outeda, P.; Huso, D.L.; Fisher, S.A.; Halushka, M.K.; Kim, H.; Qian, F.; Germino, G.G.; Watnick, T. Polycystin signaling is required for directed endothelial cell migration and lymphatic development. Cell Rep. 2014, 7, 634–644. [Google Scholar] [CrossRef]
- Jafree, D.J.; Moulding, D.; Kolatsi-Joannou, M.; Perretta_Tejedor, N.; Price, K.L.; Milmoe, N.J.; Walsh, C.L.; Correra, R.M.; Winyard, P.J.; Harris, P.C.; et al. Spatiotemporal dynamics and heterogeneity of renal lymphatics in mammalian development and cystic kidney disease. eLife 2019, 8, e48183. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Dimke, H.; Sparks, M.A.; Thomson, B.R.; Frische, S.; Coffman, T.M.; Quaggin, S.E. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J. Am. Soc. Nephrol. 2015, 26, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef]
- Ferda, J.; Hora, M.; Hes, O.; Ferdova, E.; Kreuzberg, B. Assessment of the kidney tumor vascular supply by 2-phase MDCT-angiography. Eur. J. Radiol. 2007, 62, 295–301. [Google Scholar] [CrossRef]
- Qian, C.N.; Huang, D.; Wondergem, B.; Teh, B.T. Complexity of tumor vasculature in clear cell renal cell carcinoma. Cancer 2009, 115, 2282–2289. [Google Scholar] [CrossRef]
- Anderson, H.; Yap, J.T.; Wells, P.; Miller, M.P.; Propper, D.; Price, P.; Harris, A.L. Measurement of renal tumour and normal tissue perfusion using positron emission tomography in a phase II clinical trial of razoxane. Br. J. Cancer 2003, 89, 262–267. [Google Scholar] [CrossRef]
- Schraml, P.; Athelogou, M.; Hermanns, T.; Huss, R.; Moch, H. Specific immune cell and lymphatic vessel signatures identified by image analysis in renal cancer. Mod. Pathol. 2019, 32, 1042–1052. [Google Scholar] [CrossRef]
- Moch, H.; Presti, J.C., Jr.; Sauter, G.; Buchholz, N.; Jordan, P.; Mihatsch, M.J.; Waldman, F.M. Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res. 1996, 56, 27–30. [Google Scholar] [PubMed]
- Gronwald, J.; Storkel, S.; Holtgreve-Grez, H.; Hadaczek, P.; Brinkschmidt, C.; Jauch, A.; Lubinski, J.; Cremer, T. Comparison of DNA gains and losses in primary renal clear cell carcinomas and metastatic sites: Importance of 1q and 3p copy number changes in metastatic events. Cancer Res. 1997, 57, 481–487. [Google Scholar] [PubMed]
- Schullerus, D.; Herbers, J.; Chudek, J.; Kanamaru, H.; Kovacs, G. Loss of heterozygosity at chromosomes 8p, 9p, and 14q is associated with stage and grade of non-papillary renal cell carcinomas. J. Pathol. 1997, 183, 151–155. [Google Scholar] [CrossRef]
- Reutzel, D.; Mende, M.; Naumann, S.; Storkel, S.; Brenner, W.; Zabel, B.; Decker, J. Genomic imbalances in 61 renal cancers from the proximal tubulus detected by comparative genomic hybridization. Cytogenet. Cell Genet. 2001, 93, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Rigola, M.A.; Casadevall, C.; Bernues, M.; Caballin, M.R.; Fuster, C.; Gelabert, A.; Egozcue, J.; Miro, R. Analysis of kidney tumors by comparative genomic hybridization and conventional cytogenetics. Cancer Genet. Cytogenet. 2002, 137, 49–53. [Google Scholar] [CrossRef]
- Alimov, A.; Sundelin, B.; Bergerheim, U.; Pavlenko, M.; Pisa, P.; Zetterberg, A.; Larsson, C.; Lagercrantz, S. Molecular cytogenetic characterization shows higher genetic homogeneity in conventional renal cell carcinoma compared to other kidney cancers. Int. J. Oncol. 2004, 25, 955–960. [Google Scholar] [PubMed]
- Sanjmyatav, J.; Schubert, J.; Junker, K. Comparative study of renal cell carcinoma by CGH, multicolor-FISH and conventional cytogenic banding analysis. Oncol. Rep. 2005, 14, 1183–1187. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Matsuura, K.; Karnan, S.; Tagawa, H.; Nakada, C.; Tanigawa, M.; Tsukamoto, Y.; Uchida, T.; Kashima, K.; Akizuki, S.; et al. High-resolution analysis of DNA copy number alterations and gene expression in renal clear cell carcinoma. J. Pathol. 2007, 213, 392–401. [Google Scholar] [CrossRef]
- Chen, M.; Ye, Y.; Yang, H.; Tamboli, P.; Matin, S.; Tannir, N.M.; Wood, C.G.; Gu, J.; Wu, X. Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int. J. Cancer 2009, 125, 2342–2348. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Senbabaoglu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; Shuch, B.; Micevic, G.; De Velasco, G.; Shinbrot, E.; et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016, 14, 2476–2489. [Google Scholar] [CrossRef]
- Clifford, S.C.; Walsh, S.; Hewson, K.; Green, E.K.; Brinke, A.; Green, P.M.; Gianelli, F.; Eng, C.; Maher, E.R. Genomic organization and chromosomal localization of the human CUL2 gene and the role of von Hippel-Lindau tumor suppressor-binding protein (CUL2 and VBP1) mutation and loss in renal-cell carcinoma development. Genes Chromosomes Cancer 1999, 26, 20–28. [Google Scholar] [CrossRef]
- Buchholz, B.; Eckardt, K.U. Role of oxygen and the HIF-pathway in polycystic kidney disease. Cell. Signal. 2020, 69, 109524. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Mandriota, S.; Jurgensen, J.S.; Wiesener, M.S.; Horstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.U. Expression of hypoxia-inducible factor-1 and -2 in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Schodel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT pathway in renal cell carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Brugarolas, J. Molecular genetics of clear-cell renal cell carcinoma. J. Am. Soc. Clin. Oncol. 2014, 32, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 2018, 173, 595–610.e11. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Chen, Y.B.; Wren, J.; Gonen, M.; Abdel-Wahab, O.; Heguy, A.; Liu, H.; Takeda, S.; Tickoo, S.K.; Reuter, V.E.; et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur. Urol. 2013, 63, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Testa, J.R.; Yang, H. The roles of chromatin-remodelers and epigenetic modifiers in kidney cancer. Cancer Genet. 2015, 208, 206–214. [Google Scholar] [CrossRef]
- Kim, E.; Zschiedrich, S. Renal cell carcinoma in von Hippel-Lindau disease-From tumor genetics to novel therapeutic strategies. Front. Pediatr. 2018, 6, 16. [Google Scholar] [CrossRef]
- Banumathy, G.; Cairns, P. Signaling pathways in renal cell carcinoma. Cancer Biol. Ther. 2010, 10, 658–664. [Google Scholar] [CrossRef]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Haase, V. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006, 69, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jee, K.; Kim, D.; Koh, H.; Chung, J. Cyclic AMP inhibits Akt activity by blocking the membrane localization of PDK1. J. Biol. Chem. 2001, 276, 12864–12870. [Google Scholar] [CrossRef]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef]
- Kosti, A.; de Araujo, P.R.; Li, W.Q.; Guardia, G.D.A.; Chiou, J.; Yi, C.; Ray, D.; Meliso, F.; Li, Y.M.; Delambre, T.; et al. The RNA-binding protein SERBP1 functions as a novel oncogenic factor in glioblastoma by bridging cancer metabolism and epigenetic regulation. Genome Biol. 2020, 21, 195. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Wallace, D.P.; Magenheimer, B.S.; Hempson, S.J.; Grantham, J.J.; Calvet, J.P. Calcium restriction allows cAMP cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004, 279, 40419–40430. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell. Biol. 2009, 29, 2359–2371. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1a expression and function by the mammalian target or rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1a (HIF-1a) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef]
- Trudel, M.; Lanoix, J.; Barisoni, L.; Blouin, M.J.; Desforges, M.; L’Italien, C.; D’Agati, V. C-MYC-induced apoptosis in polycystic kidney disease is Bcl-2 and p53 independent. J. Exp. Med. 1997, 186, 1873–1884. [Google Scholar] [CrossRef]
- Couillard, M.; Guillaume, R.; Tanji, N.; D’Agati, V.; Trudel, M. c-myc-induced apoptosis in polycystic kidney disease is independent of FasL/Fas interaction. Cancer Res. 2002, 62, 2210–2214. [Google Scholar]
- Wallace, D.P. Cyclic AMP-mediated cyst expansion. Biochim. Biophys. Acta 2011, 1812, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Shuch, B.; Vocke, C.D.; Metwalli, A.R.; Bratslavsky, G.; Middelton, L.; Yang, Y.; Wei, M.H.; Pautler, S.E.; Peterson, J.; et al. Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef]
- Warburg, O.; Posener, K.; Negelein, E. Metabolism of the carcinoma cell. Biochem. Z. 1924, 152, 309. [Google Scholar] [CrossRef]
- Yin, C.; Qie, S.; Sang, N. Carbon source metabolism and its regulation in cancer cells. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Okuda, H.; Kobayashi, A.; Watabe, K. Metabolic genes in cancer: Their roles in tumor progression and clinical implications. Biochim. Biophys. Acta 2010, 1805, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef]
- Grabmaier, K.; Weijert, M.C.; Verhaegh, G.W.; Schalken, J.A.; Oosterwijk, E. Strict regulation of CAIXG250/MN by HIF-1α in clear cell renal cell carcinoma. Oncogene 2004, 23, 5624–5631. [Google Scholar] [CrossRef]
- Drusian, L.; Nigro, E.A.; Mannella, V.; Pagliarini, R.; Pema, M.; Costa, A.S.; Benigni, F.; Larcher, A.; Chiaravalli, M.; Gaude, E.; et al. MTORC1 upregulation leads to accumulation of the oncometabolite fumarate in a mouse model of renal cell carcinoma. Cell Rep. 2018, 24, 1093–1104. [Google Scholar] [CrossRef]
- Lai, S.; Jiao, B.; Wang, X.; Xu, X.; Zhang, M.; Diao, T.; Zhang, G. Renal cell carcinoma originating in the free wall of simple renal cyst: Two unusual case reports with literature review. Medicine 2019, 98, e15249. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis ameliorates renal cyst growth in polycystic kidney disease. Cell Metab. 2019, 30, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Firth, J.D.; Ratcliffe, P.J. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct cis-acting sequences. J. Biol. Chem. 1995, 270, 29083–29089. [Google Scholar] [CrossRef]
- Kraus, A.; Peters, D.J.M.; Klanke, B.; Weidemann, A.; Willam, C.; Schley, G.; Kunzelmann, K.; Eckardt, K.U.; Buchholz, B. HIF-1α promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 2018, 94, 887–899. [Google Scholar] [CrossRef]
- Bernhardt, W.M.; Wiesener, M.S.; Weidemann, A.; Schmitt, R.; Weichert, W.; Lechler, P.; Campean, V.; Ong, A.C.M.; Willam, C.; Gretz, N.; et al. Involvement of hypoxia-inducible transcription factors in polycystic kidney disease. Am. J. Pathol. 2007, 170, 830–842. [Google Scholar] [CrossRef]
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Jantsch, J.; Kunzelmann, K.; et al. Hypoxia-inducible factor-1alpha causes renal cyst expansion through calcium-activated chloride secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, I.; Taheri, S.; Eroglu, E.; Sener, E.F.; Zararsiz, G.; Uzun, I.; Tufan, E.; Mehmetbeyoglu, E.; Bayramov, K.K.; Sipahioglu, M.H.; et al. Systemic succinate, hypoxia-inducible Factor-1 alpha, and IL-1β gene expression in autosomal dominant polycystic kidney disease with and without hypertension. Cardiorenal Med. 2019, 9, 370–381. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Tulk, B.M.; Edwards, J.C. NCC27, a homolog of intracellular CI-channel p64, is expressed in brush border of renal proximal tubule. Am. J. Physiol. 1998, 274, 1140–1149. [Google Scholar]
- Nesiu, A.; Cimpean, A.M.; Ceausu, R.A.; Adile, A.; Ioiart, I.; Porta, C.; Mazzanti, M.; Camerota, T.C.; Raica, M. Intracellular chloride ion channel protein-1 expression in clear cell renal cell carcinoma. Cancer Genom. Proteom. 2019, 16, 299–307. [Google Scholar] [CrossRef]
- Barbieri, F.; Wurth, R.; Pattarozzi, A.; Verduci, I.; Mazzola, C.; Cattaneo, C.; Tonelli, M.; Solari, A.; Bajetto, A.; Daga, A.; et al. Inhibition of chloride intracellular channel I (CLIC1) as biguanide class-effect to impair human glioblastoma stem cell viability. Front. Pharmacol. 2018, 9, 899. [Google Scholar] [CrossRef] [PubMed]
- Littler, D.R.; Harrop, S.J.; Fairlie, W.D.; Brown, L.J.; Pankhurst, G.J.; Pankhurst, S.; DeMaere, M.Z.; Campbell, T.J.; Bauskin, A.R.; Tonini, R.; et al. The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J. Biol. Chem. 2004, 279, 9298–9305. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, S.C.; Howell, M.W.; Cordina, N.M.; Littler, D.R.; Breit, S.N.; Curmi, P.M.; Brown, L.J. Oxidation promotes insertion of the CLIC1 chloride intracellular channel into the membrane. Eur. Biophys. J. 2009, 39, 129–138. [Google Scholar] [CrossRef]
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Bescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655. [Google Scholar] [CrossRef]
- Wang, W.; Xu, X.; Wang, W.; Shao, W.; Li, L.; Yin, W.; Xiu, L.; Mo, M.; Zhao, J.; He, Q.; et al. The expression and clinical significance of CLIC1 and HSP27 in lung adenocarcinoma. Tumour Biol. 2011, 32, 1199–1208. [Google Scholar] [CrossRef]
- Gurski, L.A.; Knowles, L.M.; Basse, P.H.; Maranchie, J.K.; Watkins, S.C.; Pilch, J. Relocation of CLIC1 promotes tumor cell invasion and colonization of fibrin. Mol. Cancer Res. 2014, 13, 273–280. [Google Scholar] [CrossRef]
- Hanaoka, K.; Devuyst, O.; Schwiebert, E.M.; Wilson, P.D.; Guggino, W.B. A role for CFTR in human autosomal dominant polycystic kidney disease. Am. J. Physiol. Cell Physiol. 1996, 270, C386–C399. [Google Scholar] [CrossRef]
- Xia, X.; Wang, J.; Liu, Y.; Yue, M. Lower cystic fibrosis transmembrane conductance regulator (CFTR) promotes the proliferation and migration of endometrial carcinoma. Med. Sci. Monit. 2017, 23, 966–974. [Google Scholar] [CrossRef]
- Pinto, C.S.; Reif, G.A.; Nivens, E.; White, C.; Wallace, D.P. Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am. J. Physiol. Ren. Physiol. 2012, 303, F1412–F1424. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Patel, B.; Choyke, P.L.; Lubensky, I.A.; Vocke, C.D.; Harris, C.; Venzon, D.; Burtis, W.J.; Linehan, W.M. Hypercalcemia in patients with metastatic renal cell carcinoma: Effect of nephrectomy and metabolic evaluation. J. Urol. 1997, 158, 733–739. [Google Scholar] [CrossRef]
- Mangolini, A.; de Stephanis, L.; Aguiari, G. Role of calcium in polycystic kidney disease: From signaling to pathology. World J. Nephrol. 2016, 5, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Sussman, C.R.; Wang, X.; Harris, P.C.; Torres, V.E. Vasopressin and disruption of calcium signaling in polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 2006, 17, 178–187. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef]
- Basten, S.G.; Willekers, S.; Vermaat, J.S.; Slaats, G.G.; Voest, E.E.; van Diest, P.J.; Giles, R.H. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2013, 2, 2. [Google Scholar] [CrossRef]
- Kathem, S.H.; Mohieldin, A.M.; Nauli, S.M. The roles of primary cilia in polycystic kidney disease. AIMS Mol. Sci. 2014, 1, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.K. Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1381–1388. [Google Scholar] [CrossRef]
- Frew, I.J.; Thoma, C.R.; Georgiev, S.; Minola, A.; Hitz, M.; Montani, M.; Moch, H.; Krek, W. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008, 27, 1747–1757. [Google Scholar] [CrossRef]
- Dere, R.; Perkins, A.L.; Bawa-Khalfe, T.; Jonasch, D.; Walker, C.L. Beta-catenin links von hippel-lindau to aurora kinase A and loss of primary cilia in renal cell carcinoma. J. Am. Soc. Nephrol. 2015, 26, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, E.W.; Walz, G.; Benzing, T. Von hippel-lindau: A tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res. 2007, 67, 4537–4540. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Seeger-Nukpezah, T.; Golemis, E.A. The role of the cilium in normal and abnormal cell cycles: Emphasis on renal cystic pathologies. Cell. Mol. Life Sci. 2013, 70, 1849–1874. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Harten, S.K.; Tran, M.G.; Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. 2006, 17, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Aurora A kinase activity influences calcium signaling in kidney cells. J. Am. Soc. Nephrol. 2011, 11, 814–827. [Google Scholar]
- Nikonova, A.S.; Plotnikova, O.V.; Serzhanova, V.; Efimov, A.; Bogush, I.; Cai, K.Q.; Hensley, H.H.; Egleston, B.L.; Klein-Szanto, A.; Seeger-Nukpezah, T.; et al. Nedd9 restrains renal cystogenesis in Pkd1−/− mice. Proc. Natl. Acad. Sci. USA 2014, 111, 12859–12864. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.S.; Deneka, A.Y.; Eckman, L.; Kopp, M.C.; Hensley, H.H.; Egleston, B.L.; Golemis, E.A. Opposing effects of inhibitors of Aurora-A and EGFR in autosomal-dominant polycystic kidney disease. Front. Oncol. 2015, 5, 228. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Gleeson, J.G. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; San Agustin, J.T.; Follit, J.A.; Rosenbaum, J.L.; Witman, G.B. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 2002, 12, R378–R380. [Google Scholar] [CrossRef]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef]
- Moyer, J.H.; Lee-Tischler, M.J.; Kwon, H.Y.; Schrick, J.J.; Avner, E.D.; Sweeney, W.E.; Godfrey, V.L.; Cacheiro, N.L.; Wilkinson, J.E.; Woychik, R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994, 264, 1329–1333. [Google Scholar] [CrossRef]
- Pazour, G.J.; Baker, S.A.; Deane, J.A.; Cole, D.G.; Dickert, B.L.; Rosenbaum, J.L.; Witman, G.B.; Besharse, J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002, 157, 103–113. [Google Scholar] [CrossRef]
- Kramer-Zucker, A.G.; Olale, F.; Haycraft, C.J.; Yoder, B.K.; Schier, A.F.; Drummond, I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 2005, 132, 1907–1921. [Google Scholar] [CrossRef]
- Obara, T.; Mangos, S.; Liu, Y.; Zhao, J.; Wiessner, S.; Kramer-Zucker, A.G.; Olale, F.; Schier, A.F.; Drummond, I.A. Polycystin-2 immunolocalization and function in zebrafish. J. Am. Soc. Nephrol. 2006, 17, 2706–2718. [Google Scholar] [CrossRef]
- Mangos, S.; Lam, P.Y.; Zhao, A.; Liu, Y.; Mudumana, S.; Vasilyev, A.; Liu, A.; Drummond, I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model Mech. 2010, 3, 354–365. [Google Scholar] [CrossRef]
- Gamberi, C.; Hipfner, D.R.; Trudel, M.; Lubell, W.D. Bicaudal C mutation causes myc and TOR pathway up-regulation and polycystic kidney disease-like phenotypes in drosophila. PLoS Genet. 2017, 13, e1006694. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Sinha, S.; Dwivedi, N.; Tao, S.; Jamadar, A.; Kakade, V.R.; O’Neil, M.; Weiss, R.H.; Enders, J.; Calvet, J.P.; Thomas, S.M.; et al. Targeting the vasopressin type-2 receptor for renal cell carcinoma therapy. Oncogene 2020, 39, 1231–1245. [Google Scholar] [CrossRef]
- Sherpa, R.T.; Mohieldin, A.M.; Pala, R.; Wachten, D.; Ostrom, R.S.; Nauli, S.M. Sensory primary cilium is a responsive cAMP microdomain in renal epithelia. Sci. Rep. 2019, 9, 6523. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Kulkarni, P.; Majid, S.; Shahryari, V.; Hashimoto, Y.; Bhat, N.S.; Shiina, M.; Deng, G.; Saini, S.; Tabatabai, Z.L.; et al. MicroRNA-203 inhibits long noncoding RNA hotair and regulates tumorigenesis through epithelial-to-mesenchymal transition pathway in renal cell carcinoma. Mol. Cancer Ther. 2018, 17, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Huang, W.; Weng, G.; Cui, P.; Liang, H.; Li, Y. LncRNA PVT1 promotes proliferation, invasion and epithelial–mesenchymal transition of renal cell carcinoma cells through downregulation of miR-16-5p. OncoTargets Ther. 2019, 12, 2563–2575. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chen, Q.; Liu, X.I.N.; Ma, M.; Yang, X.; Gong, B.; Sun, T.; Chen, J. Silencing of the long non-coding RNA GHET1 inhibits cell proliferation and migration of renal cell carcinoma through epithelial-mesenchymal transition. Oncol. Lett. 2019, 17, 3173–3180. [Google Scholar]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; Van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef]
- Patel, V.; Hajarnis, S.; Williams, D.; Hunter, R.; Huynh, D.; Igarashi, P. MicroRNAs regulate renal tubule maturation through modulation of Pkd1. J. Am. Soc. Nephrol. 2012, 23, 1941–1948. [Google Scholar] [CrossRef]
- Gilyazova, I.R.; Klimentova, E.A.; Bulygin, K.V.; Izmailov, A.A.; Bermisheva, M.A.; Galimova, E.F.; Safiullin, R.I.; Galimov, S.N.; Pavlov, V.N.; Khusnutdinova, E.K. MicroRNA-200 family expression analysis in metastatic clear cell renal cell carcinoma patients. Cancer Gene Ther. 2020, 27, 768–772. [Google Scholar] [CrossRef]
- Jiang, J.; Yi, B.; Ding, S.; Sun, J.; Cao, W.; Liu, M. Demethylation drug 5-AZA-2′-deoxycytidine-induced upregulation of miR-200c inhibits the migration, invasion and epithelial-mesenchymal transition of clear cell renal cell carcinoma in vitro. Oncol. Lett. 2016, 11, 3167–3172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hajarnis, S.; Lakhia, R.; Yheskel, M.; Williams, D.; Sorourian, M.; Liu, X.; Aboudehen, K.; Zhang, S.; Kersjes, K.; Galasso, R.; et al. MicroRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.F.; Mankaruos, M.; Scorilas, A.; Youssef, Y.; Girgis, A.; Mossad, S.; Metias, S.; Rofael, Y.; Honey, R.J.; Stewart, R.; et al. The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J. Urol. 2010, 183, 743–751. [Google Scholar] [CrossRef]
- Faragalla, H.; Youssef, Y.M.; Scorilas, A.; Khalil, B.; White, N.M.A.; Mejia-Guerrero, S.; Khella, H.; Jewett, M.A.S.; Evans, A.; Lichner, Z.; et al. The clinical utility of miR-21 as a diagnostic and prognostic marker for renal cell carcinoma. J. Mol. Diagn. 2012, 14, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Hajarnis, S.; Williams, D.; Aboudehen, K.; Yheskel, M.; Xing, C.; Hatley, M.E.; Torres, V.E.; Wallace, D.P.; Patel, V. MicroRNA-21 aggravates cyst growth in a model of polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wang, X.; Yang, X.; Ji, J.; Wang, Q.; Yue, X.; Dong, Z. Long non-coding RNA DUXAP8 enhances renal cell carcinoma progression via downregulating miR-126. Med. Sci. Monit. 2018, 24, 7340–7347. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xu, R.; Xu, X.; Zhou, Y.; Cui, L.; He, X. Downregulation of lncRNA CASC2 by microRNA-21 increases the proliferation and migration of renal cell carcinoma cells. Mol. Med. Rep. 2016, 14, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Aboudehen, K.; Farahani, S.; Kanchwala, M.; Chan, S.C.; Avdulov, S.; Mickelson, A.; Lee, D.; Gearhart, M.D.; Patel, V.; Xing, C.; et al. Long noncoding RNA Hoxb3os is dysregulated in autosomal dominant polycystic kidney disease and regulates mTOR signaling. J. Biol. Chem. 2018, 293, 9388–9398. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Mytsyk, Y.; Dosenko, V.; Skrzypczyk, M.A.; Borys, Y.; Diychuk, Y.; Kucher, A.; Kowalskyy, V.; Pasichnyk, S.; Mytsyk, O.; Manyuk, L. Potential clinical applications of microRNAs as biomarkers for renal cell Carcinoma. Cent. Eur. J. Urol. 2018, 71, 295–303. [Google Scholar]
- Ma, H.; Pan, J.S.; Jin, L.X.; Wu, J.; Ren, Y.D.; Chen, P.; Xiao, C.; Han, J. MicroRNA-17~92 inhibits colorectal cancer progression by targeting angiogenesis. Cancer Lett. 2016, 376, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Quittner-Strom, E.B.; Holland, W.L.; Patel, V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Renal Physiol. 2018, 314, F122–F131. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.; Hajarnis, S.; Patel, V. MicroRNAs and polycystic kidney disease. Drug Discov. Today Dis. Models 2013, 10, e137–e143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, G.M.; Luo, L.; Ding, X.M.; Dong, D.H.; Li, B.; Ma, X.C.; Sun, L.J. MicroRNA-126 inhibits tumor cell invasion and metastasis by downregulating ROCK1 in renal cell carcinoma. Mol. Med. Rep. 2016, 13, 5029–5036. [Google Scholar] [CrossRef][Green Version]
- Pourkarimi, E.; Greiss, S.; Gartner, A. Evidence that CED-9/Bcl2 and CED-4/Apaf-1 localization is not consistent with the current model for C. elegans apoptosis induction. Cell Death Differ. 2012, 19, 406–415. [Google Scholar] [CrossRef]
- Flybase. Available online: flybase.org (accessed on 27 February 2021).
- Schor, I.E.; Bussotti, G.; Maleš, M.; Forneris, M.; Viales, R.R.; Enright, A.J.; Furlong, E.E.M. Non-coding RNA expression, function, and variation during Drosophila embryogenesis. Curr. Biol. 2018, 28, 3547–3561.e9. [Google Scholar] [CrossRef]
- Li, K.; Tian, Y.; Yuan, Y.; Fan, X.; Yang, M.; He, Z.; Yang, D. Insights into the functions of LncRNAs in Drosophila. Int. J. Mol. Sci. 2019, 20, 4646. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Porras Marroquin, J.; Gamberi, C. Modeling renal disease “On the fly”. BioMed Res. Int. 2018, 2018, 5697436. [Google Scholar] [CrossRef]
- Wang, J.; Kean, L.; Yang, J.; Allan, A.K.; Davies, S.A.; Herzyk, P.; Dow, J.A.T. Function-informed transcriptome analysis of Drosophila renal tubule. Genome Biol. 2004, 5, R69. [Google Scholar] [CrossRef]
- Chien, S.; Reiter, L.T.; Bier, E.; Gribskov, M. Homophila: Human disease gene cognates in Drosophila. Nucleic Acids Res. 2002, 30, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, J.; Benoit, P.; Gamberi, C.; Paliouras, M.; Simonelig, M.; Lasko, P. Bicaudal-C recruits CCR4-NOT deadenylase to target mRNAs and regulates oogenesis, cytoskeletal organization, and its own expression. Dev. Cell 2007, 13, 691–704. [Google Scholar] [CrossRef]
- Gamberi, C.; Lasko, P. The Bic-C family of developmental translational regulators. Comp. Funct. Genom. 2012, 2012, 141386. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Chingle, R.; Lubell, W.D.; Gamberi, C. Cyst reduction in a polycystic kidney disease Drosophila model using Smac mimics. Biomedicines 2019, 7, 82. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Selber-Hnatiw, S.; Gamberi, C. Drug discovery and chemical probing in Drosophila. Genome 2020, 64, 147–159. [Google Scholar] [CrossRef]
- Li, L.; Yang, Y.; Xue, L. Regulatory functions of Pax gene family in Drosophila development. Yi Chuan 2010, 32, 115–121. [Google Scholar] [CrossRef]
- Li, C.G.; Chantry, A.; Stayner, C.; Horsfield, J.; Eccles, M.R. SMAD proteins directly suppress PAX2 transcription downstream of transforming growth factor-beta 1 (TGF-β1) signalling in renal cell carcinoma. Oncotarget 2018, 9, 26852–26867. [Google Scholar]
- Zhang, H.; Stallock, J.P.; Ng, J.C.; Reinhard, C.; Neufeld, T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000, 14, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Myc function in Drosophila. Cold Spring Harb. Perspect. Med. 2013, 3, a014324. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Neufeld, T.P.; Pan, D. Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev. Biol. 2000, 221, 404–418. [Google Scholar] [CrossRef]
- Bader, H.L.; Hsu, T. Systemic VHL gene functions and the VHL disease. FEBS Lett. 2012, 586, 1562–1569. [Google Scholar] [CrossRef]
- Adryan, B.; Decker, H.J.; Papas, T.S.; Hsu, T. Tracheal development and the von Hippel-Lindau tumor suppressor homolog in Drosophila. Oncogene 2000, 19, 2803–2811. [Google Scholar] [CrossRef]
- Hsouna, A.; Nallamothu, G.; Kose, N.; Guinea, M.; Dammai, V.; Hsu, T. Drosophila von Hippel-Lindau tumor suppressor gene function in epithelial tubule morphogenesis. Mol. Cell. Biol. 2010, 30, 3779–3794. [Google Scholar] [CrossRef]
- Hsu, T. Complex cellular functions of the von Hippel-Lindau tumor suppressor gene: Insights from model organisms. Oncogene 2012, 31, 2247–2257. [Google Scholar] [CrossRef]
- Ghabrial, A.; Luschnig, S.; Metzstein, M.M.; Krasnow, M.A. Branching morphogenesis of the Drosophila tracheal system. Annu. Rev. Cell Dev. Biol. 2003, 19, 623–647. [Google Scholar] [CrossRef] [PubMed]
- Dammai, V.; Adryan, B.; Lavenburg, K.R.; Hsu, T. Drosophila awd, the homolog of human nm23, regulates FGF receptor levels and functions synergistically with shi/dynamin during tracheal development. Genes Dev. 2003, 17, 2812–2824. [Google Scholar] [CrossRef] [PubMed]
- Duchi, S.; Fagnocchi, L.; Cavaliere, V.; Hsouna, A.; Gargiulo, G.; Hsu, T. Drosophila VHL tumor-suppressor gene regulates epithelial morphogenesis by promoting microtubule and aPKC stability. Development 2010, 137, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Ignesti, M.; Andrenacci, D.; Fischer, B.; Cavaliere, V.; Gargiulo, G. Comparative expression profiling of wild type Drosophila Malpighian tubules and von Hippel-Lindau haploinsufficient mutant. Front. Physiol. 2019, 10, 619. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Caretti, E.; Tricarico, R.; Devarajan, K.; Cheung, M.; Sementino, E.; Menges, C.W.; Nicolas, E.; Vanderveer, L.A.; Howard, S.; et al. Haploinsufficiency in tumor predisposition syndromes: Altered genomic transcription in morphologically normal cells heterozygous for VHL or TSC mutation. Oncotarget 2017, 8, 17628–17642. [Google Scholar] [CrossRef]
- Peri, S.; Devarajan, K.; Yang, D.H.; Knudson, A.G.; Balachandran, S. Meta-analysis identifies NF-B as a therapeutic target in renal cancer. PLoS ONE 2013, 8, e76746. [Google Scholar] [CrossRef]
- Bangi, E.; Ang, C.; Smibert, P.; Uzilov, A.V.; Teague, A.G.; Antipin, Y.; Chen, R.; Hecht, C.; Gruszczynski, N.; Yon, W.J.; et al. A personalized platform identifies trametinib plus zoledronate for a patient with KRAS-mutant metastatic colorectal cancer. Sci. Adv. 2019, 5, eaav6528. [Google Scholar] [CrossRef]
- Mortimer, N.T.; Moberg, K.H. Regulation of Drosophila embryonic tracheogenesis by dVHL and hypoxia. Dev. Biol. 2009, 329, 294–305. [Google Scholar] [CrossRef]
- Centanin, L.; Dekanty, A.; Romero, N.; Irisarri, M.; Gorr, T.A.; Wappner, P. Cell autonomy of HIF effects in Drosophila: Tracheal cells sense hypoxia and induce terminal branch sprouting. Dev. Cell 2008, 14, 547–558. [Google Scholar] [CrossRef]
- Li, Y.; Padmanabha, D.; Gentile, L.B.; Dumur, C.; Beckstead, R.B.; Baker, K.D. HIF- and non-HIF-regulated hypoxic responses require the estrogen-related in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003230. [Google Scholar] [CrossRef] [PubMed]
- Sonnenfeld, M.; Ward, M.; Nystrom, G.; Mosher, J.; Stahl, S.; Crews, S. The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development 1997, 124, 4571–4582. [Google Scholar]
- Mukherjee, T.; Kim, W.S.; Mandal, L.; Banerjee, U. Interaction between Notch and Hif-in development and survival of Drosophila blood cells. Science 2011, 332, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.K.; Keyes, L.; Johnson, E.; Heller, J.; Ryner, L.; Karim, F.; Krasnow, M.A. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 2002, 108, 865–876. [Google Scholar] [CrossRef]
- Igaki, T. Correcting developmental errors by apoptosis: Lessons from Drosophila JNK signaling. Apoptosis 2009, 14, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Ratheesh, A.; Belyaeva, V.; Siekhaus, D.E. Drosophila immune cell migration and adhesion during embryonic development and larval immune responses. Curr. Opin. Cell Biol. 2015, 36, 71–79. [Google Scholar] [CrossRef]
- Read, R.D.; Cavenee, W.K.; Furnari, F.B.; Thomas, J.B. A Drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 2009, 5, e1000374. [Google Scholar] [CrossRef] [PubMed]
- Buchon, N.; Broderick, N.A.; Kuraishi, T.; Lemaitre, B. Drosophila EGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC Biol. 2010, 8, 152. [Google Scholar] [CrossRef]
- Volkenhoff, A.; Hirrlinger, J.; Kappel, J.M.; Klambt, C.; Schirmeier, S. Live imaging using a FRET glucose sensor reveals glucose delivery to all cell types in the Drosophila brain. J. Insect Physiol. 2018, 106, 55–64. [Google Scholar] [CrossRef]
- Mohrmann, L.; Langenberg, K.; Krijgsveld, J.; Kal, A.J.; Heck, A.J.R.; Verrijzer, C.P. Differential targeting of two distinct SWI/SNF-related Drosophila chromatin-remodeling complexes. Mol. Cell Biol. 2004, 24, 3077–3088. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Muller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Foglizzo, M.; Middleton, A.J.; Burgess, A.E.; Crowther, J.M.; Dobson, R.C.J.; Murphy, J.M.; Day, C.L.; Mace, P.D. A bidentate Polycomb Repressive-Deubiquitinase complex is required for efficient activity on nucleosomes. Nat. Commun. 2018, 9, 3932. [Google Scholar] [CrossRef]
- Wang, C.I.; Alekseyenko, A.A.; LeRoy, G.; Elia, A.E.H.; Gorchakov, A.A.; Britton, L.M.P.; Elledge, S.J.; Kharchenko, P.V.; Garcia, B.A.; Kuroda, M.I. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat. Struct. Mol. Biol. 2013, 20, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Oldham, S.; Montagne, J.; Radimerski, T.; Thomas, G.; Hafen, E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000, 14, 2689–2694. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila myc regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef]
- Goberdhan, D.C.I.; Paricio, N.; Goodman, E.C.; Miodzik, M.; Wilson, C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 1999, 13, 3244–3258. [Google Scholar] [CrossRef]
- Huang, H.; Potter, C.J.; Tao, W.; Li, D.M.; Brogiolo, W.; Hafen, E.; Sun, H.; Xu, T. PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development 1999, 126, 5365–5372. [Google Scholar]
- Yamada, Y.; Davis, K.D.; Coffman, C.R. Programmed cell death of primordial germ cells in Drosophila is regulated by p53 and the Outsiders monocarboxylate transporter. Development 2008, 135, 207–216. [Google Scholar] [CrossRef]
- Fan, Y.; Lee, T.V.; Xu, D.; Chen, Z.; Lamblin, A.F.; Steller, H.; Bergmann, A. Dual roles of Drosophila p53 in cell death and cell differentiation. Cell Death Differ. 2010, 17, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Shlevkov, E.; Morata, G. A dp53/JNK-dependent feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2012, 19, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Rougeot, J.; Renard, M.; Randsholt, N.B.; Peronnet, F.; Mouchel-Vielh, E. The Elongin complex antagonizes the chromatin factor corto for vein versus intervein cell identity in Drosophila wings. PLoS ONE 2013, 8, e77592. [Google Scholar] [CrossRef] [PubMed]
- Minguet, J.; Smith, K.H.; Bramlage, C.P.; Bramlage, P. Targeted therapies for treatment of renal cell carcinoma: Recent advances and future perspectives. Cancer Chemother. Pharmacol. 2015, 76, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin. Cancer Res. 2008, 14, 1286–1290. [Google Scholar] [CrossRef]
- Voss, M.H.; Hakimi, A.A.; Pham, C.G.; Brannon, A.R.; Chen, Y.B.; Cunha, L.F.; Akin, O.; Liu, H.; Takeda, S.; Scott, S.N.; et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin. Cancer Res. 2014, 20, 1955–1964. [Google Scholar] [CrossRef]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef]
- Lim, S.M.; Park, H.S.; Kim, S.; Kim, S.; Ali, S.M.; Greenbowe, J.R.; Yang, I.S.; Kwon, N.J.; Lee, J.L.; Ryu, M.H.; et al. Next-generation sequencing reveals somatic mutations that confer exceptional response to everolimus. Oncotarget 2016, 7, 10547–10556. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wuthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2006, 21, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wahl, P.R.; Le Hir, M.; Wackerle-Men, Y.; Wuthrich, R.P.; Serra, A.L. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press. Res. 2007, 30, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Boletta, A.; Chapman, A.; Gattone, V.; Pei, Y.; Qian, Q.; Wallace, D.P.; Weimbs, T.; Wuthrich, R.P. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1312–1329. [Google Scholar] [CrossRef]
- Zaza, G.; Tomei, P.; Ria, P.; Granata, S.; Boschiero, L.; Lupo, A. Systemic and nonrenal adverse effects occurring in renal transplant patients treated with mTOR inhibitors. Clin. Dev. Immunol. 2013, 2013, 403280. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Granata, S.; Tomei, P.; Masola, V.; Gambaro, G.; Lupo, A. mTOR inhibitors and renal allograft: Yin and Yang. J. Nephrol. 2014, 27, 495–506. [Google Scholar] [CrossRef]
- Rizzo, M.; Porta, C. Sunitinib in the treatment of renal cell carcinoma: An update on recent evidence. Ther. Adv. Urol. 2017, 9, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Facchini, G.; Rossetti, S.; Berretta, M.; Cavaliere, C.; Scagliarini, S.; Vitale, M.G.; Ciccarese, C.; Di Lorenzo, G.; Palesandro, E.; Conteduca, V.; et al. Second line therapy with axitinib after only prior sunitinib in metastatic renal cell cancer: Italian multicenter real world SAX study final results. J. Transl. Med. 2019, 17, 296. [Google Scholar] [CrossRef]
- Rini, B.I. Sorafenib. Exp. Opin. Pharmacother. 2006, 7, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Jäger, D.; Ma, J.H.; Mardiak, J.; Ye, D.W.; Korbenfeld, E.; Zemanova, M.; Ahn, H.; Guo, J.; Leonhartsberger, N.; Stauch, K.; et al. Sorafenib treatment of advanced renal cell carcinoma patients in daily practice: The large international PREDICT study. Clin. Genitourin. Cancer 2015, 13, 156–164.e1. [Google Scholar] [CrossRef]
- Roviello, G.; Corona, S.P.; Bozza, G.; Aieta, M.; Generali, D.; Rodriquenz, M.G.; Mileo, A.M.; Imperatori, M.; Ianza, A.; Conca, R.; et al. Lenvatinib for the treatment of renal cell carcinoma. Expert Opin. Investig. Drugs 2018, 27, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- De Lisi, D.; De Giorgi, U.; Lolli, C.; Schepisi, G.; Conteduca, V.; Menna, C.; Tonini, G.; Santini, D.; Farolfi, A. Lenvatinib in the management of metastatic renal cell carcinoma: A promising combination therapy? Expert Opin. Drug Metab. Toxicol. 2018, 14, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Semrad, T.J.; Groshen, S.; Luo, C.; Pal, S.; Vaishampayan, U.; Joshi, M.; Quinn, D.I.; Mack, P.C.; Gandara, D.R.; Lara, P.N. Randomized phase 2 study of Trebananib (AMG 386) with or without continued anti-vascular endothelial growth factor therapy in patients with renal cell carcinoma who have progressed on Bevacizumab, Pazopanib, Sorafenib, or Sunitinib—Results of NCI/CTEP protocol 9048. Kidney Cancer 2019, 3, 51–61. [Google Scholar] [PubMed]
- Wang, X.; Solban, N.; Khanna, P.; Callea, M.; Song, J.; Alsop, D.C.; Pearsall, R.S.; Atkins, M.B.; Mier, J.W.; Signoretti, S.; et al. Inhibition of ALK1 signaling with dalantercept combined with VEGFR TKI leads to tumor stasis in renal cell carcinoma. Oncotarget 2016, 7, 41857–41869. [Google Scholar] [CrossRef]
- Voss, M.H.; Bhatt, R.S.; Vogelzang, N.J.; Fishman, M.; Alter, R.S.; Rini, B.I.; Beck, J.T.; Joshi, M.; Hauke, R.; Atkins, M.B.; et al. A phase 2, randomized trial evaluating the combination of dalantercept plus axitinib in patients with advanced clear cell renal cell carcinoma. Cancer 2019, 125, 2400–2408. [Google Scholar] [CrossRef]
- Massari, F.; Santoni, M.; Ciccarese, C.; Santini, D.; Alfieri, S.; Martignoni, G.; Brunelli, M.; Piva, F.; Berardi, R.; Montironi, R.; et al. PD-1 blockade therapy in renal cell carcinoma: Current studies and future promises. Cancer Treat. Rev. 2015, 41, 114–121. [Google Scholar] [CrossRef]
- Pardoll, D.M. Blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Thompson, R.H.; Dong, H.; Kwon, E.D. Implications of B7-H1 expression in clear cell carcinoma of the kidney for prognostication and therapy. Clin. Cancer Res. 2007, 13, 709s–715s. [Google Scholar] [CrossRef]
- Callea, M.; Genega, E.M.; Gupta, M.; Cheng, S.; Fay, A.P.; Song, J.; Carvo, I.; Bhatt, R.S.; McDermott, D.F.; Atkins, M.B.; et al. PD-L1 expression in primary clear cell renal cell carcinomas (ccRCCs) and their metastases. J. Clin. Oncol. 2014, 32, 4585. [Google Scholar] [CrossRef]
- Jilaveanu, L.B.; Shuch, B.; Zito, C.R.; Parisi, F.; Barr, M.; Kluger, Y.; Chen, L.; Kluger, H.M. PD-L1 expression in clear cell renal cell carcinoma: An analysis of nephrectomy and sites of metastases. J. Cancer 2014, 5, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Kammerer-Jacquet, S.F.; Deleuze, A.; Saout, J.; Mathieu, R.; Laguerre, B.; Verhoest, G.; Dugay, F.; Belaud-Rotureau, M.A.; Bensalah, K.; Rioux-Leclercq, N. Targeting the PD-1/PD-L1 pathway in renal cell carcinoma. Int. J. Mol. Sci. 2019, 20, 1692. [Google Scholar] [CrossRef] [PubMed]
- Deleuze, A.; Saout, J.; Dugay, F.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Crouzet, L.; Laguerre, B.; Belaud-Rotureau, M.A.; et al. Immunotherapy in renal cell carcinoma: The future is now. Int. J. Mol. Sci. 2020, 21, 2532. [Google Scholar] [CrossRef]
- Chen, D.J.; Huerta, S. Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009, 20, 646–658. [Google Scholar] [CrossRef]
- Van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem. Biophys. Res. Commun. 2003, 304, 487–497. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Datta, P.; Fan, X.J.; Fernandes-Alnemri, T.; Huang, Z.; Alnemri, E.S. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J. Biol. Chem. 2000, 275, 36152–36157. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Hegde, R.; Saleh, A.; Datta, P.; Shiozaki, E.; Chai, J.; Lee, R.A.; Robbins, P.D.; Fernandes-Alnemri, T.; Shi, Y.; et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001, 410, 112–116. [Google Scholar] [CrossRef]
- Lalaoui, N.; Vaux, D.L. Recent advances in understanding inhibitor of apoptosis proteins. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Cong, H.; Xu, L.; Wu, Y.; Qu, Z.; Bian, T.; Zhang, W.; Xing, C.; Zhuang, C. Inhibitor of apoptosis protein (IAP) antagonists in anticancer discovery: Current status and perspectives. J. Med. Chem. 2019, 62, 5750–5772. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Nakanishi, H.; Yamamoto, K.; Li, Y.N.; Matsubara, H.; Mikami, K.; Okihara, K.; Kawauchi, A.; Bonavida, B.; Miki, T. Downregulation of Smac/DIABLO expression in renal cell carcinoma and its prognostic significance. J. Clin. Oncol. 2005, 23, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Mahotka, C.; Heikaus, S.; Shibata, T.; Wethkamp, N.; Liebmann, J.; Suschek, C.V.; Guo, Y.; Gabbert, H.E.; Gerharz, C.D.; et al. Disturbed balance of expression between XIAP and Smac/DIABLO during tumour progression in renal cell carcinomas. Br. J. Cancer 2004, 91, 1349–1357. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kempkensteffen, C.; Hinz, S.; Christoph, F.; Krause, H.; Magheli, A.; Schrader, M.; Schostak, M.; Miller, K.; Weikert, S. Expression levels of the mitochondrial IAP antagonists Smac/DIABLO and Omi/HtrA2 in clear-cell renal cell carcinomas and their prognostic value. J. Cancer Res. Clin. Oncol. 2008, 134, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Millet-Boureima, C. Cyst Reduction in a First-in-Kind Drosophila Model of Polycystic Kidney Disease. Master’s Thesis, Concordia University, Montreal, QC, Canada, 2 December 2019. [Google Scholar]
- Fan, L.X.; Zhou, X.; Sweeney, W.E.; Wallace, D.P.; Avner, E.D.; Grantham, J.J.; Li, X. Smac-mimetic-induced epithelial cell death reduces the growth of renal cysts. J. Am. Soc. Nephrol. 2013, 24, 2010–2022. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Magenheimer, B.S.; Xia, S.; Johnson, T.; Wallace, D.P.; Calvet, J.P.; Li, R. A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat. Med. 2008, 14, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Igaki, T.; Kanda, H.; Yamamoto-Gotom, Y.; Kanuka, H.; Kuranaga, E.; Aigaki, T.; Miura, M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002, 21, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Igaki, T.; Kanuka, H.; Yagi, T.; Miura, M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 2002, 277, 28372–28375. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Yan, M.; Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002, 12, 1263–1268. [Google Scholar] [CrossRef]
- Andersen, D.S.; Colombani, J.; Palmerini, V.; Chakrabandhu, K.; Boone, E.; Rothlisberger, M.; Toggweiler, J.; Basler, J.; Mapelli, M.; Hueber, A.O.; et al. The Drosophila TNF receptor Grindelwald couple loss of cell polarity and neoplastic growth. Nature 2015, 522, 482–486. [Google Scholar] [CrossRef]
- Dosquet, C.; Schaetz, A.; Faucher, C.; Lepage, C.; Wautier, J.L.; Richard, F.; Cabane, J. Tumor necrosis factor-a, interleukin-1b and interleukin-6 in patients with renal cell carcinoma. Eur. J. Cancer 1994, 30, 162–167. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikemoto, S.; Narita, K.; Sugimura, K.; Wada, S.; Yasumoto, R.; Kishimoto, T.; Nakatani, T. Interleukin-6, tumor necrosis factor alpha and interleukin-1beta in patients with renal cell carcinoma. Br. J. Cancer 2002, 86, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Mizuno, R.; Kosaka, T.; Saya, H.; Oya, M.; Okada, Y. Expression of TNF- and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int. J. Cancer 2015, 136, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Chuang, M.J.; Sun, K.H.; Tang, S.J.; Deng, M.W.; Wu, Y.H.; Sung, J.S.; Cha, T.L.; Sun, G.H. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci. 2008, 99, 905–913. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Gaspar do Amaral, F. Melatonin as a hormone: New physiological and clinical insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hu, C.; Zhang, P.; Jiang, H.; Chen, J. Melatonin preconditioning is an effective strategy for mesenchymal stem cell-based therapy for kidney disease. J. Cell Mol. Med. 2019, 24, 25–33. [Google Scholar] [CrossRef]
- Dilman, V.M.; Anisimov, V.N.; Ostroumova, M.N.; Khavinson, V.K.; Morozov, V.G. Increase in lifespan of rats following polypeptide pineal extract treatment. Exp. Pathol. 1979, 17, 539–545. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Mylnikov, S.V.; Oparina, T.I.; Khavinson, V.K. Effect of melatonin and pineal peptide preparation epithalamin on life span and free radical oxidation in Drosophila melanogaster. Mech. Ageing Dev. 1997, 97, 81–91. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Zavarina, N.Y.; Zabezhinski, M.A.; Popovich, I.G.; Zimina, O.A.; Shtylick, A.V.; Arutjunyan, A.V.; Oparina, T.I.; Prokopenko, V.M.; Mikhalski, A.I.; et al. Melatonin increases both life span and tumor incidence in female CBA mice. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B311–B323. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Popovich, I.G.; Zabezhinski, M.A.; Anisimov, S.V.; Venushkin, G.M.; Vinogradova, I.A. Melatonin as antioxidant, geroprotector and anticarcinogen. Biochim. Biophys. Acta 2006, 1757, 573–589. [Google Scholar] [CrossRef]
- Su, S.; Hsieh, M.J.; Yang, W.E.; Chung, W.H.; Reiter, R.J.; Yang, S.F. Cancer metastasis: Mechanisms of inhibition by melatonin. J. Pineal Res. 2017, 62, e12370. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Sharifi, M.; Reiter, R.J.; Davoodabadi, A.; Asemi, Z. Melatonin and non-small cell lung cancer: New insights into signaling pathways. Cancer Cell Int. 2019, 19, 131. [Google Scholar] [CrossRef]
- Reiter, R.J. Mechanisms of cancer inhibition by melatonin. J. Pineal Res. 2004, 37, 213–214. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Zhou, Y.; Meng, X.; Zhang, J.J.; Xu, D.P.; Li, H.B. Melatonin for the prevention and treatment of cancer. Oncotarget 2017, 8, 39896–39921. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a full service and anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Lin, K.C.; Wallace, C.G.; Chen, Y.T.; Yang, C.C.; Leu, S.; Chen, Y.C.; Sun, C.K.; Tsai, T.H.; Chen, Y.L.; et al. Additional benefit of combined therapy with melatonin and apoptotic adipose-derived mesenchymal stem cell against sepsis-induced kidney injury. J. Pineal Res. 2014, 57, 16–32. [Google Scholar] [CrossRef]
- Saberi, K.; Pasbakhsh, P.; Omidi, A.; Borhani-Haghighi, M.; Nekoonam, S.; Omidi, N.; Ghasemi, S.; Kashani, I.R. Melatonin preconditioning of bone-marrow derived mesenchymal stem cells promotes their engraftment and improves renal regeneration in a rat model of chronic kidney disease. J. Mol. Hist. 2019, 50, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin synthesis and function: Evolutionary history in animals and plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Lee, L.M.; Lee, W.J.; Chu, C.Y.; Tan, P.; Yang, Y.C.; Chen, W.Y.; Yang, S.F.; Hsiao, M.; Chien, M.H. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-DNA-binding activity. J. Pineal Res. 2016, 60, 277–290. [Google Scholar] [CrossRef]
- Park, E.J.; Woo, S.M.; Min, K.J.; Kwon, T.K. Transcriptional and post-transcriptional regulation of Bim controls apoptosis in melatonin-treated human renal cancer Caki cells. J. Pineal Res. 2014, 56, 97–106. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Hosseinzadeh, A.; Juybari, K.B.; Mehrzadi, S. Melatonin and urological cancers: A new therapeutic approach. Cancer Cell Int. 2020, 20, 444. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Rozencwaig, R.; Polyak, F.; Gamberi, C. Cyst reduction by melatonin in a novel Drosophila model of polycystic kidney disease. Molecules 2020, 25, 5477. [Google Scholar] [CrossRef]
- Pei, Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol. Med. 2001, 7, 151–156. [Google Scholar] [CrossRef]
- Harris, P.C. What is the role of somatic mutation in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1073–1076. [Google Scholar] [CrossRef]
- Eccles, M.R.; Stayner, C.A. Polycystic kidney disease—Where gene dosage counts. F1000Prime Rep. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Xu, D.; Mei, C. The association between autosomal dominant polycystic kidney disease and cancer. Int. Urol. Nephrol. 2019, 51, 93–100. [Google Scholar] [CrossRef]
- Shim, K.E.; Lee, C.; Kim, J.U.; Choi, G.H.; Kwak, K.M.; Kim, S.H.; Kim, H.; Yoon, J.W.; Shin, T.Y.; Jeong, C.W.; et al. Comprehensive analysis of mutations of renal cell carcinoma in an autosomal dominant polycystic kidney disease patient. Medicine 2020, 99, e20071. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.B. An hereditary tumor in the fruit fly, Drosophila. Cancer Res. 1918, 3, 279–301. [Google Scholar]
- Stark, M.B. A benign tumor that is hereditary in Drosophila. Proc. Natl. Acad. Sci. USA 1919, 5, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.B. An hereditary tumor. J. Exp. Zool. 1919, 27, 509–529. [Google Scholar] [CrossRef]
- Wilson, I.T. Two new hereditary tumors in Drosophila. Genetics 1924, 9, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Eichenlaub, T.; Cohen, S.M. Cancer in Drosophila: Imaginal discs as a model for epithelial tumor formation. Curr. Top. Dev. Biol. 2016, 116, 181–199. [Google Scholar]
- Pagliarini, R.A.; Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Tipping, M.; Perrimon, N. Drosophila as a model for context-dependent tumorigenesis. J. Cell Physiol. 2014, 229, 27–33. [Google Scholar] [PubMed]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef]
- Brumby, A.M.; Richardson, H.E. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef]
- Grifoni, D.; Garoia, F.; Schimanski, C.C.; Schmitz, G.; Laurenti, E.; Galle, P.R.; Pession, A.; Cavicchi, S.; Strand, D. The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 2004, 23, 8688–8694. [Google Scholar] [CrossRef]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef]
- Fahey-Lozano, N.; La Marca, J.E.; Portela, M.; Richardson, H.E. Drosophila models of cell polarity and cell competition in tumourigenesis. Adv. Exp. Med. Biol. 2019, 1167, 37–64. [Google Scholar]
- Eichenlaub, T.; Villadsen, R.; Freitas, F.C.P.; Andrejeva, D.; Aldana, B.I.; Nguyen, H.T.; Petersen, O.W.; Gorodkin, J.; Herranz, H.; Cohen, S.M. Warburg effect metabolism drives neoplasia in a Drosophila genetic model of epithelial cancer. Curr. Biol. 2018, 28, 3220–3228. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Cagan, R. Drosophila as a tool for personalized medicine: A primer. Per. Med. 2010, 7, 621–632. [Google Scholar] [CrossRef]
- Sonoshita, M.; Cagan, R.L. Modeling human cancers in Drosophila. Curr. Top. Dev. Biol. 2017, 121, 287–309. [Google Scholar] [PubMed]
- Bangi, E. A Drosophila based cancer drug discovery framework. Adv. Exp. Med. Biol. 2019, 1167, 237–248. [Google Scholar] [PubMed]





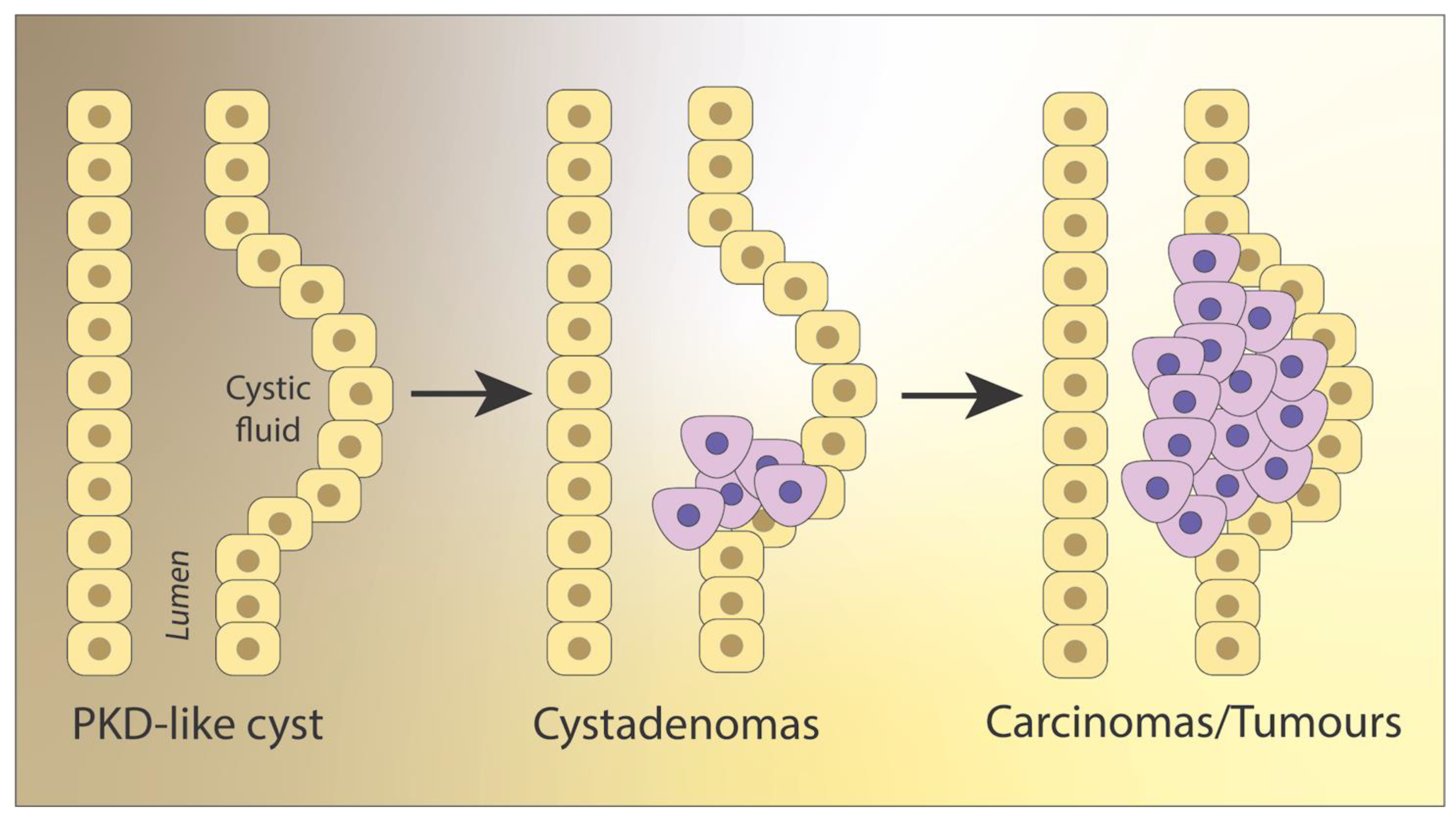{kind=link}
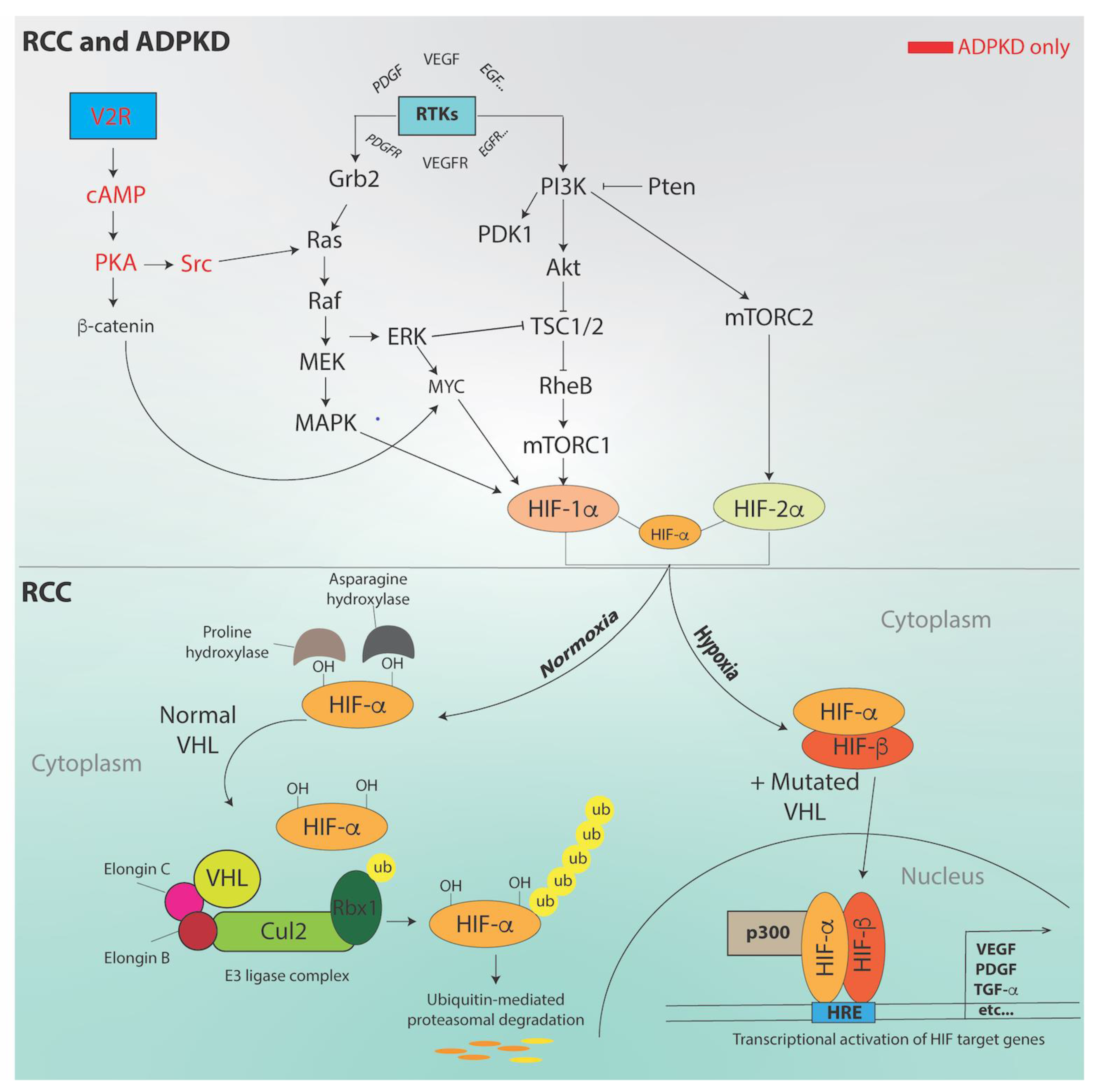{kind=link}
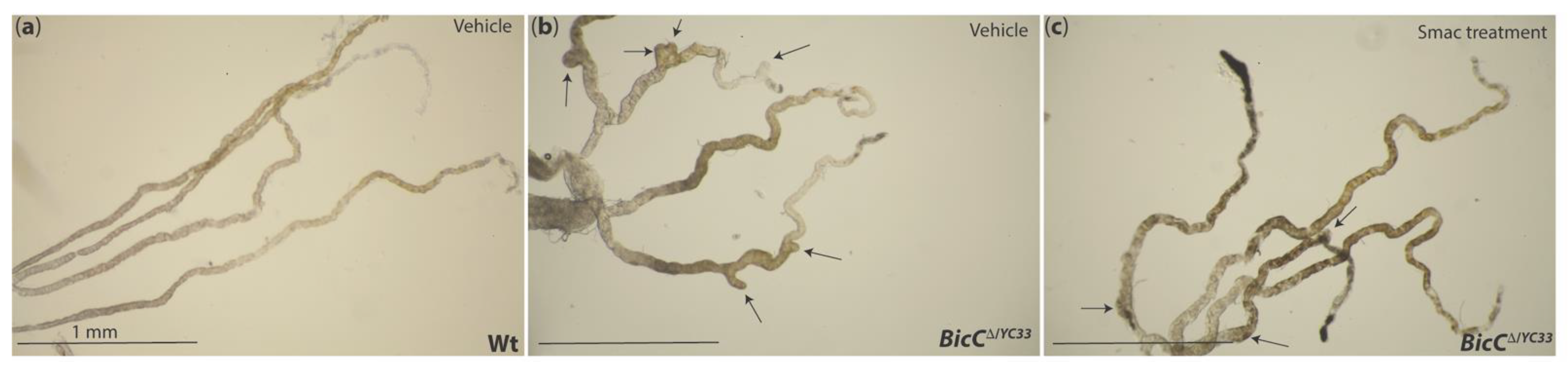{kind=link}
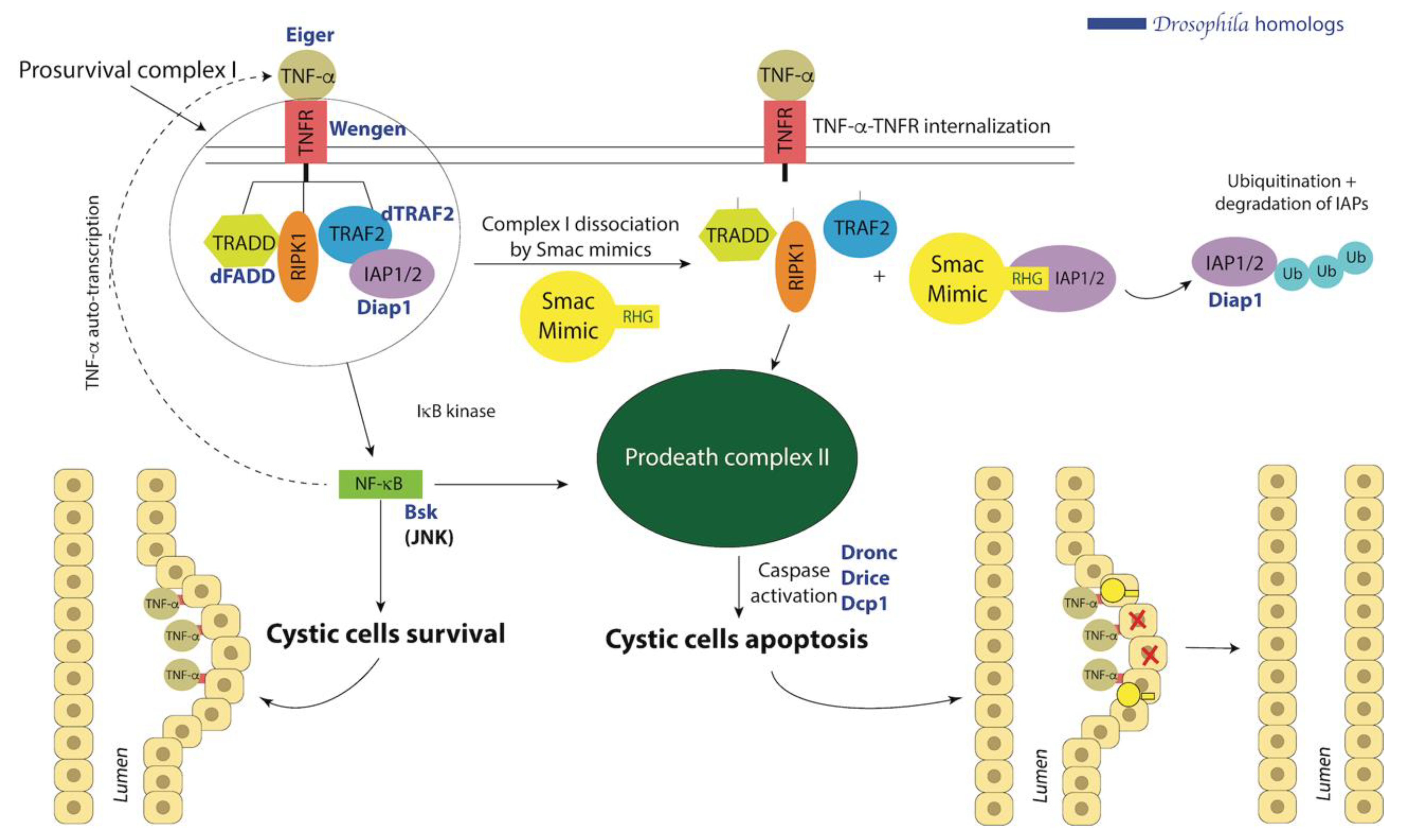{kind=link}
| RCC Subtypes | Prevalence | Description | Gene Mutations | References |
|---|---|---|---|---|
| Clear cell RCC | 70–90% of all RCC 1–3% of all malignant visceral neoplasms | Tumor cells with clear cytoplasm | VHL, PBRM1, SETD2, BAP1, MTOR, TCEB1, PIK3CA, KDM5C. TP53, PTEN | [5] |
| Papillary RCC Type 1 | 10–15% | Papillae lined with one layer of tumor cells with low grade nuclei and partially clear cytoplasm | MET | [5,20,21] |
| Papillary RCC Type 2 | 10–15% | Abundant eosinophilic cytoplasm and large pseudostratified cells with high grade nuclei | CDKN2A, SETD2, NRF2, FH | [20,21,22] |
| Chromophobe | 5–7% | Large cells, polygonal reticulated cytoplasm, distinct cell borders, atypical nuclei with perinuclear halo. Contain fine eosinophilic granules. | TP53, PTEN, TSC1 | [21,23,24,25] |
| Multilocular Cystic RCC (Cystic-solid) | 1–5% | Multiseptated cystic space, cysts lined by cuboidal clear cells or flattened epithelium with septa and clear cytoplasm | GIGYF2, FGFR3, SETD2, BCR, KMT2C, TSC2 | [26,27] |
| Bellini Duct Carcinoma | Less than 1% of all malignant kidney tumors | Malignant tumor containing metanephric, stromal and epithelial derivatives | CDKN2A deletion, SLC gene altered | [28] |
| Von Hippel Lindau disease | 1% of RCC | Visceral cysts in kidney, pancreas, epididymis | VHL, MYC as potential target of 8q amplification. | [29] |
| Wilm’s tumors (nephroblastoma) | 5–6% of kidney cancer in children | Triphasic. Composed of epithelial, blastemal, and stromal elements | WT1, CTNNB1, AMER1 Predisposition genes identified including TRIM28, FBXWJ, NYNRIN, KDM3B | [30,31] |
| Tumor Grade | Morphology | Prevalence | Description |
|---|---|---|---|
| G1 |  | Very rare | Round or uniform nuclei with barely visible nucleoli |
| G2 |  | 40% | Somewhat irregular nuclear contours with nucleoli visible only at 400× |
| G3 |  | 30–40% | Moderate to prominent irregular nuclear contours with nucleoli visible at 100× |
| G4 |  | 15% | Multilobular and grotesque nuclei with large and prominent nucleoli |
| Non-Coding RNA | Expression | Role | Model | References |
|---|---|---|---|---|
| miR-200 family | Downregulated in cystic kidneys | Promote cyst formation by binding to Pkd1 | Ksp/Cre; DicerF/F mutant mice | [174] |
| Downregulated in ccRCC | Inhibits EMT | ccRCC tissues from patients | [175,176] | |
| miR-17~92 cluster | Upregulated in ADPKD | Targets Pparα to promote cyst formation | Ksp/Cre;Pkd1F/F (PKD1-KO) and Pkhd1/Cre;Pkd2F/F (Pkd2-KO) mice kidneys | [177] |
| Upregulated in ccRCC | Promote tumor cell proliferation | Kidney tissues from ccRCC patients | [178] | |
| miR-21 | Upregulated in RCC | Prognostic marker | RCC tissues from patients | [179] |
| Upregulated in PKD | Promote cyst formation | Pkhd1/Cre;Pkd2F/F, Ksp/Cre;Pkd1F/F, Ksp/Cre;Hnf-1βF/F | [180] | |
| DUXAP8 | Upregulated in RCC | Decreases miR-126 | Renal cancer cell lines A498, 786-O | [181] |
| GHET1 | Upregulated in RCC | Promotes EMT | RCC tissues form patients, 786-O and A-498 cells | [172] |
| PVT1 | Upregulated in RCC | Decreases miR-16-5p expression | ccRCC tissues from patients, renal cancer cell lines A498, 786-O, ACHN, Caki-1 | [171] |
| HOTAIR | Upregulated in RCC | Attenuated by miR-203 | ccRCC from patients, renal cancer cell lines ACHN, Caki-1 | [170] |
| CASC2 | Downregulated in RCC | Targeted by miR-21 | RCC tissues from patients, renal cancer cell lines A498, 786-O | [182] |
| Hosb3os | Downregulated in ADPKD | Negatively regulates mTOR signaling | Ksp/Cre;Pkd1f/f and Pkhd1/Cre;Pkd2f/f mice, mIMCD3 cells | [183] |
| Human RCC Gene | Drosophila Homolog | Mutant Phenotype(s) | Molecular and Cellular Function(s) | Reference(s) |
|---|---|---|---|---|
| VHL | dVHL | Cell migration and polarity defective | Regulates embryonic morphogenesis of trachea and follicular epithelium; promotes endocytic vesicle transport | [210,219] |
| HIF-α | sima | Exacerbated hyperlactatemic phenotype | During development, directs hypoxia-driven terminal branching of trachea | [220,221] |
| HIF-1β | Tango | CNS midline and tracheal defects | Role in CNS midline development and tracheal tubule formation; possibly a cytosolic sink to sequester Sima and prevent interaction with Notch | [222,223] |
| VEGF | Pvf1 Pvf2 Pvf3 | Severe defects in hemocyte migration | Regulation of blood cell migration and activation of the canonical Ras/Raf/MAPK cascase, PI3K, TORC1, Rho family of small GTPases, and JNK cascade | [224,225,226] |
| EGFR | dEGFR | Glial hyperplasia, CNS morphogenesis defects, lethal | Growth regulation, cell survival and proliferation, developmental patterning | [227,228] |
| GLUT1 | dGlut1 | - | Glucose transport/uptake in neurons | [229] |
| PBRM1 | polybromo | - | Chromatin remodeling with Brahma complex; regulation of gene transcription | [230] |
| BAP1 | Calypso | - | With ASX, forms a Polycomb group (PcG) protein complex called PR-DUB involved in deubiquitination; required for efficient activity on nucleosomes | [231,232] |
| SETD2 | Set2 | - | Encodes an essential histone methyltransferase that functions with CG4747 to facilitate targeting of the male-specific lethal (MSL) complex to active genes | [233] |
| mTOR | dTor | Reduced nucleolar size, lipid vesicle aggregation in larval fat body, cell type-specific pattern of cell cycle arrest | Regulates cellular growth; amino acid and nutritional sensing | [204,234] |
| c-Myc | dMyc | Smaller cell size, body size, and viability | Regulates cell cycle, stem cell differentiation, cell proliferation, embryo and adult size | [205,235] |
| PTEN | dPTEN | Larger eyes and heads, faster proliferation | Encodes a negative effector of insulin signaling; control of cell size, proliferation and apoptosis | [207,236,237] |
| TP53 | Dp53 | Massive cell death in developing eye; apoptotic defect of primordial germ cells | Adaptive responses to genotoxic stress; inhibits cell differentiation; activates canonical caspase-dependent apoptosis pathway during stress | [238,239,240] |
| TCEB1 | EloB | Vein truncation in wings | Essential gene that facilitates Elongin complex assembly and stability; important for wing development and identity | [241] |
| Drugs | Targets | References |
|---|---|---|
| Sunitinib | Inhibits several receptor tyrosine kinases, including VEGFR types 1 and 2, PDGFRs, and more. | [253] |
| Axitinib | Potent inhibitor of VEGFR1,2 and 3. Axitinib had a higher progression free survival (defined as the range between the date of the first dose and the date of disease progression or death) compared to sorafenib. Axitinib established the utility of second-generation angiogenesis inhibitors with broader activity to overcome sunitinib resistance. | [254] |
| Sorafenib | Multi-targeted tyrosine kinase inhibitor of VEGFR2, VEGFR3, and PDGFR-β. | [255,256] |
| Lenvatinib | Selectively targets VEGFR1, 2, and 3, FGFRs, and PDGFR-α. Also effective towards non-VEGFR pathways. | [257] |
| Lenvatinib + everolimus | Combination exhibited longer progression free survival than everolimus (but not lenvatinib) alone. | [258,259] |
| Trebananib | Fusion protein disrupting the interaction of angiopoietin 1 and 2 with receptor Tie2. Did not show promising effects in anti-angiogenesis resistant RCC. | [260] |
| Dalantercept | Antagonist of Activin receptor-like kinase (ALK)-1/Bone morphogenetic protein (BMP)-9 signaling for the treatment of metastatic RCC. Combination with axitinib in heavily pretreated ccRCC patients did not seem to improve. | [261,262] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millet-Boureima, C.; He, S.; Le, T.B.U.; Gamberi, C. Modeling Neoplastic Growth in Renal Cell Carcinoma and Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 3918. https://doi.org/10.3390/ijms22083918
Millet-Boureima C, He S, Le TBU, Gamberi C. Modeling Neoplastic Growth in Renal Cell Carcinoma and Polycystic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(8):3918. https://doi.org/10.3390/ijms22083918
Chicago/Turabian StyleMillet-Boureima, Cassandra, Stephanie He, Thi Bich Uyen Le, and Chiara Gamberi. 2021. "Modeling Neoplastic Growth in Renal Cell Carcinoma and Polycystic Kidney Disease" International Journal of Molecular Sciences 22, no. 8: 3918. https://doi.org/10.3390/ijms22083918
APA StyleMillet-Boureima, C., He, S., Le, T. B. U., & Gamberi, C. (2021). Modeling Neoplastic Growth in Renal Cell Carcinoma and Polycystic Kidney Disease. International Journal of Molecular Sciences, 22(8), 3918. https://doi.org/10.3390/ijms22083918