
Substructural Approach for Assessing the Stability of Higher Fullerenes



Abstract
1. Introduction
2. Stability Criteria for Fullerenes
- In stable carbon clusters (fullerenes), each atom has a coordination number 3.
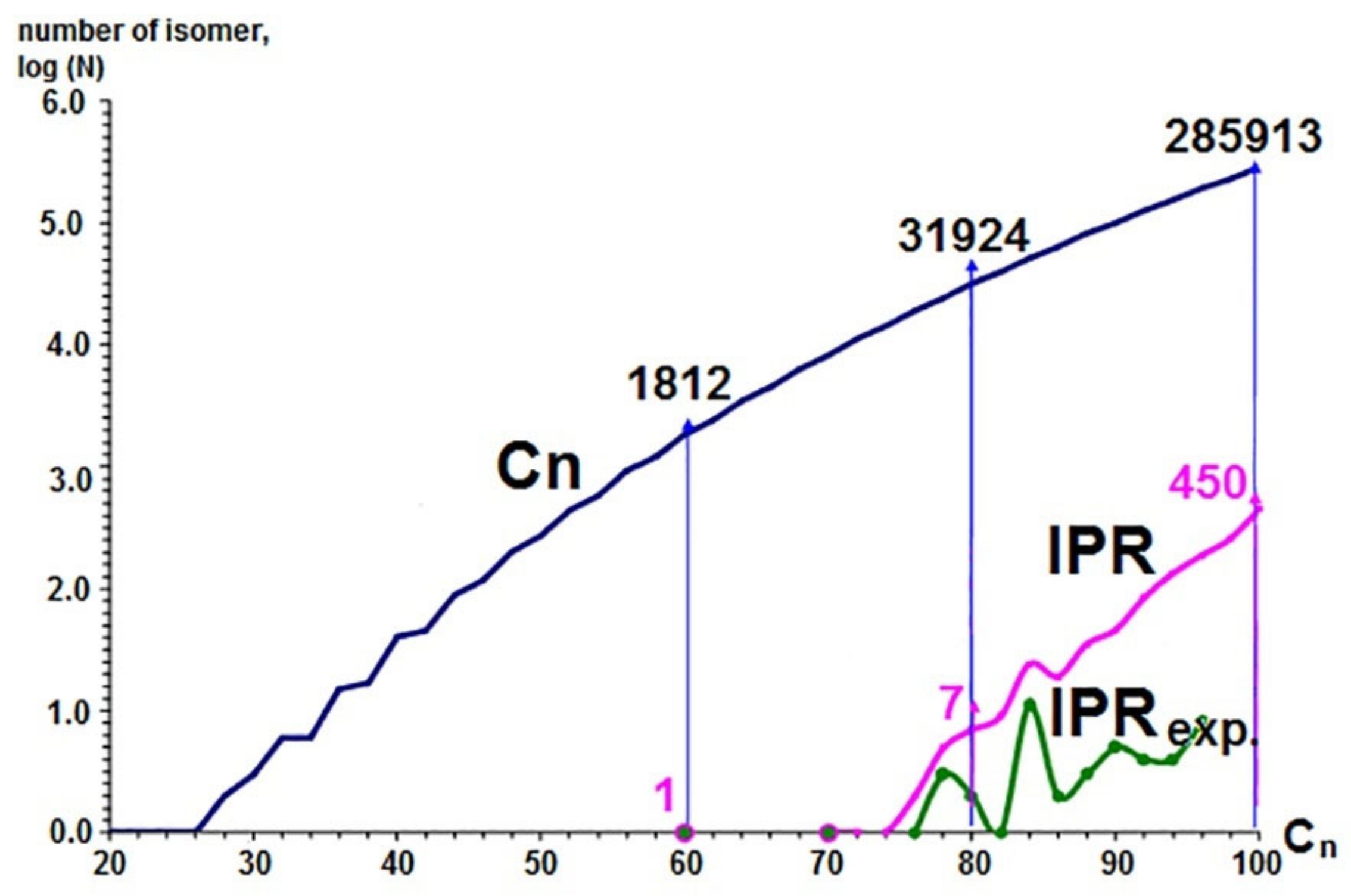
- Clusters containing only five- and six-membered rings (pentagons and hexagons, respectively) are more stable.
- Five-member cycles in carbon clusters must be isolated (the Isolated Pentagon Rule).
- More symmetric carbon clusters are more stable, which leads to a uniform distribution of the curvature of the cluster surface.
- It is necessary to have a closed electronic shell for a stable cluster.
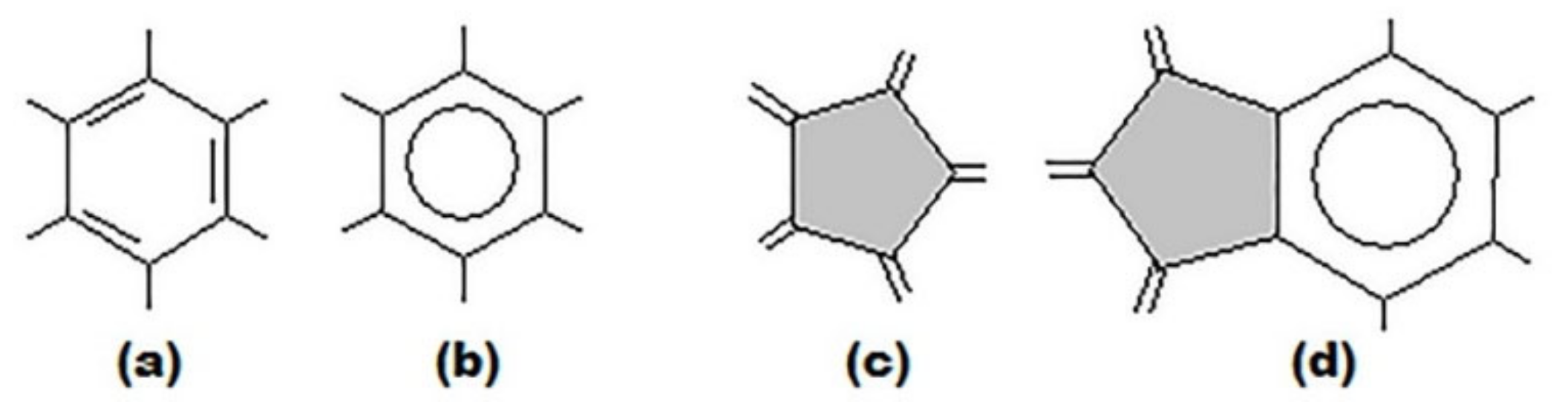
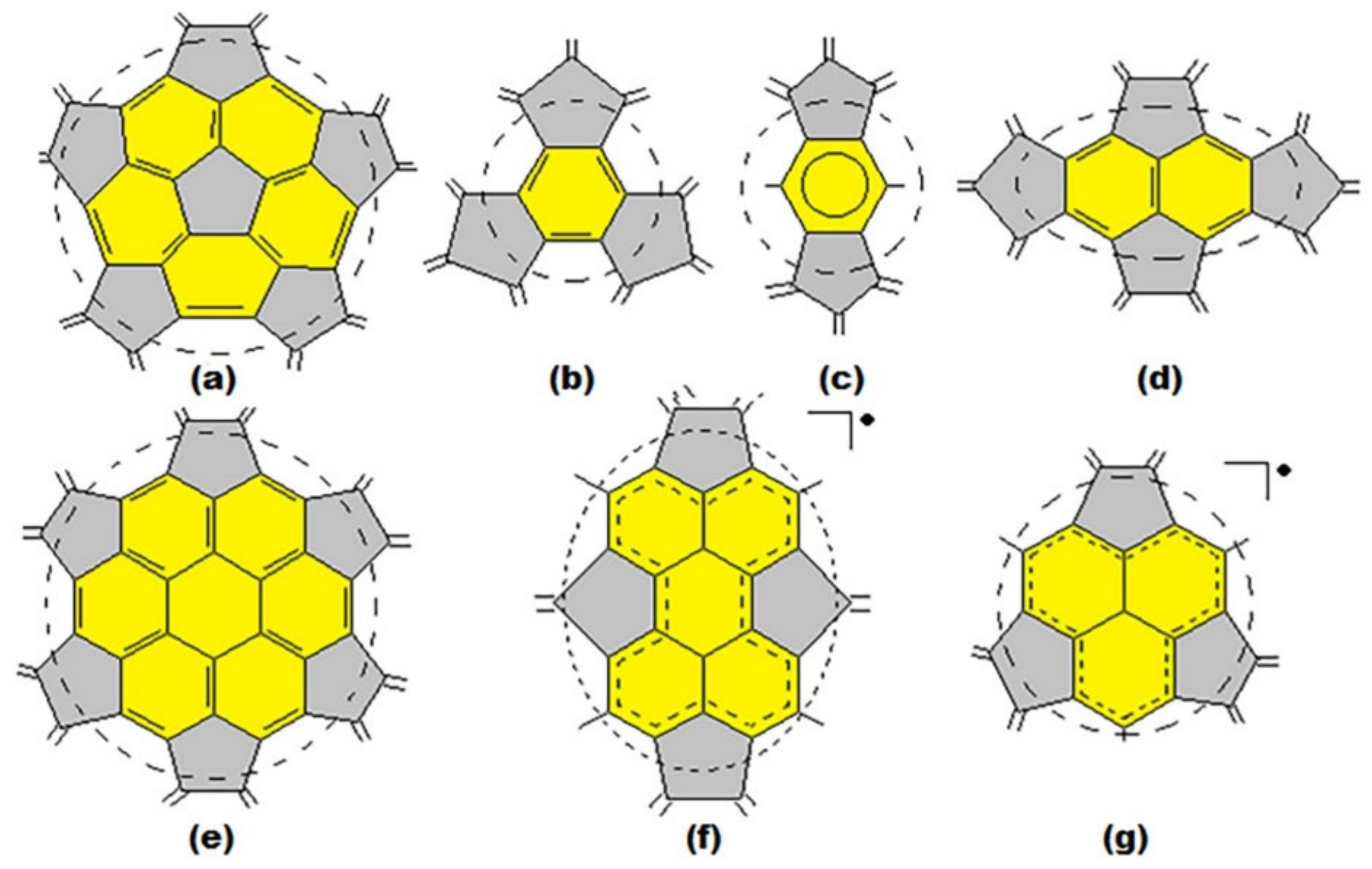
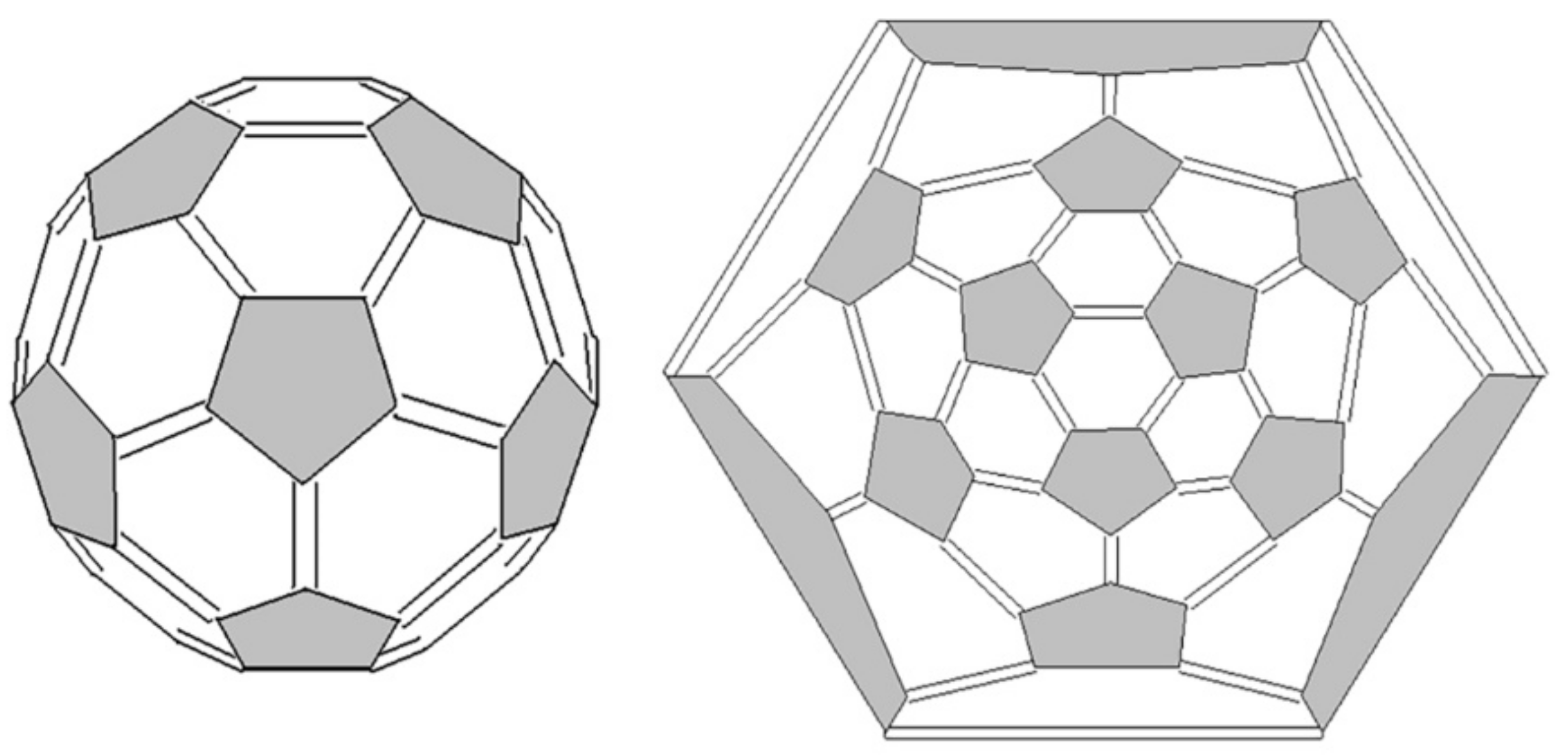
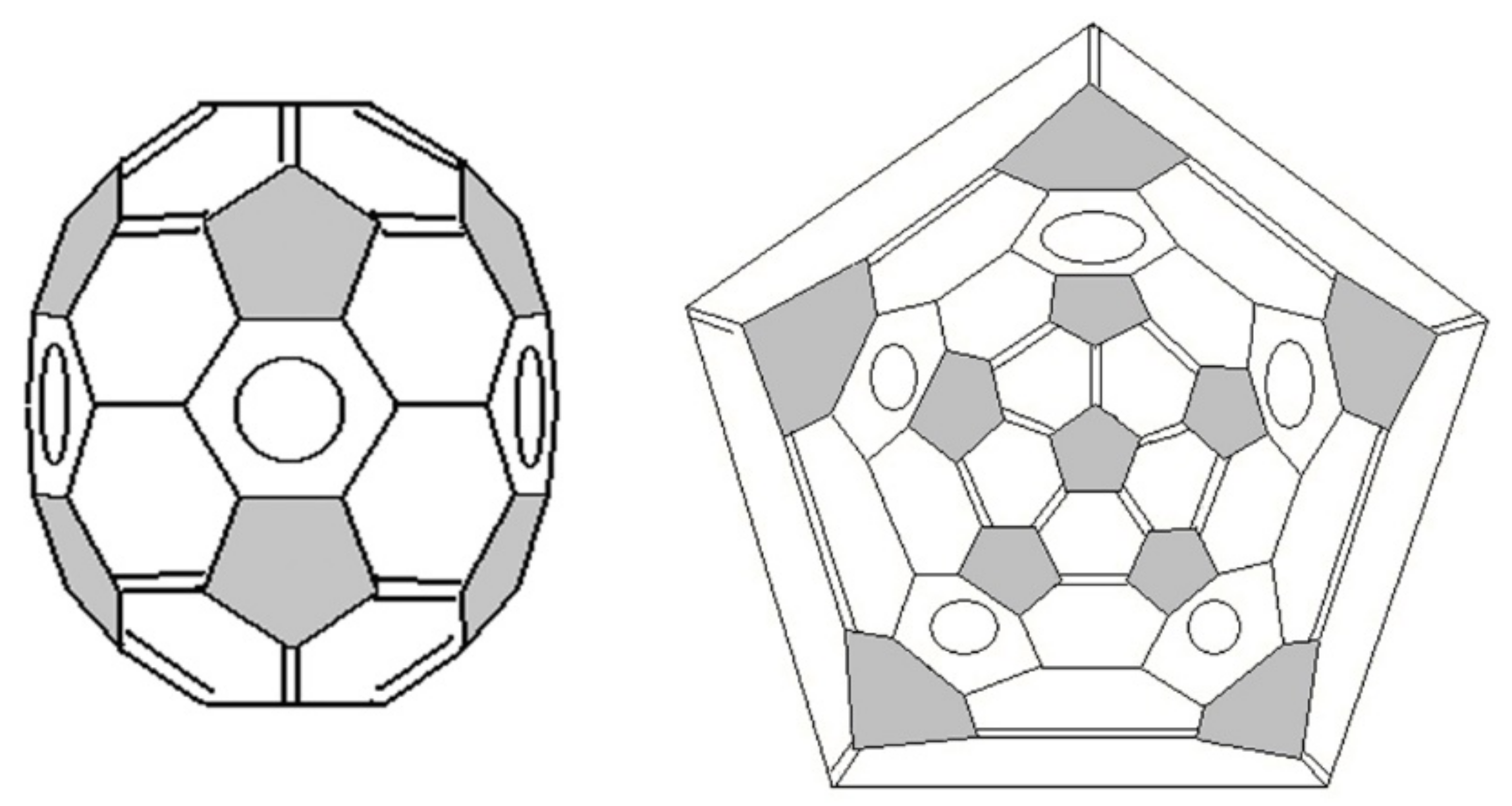
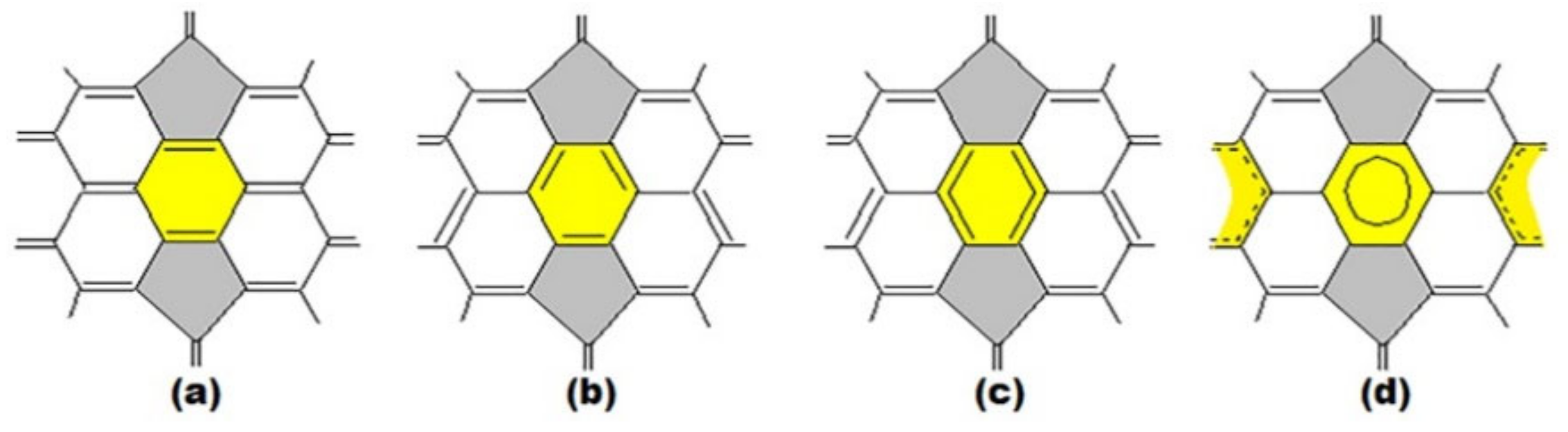
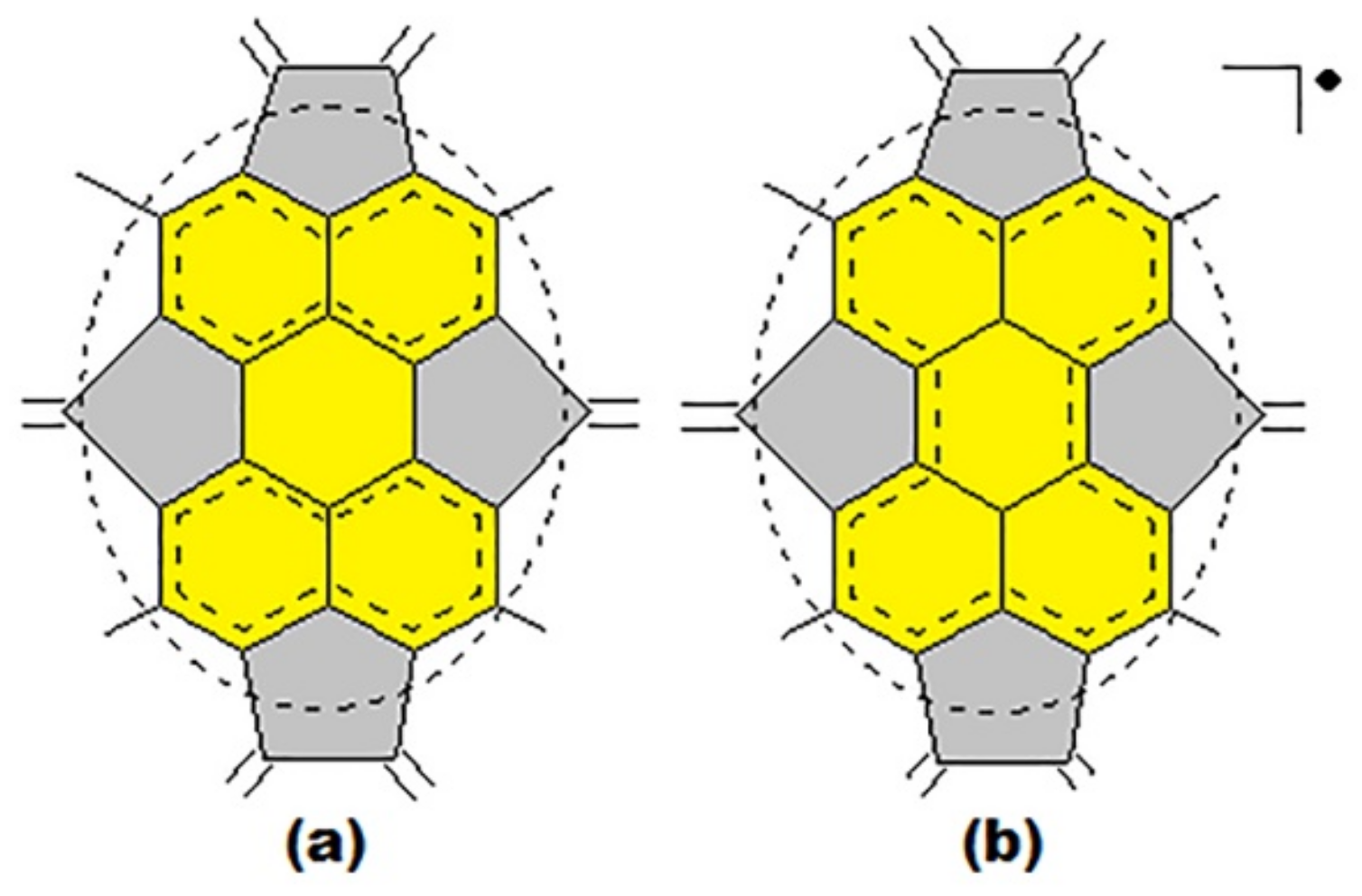
3. Basic Principles of the Substructural Approach to Modeling the Structure and Stability of Higher Fullerenes
- Stage 1. Creation of structural formula of a fullerene molecule.
- Stage 2. Identification of substructures, i.e., fragments, of a fullerene molecule that are characterized by their geometry and electronic parameters.
- Stage 3. At last, carrying out quantum-chemical calculations of a given molecule.
4. Methodological Features of Quantum Chemical Research of the Electronic Structure and Stability of Higher Fullerenes
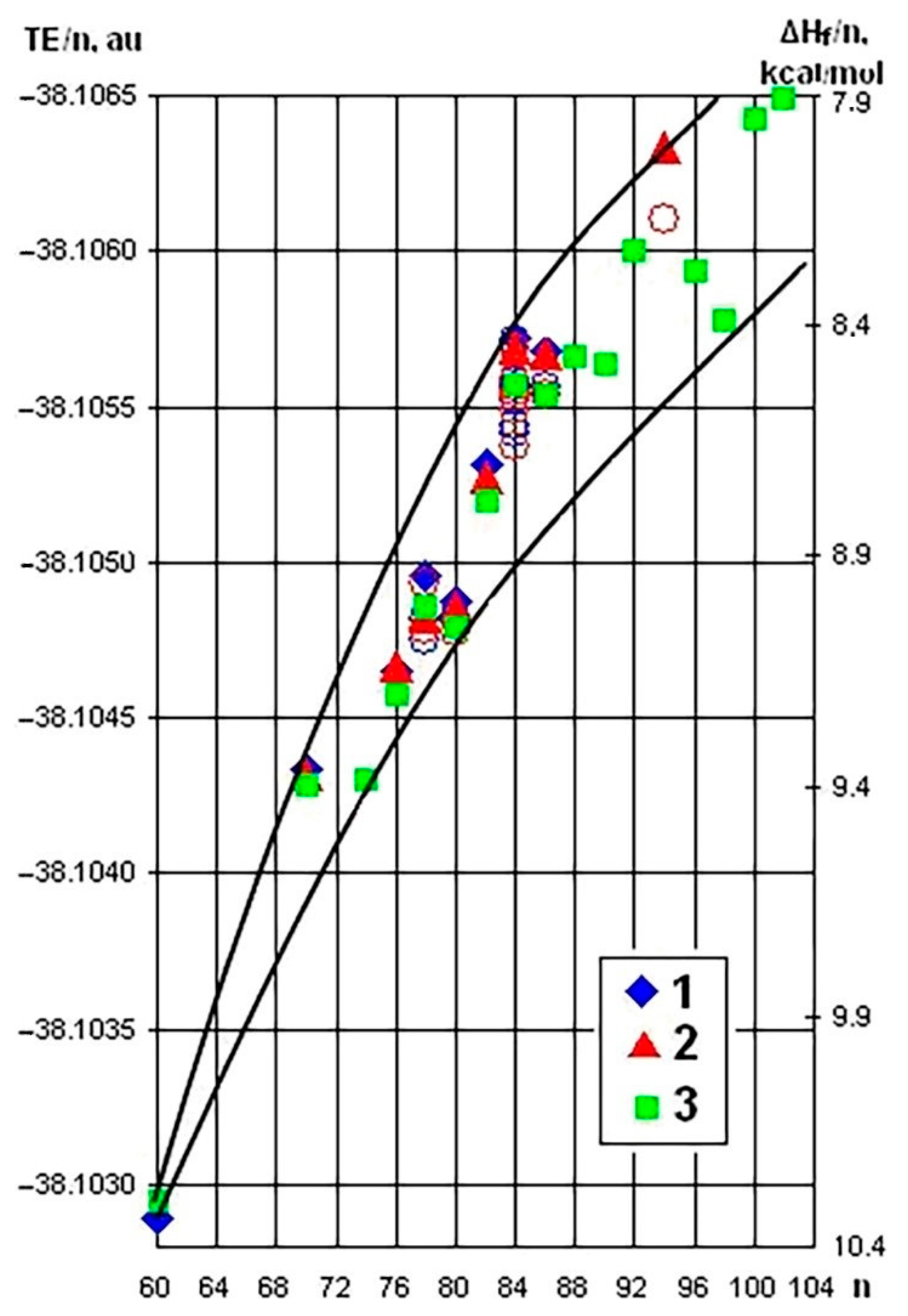
5. Applying the Substructural Approach to Modeling the Structure and Stability of Higher Fullerenes
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Osawa, E. Superaromaticity. Kagaku 1970, 25, 854–863. [Google Scholar]
- Yoshida, Z.; Osawa, E. Aromaticity; Chemical Monograph Series 22; Kagaku-Dojin: Kyoto, Japan, 1971; pp. 174–178. [Google Scholar]
- Bochvar, D.A.; Gal‘pern, E.G. About hypothetical systems: Carbododecahedron, s-icosahedron and carbo-s-icosahedron. Dokl. Akad. Nauk SSSR 1973, 209, 610–612. [Google Scholar]
- Kroto, H.W. The stability of the fullerenes Cn, with n = 24, 28, 32, 36, 50, 60 and 70. Nature 1987, 329, 529–531. [Google Scholar] [CrossRef]
- Schmalz, T.G.; Seitz, W.A.; Klein, D.J.; Hite, G.E. Elemental carbon cages. J. Am. Chem. Soc. 1988, 110, 1113–1127. [Google Scholar] [CrossRef]
- Raghavachari, K.; McMichael Rohlfing, C. Isomers of C78. Competition between electronic and steric factors. Chem. Phys. Lett. 1993, 208, 436–440. [Google Scholar] [CrossRef]
- Liu, X.; Schmalz, T.G.; Klein, D.J. Favorable structures for higher fullerenes. Chem. Phys. Lett. 1992, 188, 550–554. [Google Scholar] [CrossRef]
- Taylor, R. Rationalisation of the most stable isomer of a fullerene Cn. J. Chem. Soc. Perkin Trans. 2 1992, 3–4. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Ori, O.; László, I. Isomers of the C84 fullerene: A theoretical consideration within energetic, structural, and topological approaches. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 100–110. [Google Scholar] [CrossRef]
- Dobrynin, A.A.; Ori, O.; Putz, M.V.; Vesnin, A.Y. Generalized topological efficiency–case study with C84 fullerene. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 545–550. [Google Scholar] [CrossRef]
- Brinkmann, G.; Goedgebeur, J.; McKay, B.D. The generation of fullerenes. J. Chem. Inf. Model. 2012, 52, 2910–2918. [Google Scholar] [CrossRef]
- Fowler, P.; Manolopoulos, D. An Atlas of Fullerenes; Dover Publ. Inc.: Mineola, NY, USA, 2006. [Google Scholar]
- Diener, M.D.; Alford, J.M. Isolation and properties of small-bandgap fullerenes. Nature 1998, 393, 668–671. [Google Scholar] [CrossRef]
- Anderson, M.R.; Dorn, H.C.; Stevenson, S.A. Making connections between metallofullerenes and fullerenes: Electrochemical investigations. Carbon N. Y. 2000, 38, 1663–1670. [Google Scholar] [CrossRef]
- Aihara, J.I. Reduced HOMO-LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Aihara, J.I. Weighted HOMO-LUMO energy separation as an index of kinetic stability for fullerenes. Theor. Chem. Acc. 1999, 102, 134–138. [Google Scholar] [CrossRef]
- Aihara, J.I. Correlation found between the HOMO-LUMO energy separation and the chemical reactivity at the most reactive site for isolated-pentagon isomers of fullerenes. Phys. Chem. Chem. Phys. 2000, 2, 3121–3125. [Google Scholar] [CrossRef]
- Aihara, J.I. Many reactive fullerenes tend to form stable metallofullerenes. J. Phys. Chem. A 2002, 106, 11371–11374. [Google Scholar] [CrossRef]
- Cai, W.; Shao, N.; Shao, X.; Pan, Z. Structural analysis of carbon clusters by using a global optimization algorithm with Brenner potential. J. Mol. Struct. THEOCHEM 2004, 678, 113–122. [Google Scholar] [CrossRef]
- Austin, S.J.; Fowler, P.W.; Manolopoulos, D.E.; Orlandi, G.; Zerbetto, F. Structural motifs and the stability of fullerenes. J. Phys. Chem. 1995, 99, 8076–8081. [Google Scholar] [CrossRef]
- Haddon, R.C. Chemistry of the fullerenes: The manifestation of strain in a class of continuous aromatic molecules. Science 2010, 261, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K. Enumeration of stereo, position and chiral isomers of polysubstituted giant fullerenes: Applications to C180 and C240. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 687–696. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Ori, O.; Cataldo, F.; Ashrafi, A.R.; Putz, M.V. Face colorings and chiral face colorings of icosahedral giant fullerenes: C80 to C240. Fuller. Nanotub. Carbon Nanostruct. 2021, 29, 1–12. [Google Scholar] [CrossRef]
- Dunk, P.W.; Kaiser, N.K.; Mulet-gas, M.; Rodr, A.; Poblet, J.M.; Shinohara, H.; Hendrickson, C.L.; Marshall, A.G.; Kroto, H.W. The smallest stable fullerene, M@C28 (M=Ti,Zr,U): Stabilization and growth from carbon vapor. J. Am. Chem. Soc. 2012, 134, 9380–9389. [Google Scholar] [CrossRef]
- Chuvilin, A.; Kaiser, U.; Bichoutskaia, E.; Besley, N.A.; Khlobystov, A.N. Direct transformation of graphene to fullerene. Nat. Chem. 2010, 2, 450–453. [Google Scholar] [CrossRef]
- Berné, O.; Tielens, A.G.G.M. Formation of buckminsterfullerene (C60) in interstellar space. Proc. Natl. Acad. Sci. USA 2012, 109, 401–406. [Google Scholar] [CrossRef]
- Baronnet, J.M.; Ershov-Pavlov, E.A.; Megy, S. Plasma parameters of an argon DC arc with graphite electrodes. J. Phys. D Appl. Phys. 1999, 32, 2552–2559. [Google Scholar] [CrossRef]
- Saunders, M.; Jiménez-Vázquez, H.A.; Cross, R.J.; Poreda, R.J. Stable compounds of helium and neon: He@C60 and Ne@C60. Science 1993, 259, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.; Jiménez-Vázquez, H.A.; Cross, R.J.; Mroczkowski, S.; Freedberg, D.I.; Anet, F.A.L. Probing the interior of fullerenes by 3He NMR spectroscopy of endohedral 3He@C60 and 3He@C70. Nature 1994, 367, 256–258. [Google Scholar] [CrossRef]
- Bühl, M.; Thiel, W. Ab initio helium NMR chemical shifts of endohedral fullerene compounds He@Cn (n = 32-180). Chem. Phys. Lett. 1995, 233, 585–589. [Google Scholar] [CrossRef]
- Rubin, Y.; Jarrosson, T.; Wang, G.W.; Bartberger, M.D.; Houk, K.N.; Schick, G.; Saunders, M.; Cross, R.J. Insertion of helium and molecular hydrogen through the orifice of an open fullerene. Angew. Chem. Int. Ed. 2001, 40, 1543–1546. [Google Scholar] [CrossRef]
- Cross, R.J.; Khong, A.; Saunders, M. Using cyanide to put noble gases inside C60. J. Org. Chem. 2003, 68, 8281–8283. [Google Scholar] [CrossRef] [PubMed]
- Khamatgalimov, A.R. Structure and Stability of Hogher Fullerenes in C60-C86 Row; Arbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences: Kazan, Russia, 2016. (In Russian) [Google Scholar]
- Khamatgalimov, A.R. Geometric and Electronic Structure of Fullerene Molecules C72, C74, and C82. Ph.D. Thesis, Kazan State Technological University, Kazan, Russia, 2003. (In Russian). [Google Scholar]
- Kovalenko, V.I.; Khamatgalimov, A.R. Regularities in the molecular structures of stable fullerenes. Russ. Chem. Rev. 2006, 75, 981–988. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Molecular structures of unstable isolated-pentagon-rule fullerenes C72-C86. Russ. Chem. Rev. 2016, 85, 836–853. [Google Scholar] [CrossRef]
- Kovalenko, V.I. The Pentagonal Pole Model of Fullerenes C60 and C70. In Proceedings of the Structure and Dynamics of Molecular Systems, Yalchik, Russia, 23–27 June 1997; pp. 88–91. (In Russian). [Google Scholar]
- Kovalenko, V.I.; Semyashova, M.V. Delocalization of bonds in hexagon of fullerenes. Chem. Comput. Simul. Butl. Commun. 2000, 3, 41–43. [Google Scholar]
- Hedberg, K.; Hedberg, L.; Bethune, D.S.; Brown, C.A.; Dorn, H.C.; Johnson, R.D.; De Vries, M. Bond lengths in free molecules of buckminsterfullerene, C60, from gas-phase electron diffraction. Science 1991, 254, 410–412. [Google Scholar] [CrossRef]
- Hedberg, K.; Hedberg, L.; Bühl, M.; Bethune, D.S.; Brown, C.A.; Johnson, R.D. Molecular structure of free molecules of the fullerene C70 from gas- phase electron diffraction. J. Am. Chem. Soc. 1997, 119, 5314–5320. [Google Scholar] [CrossRef]
- Baker, J.; Fowler, P.W.; Lazzeretti, P.; Malagoli, M.; Zanasi, R. Structure and properties of C70. Chem. Phys. Lett. 1991, 184, 182–186. [Google Scholar] [CrossRef]
- García-Domenech, R.; Gálvez, J.; de Julián-Ortiz, J.V.; Pogliani, L. Some new trends in chemical graph theory. Chem. Rev. 2008, 108, 1127–1169. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Hirsch, A. Spherical aromaticity of fullerenes. Chem. Rev. 2001, 101, 1153–1184. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.; Brettreich, M. Fullerenes: Chemistry and Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 9783527603497. [Google Scholar]
- Tomanek, D.; Frederick, N. Cn Fullerenes. Available online: http://www.nanotube.msu.edu/fullerene/fullerene-isomers.html (accessed on 7 February 2013).
- Krief, P.; Becker, J.Y.; Ellern, A.; Khodorkovsky, V.; Neilands, O.; Shapiro, L. s-Indacene-1,3,5,7(2H,6H)-tetraone (‘Janus dione’) and 1-3-dioxo-5,6-indane-dicarboxylic acid: Old and new 1,3-indandione derivatives. Synthesis 2004, 15, 2509–2512. [Google Scholar] [CrossRef]
- Liskin, D.V.; Valente, E.J. A dipseudoacid, C16H18O6. J. Mol. Struct. 2008, 878, 149–154. [Google Scholar] [CrossRef]
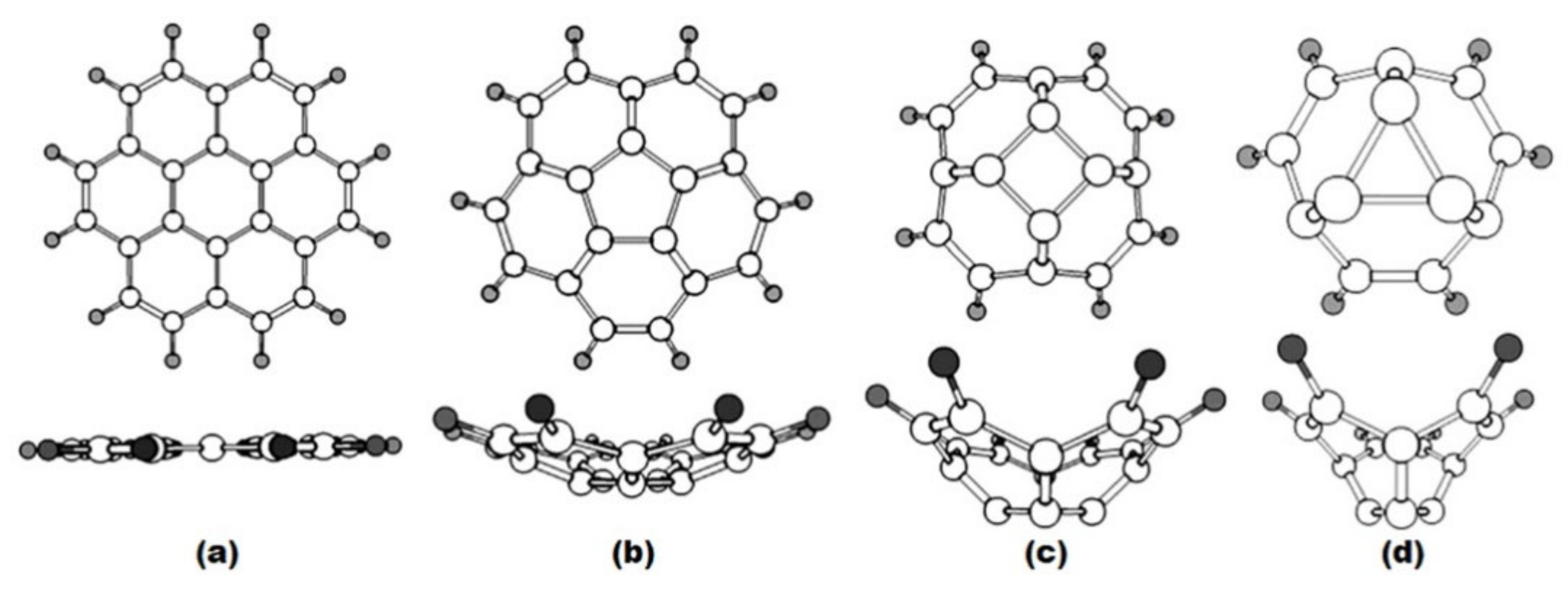
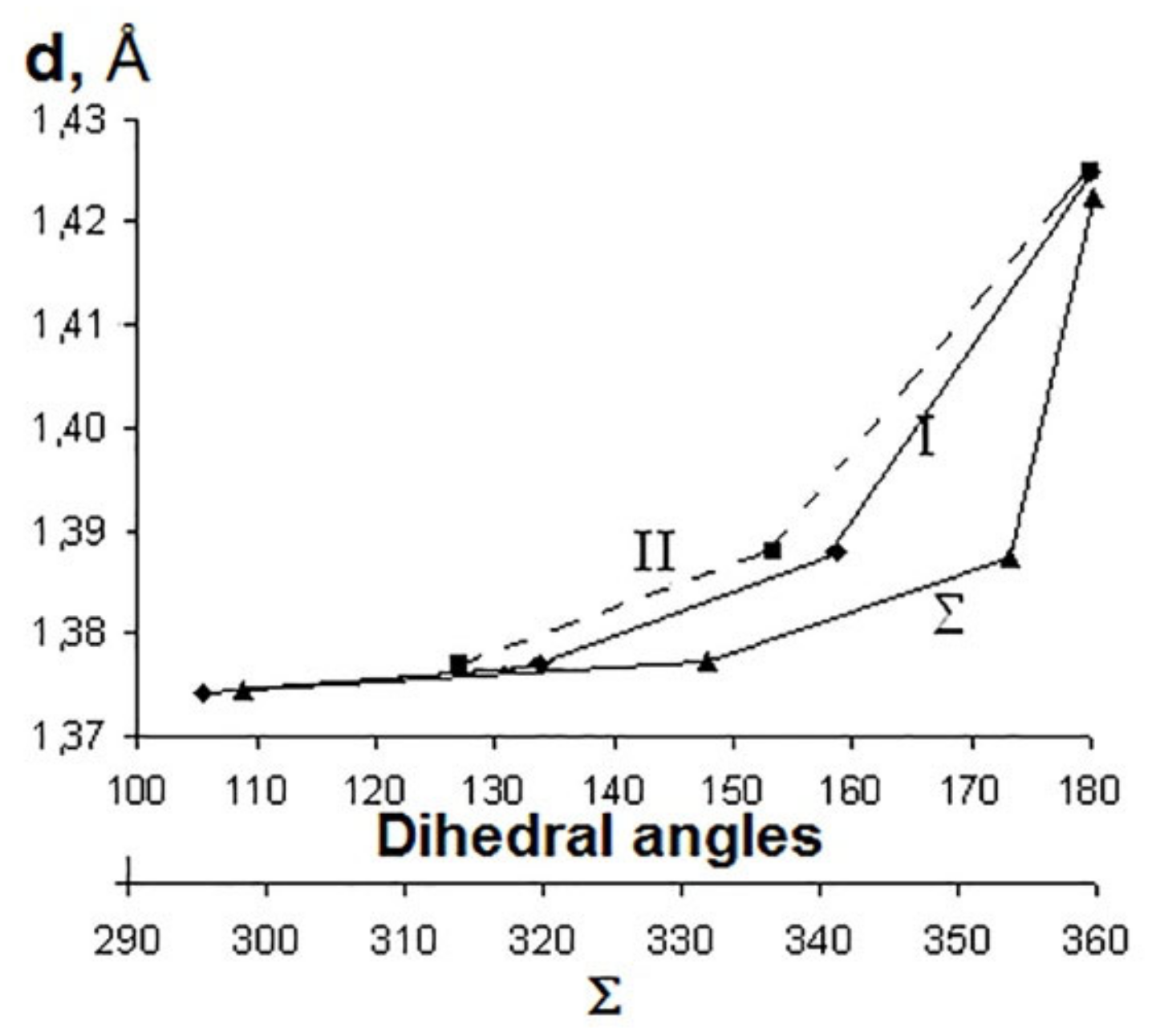
- Khamatgalimov, A.R.; Korolev, S.S.; Kovalenko, V.I. Search for optimal dihedral angles in fullerene: Geometry of structures with six-, five-, four- and three-membered rings surrounded by hexagons. Bull. Bashkir Univer. 2008, 3, 768–771. (In Russian) [Google Scholar]
- Martin, J.M.L. The vibrational spectra of corannulene and coronene. A density functional study. Chem. Phys. Lett. 1996, 262, 97–104. [Google Scholar] [CrossRef]
- Hedberg, L.; Hedberg, K.; Cheng, P.C.; Scott, L.T. Gas-phase molecular structure of corannulene, C20H10. An electron-diffraction study augmented by ab initio and normal coordinate calculations. J. Phys. Chem. A 2000, 104, 7689–7694. [Google Scholar] [CrossRef]
- Petrukhina, M.A.; Andreini, K.W.; Mack, J.; Scott, L.T. X-ray quality geometries of geodesic polyarenes from theoretical calculations: What levels of theory are reliable? J. Org. Chem. 2005, 70, 5713–5716. [Google Scholar] [CrossRef]
- Kataeva, O.; Khrizanforov, M.; Budnikova, Y.; Islamov, D.; Burganov, T.; Vandyukov, A.; Lyssenko, K.; Mahns, B.; Nohr, M.; Hampel, S.; et al. Crystal growth, dynamic and charge transfer properties of new coronene charge transfer complexes. Cryst. Growth Des. 2016, 16, 331–338. [Google Scholar] [CrossRef]
- Gan, L.H.; Liu, J.; Hui, Q.; Shao, S.Q.; Liu, Z.H. General geometrical rule for stability of carbon polyhedra. Chem. Phys. Lett. 2009, 472, 224–227. [Google Scholar] [CrossRef]
- Sidorov, L.N.; Yurovskaya, M.A.; Borschevsky, A.Y.; Trushkov, I.V.; Ioffe, I.N. Fullerenes; Exam Publishing: Moscow, Russia, 2005. [Google Scholar]
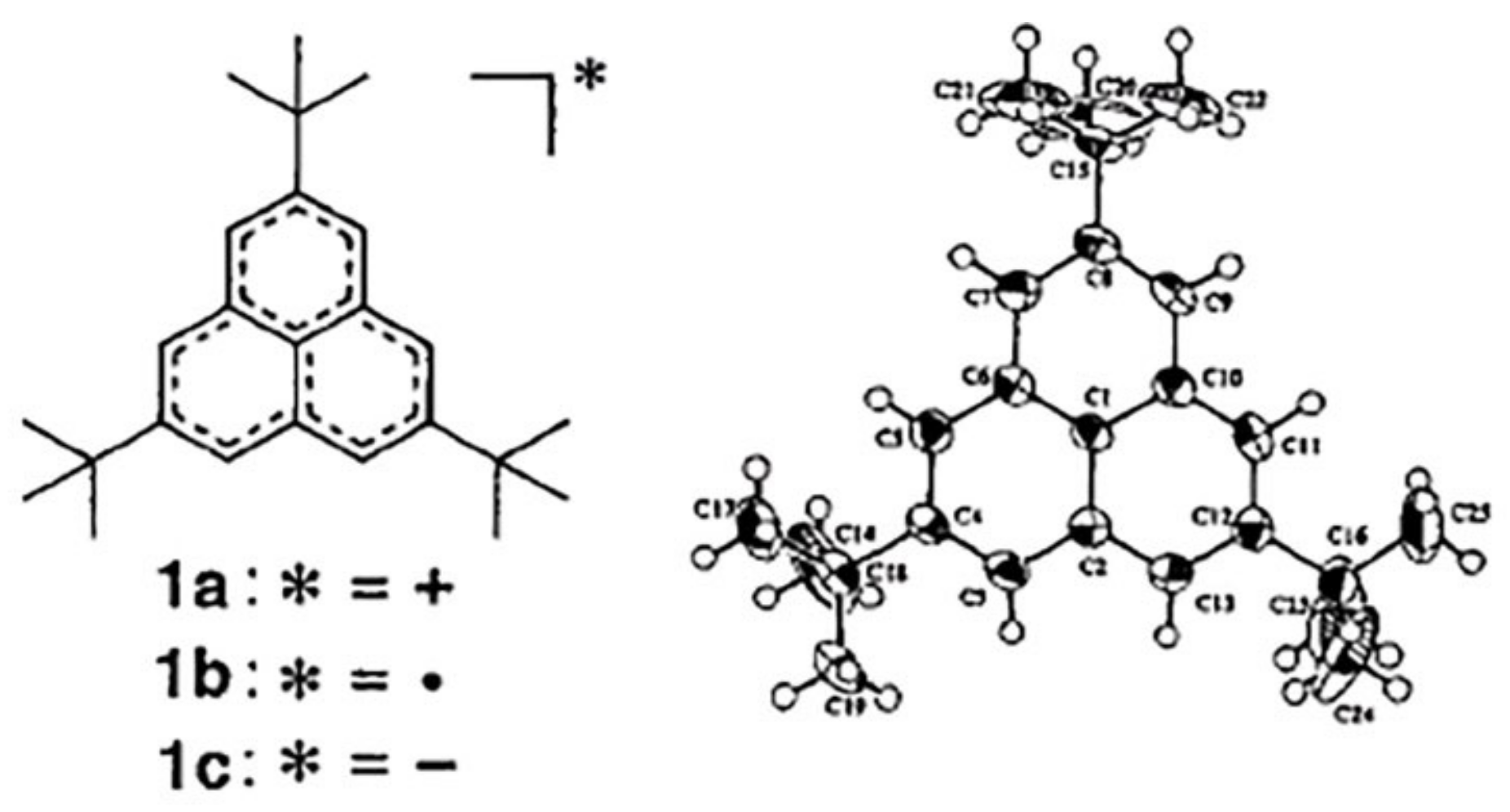
- Morita, Y.; Nishida, S. Phenalenyls, Cyclopentadienyls, and Other Carbon-Centered Radicals. In Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Hicks, R.G., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2011. [Google Scholar]
- Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; et al. A stable neutral hydrocarbon radical: Synthesis, crystal structure, and physical properties of 2,5,8-tri-tert-butyl-phenalenyl [12]. J. Am. Chem. Soc. 1999, 121, 1619–1620. [Google Scholar] [CrossRef]
- Kubo, T.; Goto, Y.; Uruichi, M.; Yakushi, K.; Nakano, M.; Fuyuhiro, A.; Morita, Y.; Nakasuji, K. Synthesis and characterization of acetylene-linked bisphenalenyl and metallic-like behavior in its charge-transfer complex. Chem. Asian J. 2007, 2, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
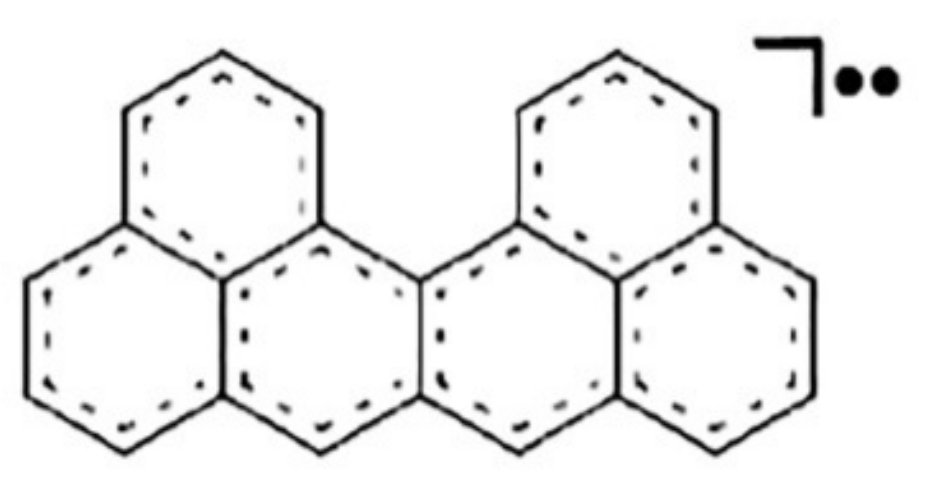
- Melle-Franco, M. Uthrene, a radically new molecule? Chem. Commun. 2015, 51, 5387–5390. [Google Scholar] [CrossRef] [PubMed]
- Khamatgalimov, A.R.; Melle-Franco, M.; Gaynullina, A.A.; Kovalenko, V.I. Ythrene: From the real radical fullerene substructure to hypothetical (yet?) radical molecules. J. Phys. Chem. C 2019, 123, 1954–1959. [Google Scholar] [CrossRef]
- Kovalenko, V.I.; Khamatgalimov, A.R. Open-shell fullerene C74: Phenalenyl-radical substructures. Chem. Phys. Lett. 2003, 377, 263–268. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Stability of isolated-pentagon-rule isomers of fullerene C76. Fuller. Nanotub. Carbon Nanostruct. 2015, 23, 148–152. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Stabilization of IPR open-shell fullerenes C74 (D3h) and C76 (Td) in radical addition reactions. Fuller. Nanotub. Carbon Nanostruct. 2017, 25, 128–132. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Radical IPR Fullerenes C74 (D3h) and C76 (Td): Dimer, trimer, etc. Experiments and theory. J. Phys. Chem. C 2018, 122, 3146–3151. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Polymeric forms of radical fullerenes C74 (D3h) and C76 (Td). Butl. Comm. 2018, 53, 63–70. (In Russian) [Google Scholar]
- Khamatgalimov, A.R.; Gaynullina, A.A.; Kovalenko, V.I. Phenalenyl-like substructures in fullerene molecules. J. Mater. Sci. Eng. 2018, 7, 81. [Google Scholar]
- Yeretzian, C.; Wiley, J.B.; Holczer, K.; Su, T.; Nguyen, S.; Kaner, R.B.; Whetten, R.L. Partial separation of fullerenes by gradient sublimation. J. Phys. Chem. 1993, 97, 10097–10101. [Google Scholar] [CrossRef]
- Shustova, N.B.; Kuvychko, I.V.; Bolskar, R.D.; Seppelt, K.; Strauss, S.H.; Popov, A.A.; Boltalina, O.V. Trifluoromethyl derivatives of insoluble small-HOMO LUMO-gap hollow higher fullerenes. NMR and DFT structure elucidation of C2-(C74-D3h)(CF3)12, Cs-(C76-Td(2))(CF3)12, C2-(C78-D3h(5))(CF3)12. J. Am. Chem. Soc. 2006, 128, 15793–15798. [Google Scholar] [CrossRef]
- Shustova, N.B.; Newell, B.S.; Miller, S.M.; Anderson, O.P.; Bolskar, R.D.; Seppelt, K.; Popov, A.A.; Boltalina, O.V.; Strauss, S.H. Discovering and verifying elusive fullerene cage isomers: Structures of C2-p11-(C74-D3h)(CF3)12 and C2-p11-(C78-D3h(5))(CF3)12. Angew. Chem. Int. Ed. 2007, 46, 4111–4114. [Google Scholar] [CrossRef]
- Kovalenko, V.I.; Khamatgalimov, A.R. Electronic Structure and Stability of Higher Fullerenes. In NATO Security through Science Series A: Chemistry and Biology; Springer: Dordrecht, The Netherlands, 2007; pp. 437–441. ISBN1 1402055129. ISBN2 9781402055126. [Google Scholar]
- Fowler, P.W.; Soncini, A. Visualising aromaticity of bowl-shaped molecules. Phys. Chem. Chem. Phys. 2011, 13, 20637–20643. [Google Scholar] [CrossRef]
- Soncini, A.; Viglione, R.G.; Zanasi, R.; Fowler, P.W.; Jenneskens, L.W. Efficient mapping of ring currents in fullerenes and other curved carbon networks. Comptes Rendus Chim. 2006, 9, 1085–1093. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Endohedral higher metal-containing fullerenes: Structures and properties. Ross. Khimicheskij Zhurnal (Zhurnal Ross. Khimicheskogo Obs. Im. D.I. Mendeleeva) 2004, 48, 28–36. [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. The Typical Reasons of Fullerenes Instability on the of C72 and C74 Example. In Proceedings of the Structure and Dynamics of Molecular Systems, Yalchik, Russia, 23–29 June 2003; pp. 186–189. (In Russian). [Google Scholar]
- Khamatgalimov, A.R.; Korolev, S.S.; Kovalenko, V.I. The Reasons of Instability of Isomer 4 (D3h) of C78 Fullerene. In Proceedings of the Nanoparticles in Condensed Matter, Minsk, Belarus, 22–25 April 2008; BGU Publ.: Minsk, Belarus, 2008; pp. 54–59. (In Russian). [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. Stabilization of the C74 fullerene molecule in the form of fullerene hydrides C74H2. Bull. Bashkir Univ. 2008, 3, 772–775. (In Russian) [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. Electronic structure and stability of C80 fullerene IPR isomers. Fuller. Nanotub. Carbon Nanostruct. 2011, 19, 599–604. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Electronic structure and stability of fullerene C82 isolated-pentagon-rule isomers. J. Phys. Chem. A 2011, 115, 12315–12320. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Deformation and thermodynamic instability of a C84 fullerene cage. Russ. J. Phys. Chem. A 2010, 84, 636–641. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. 24 IPR isomers of fullerene C84: Cage deformation as geometrical characteristic of local strains. Int. J. Quantum Chem. 2012, 112, 1055–1065. [Google Scholar] [CrossRef]
- Kovalenko, V.I.; Tuktamysheva, R.A.; Khamatgalimov, A.R. Electronic structures of some of C84 fullerene isomers and the structures of their perfluoroalkyl derivatives. Russ. J. Phys. Chem. A 2014, 88, 103–107. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. Electronic structure and stability of C86 fullerene Isolated-Pentagon-Rule isomers. Int. J. Quantum Chem. 2011, 111, 2966–2971. [Google Scholar] [CrossRef]
- Tuktamysheva, R.A.; Khamatgalimov, A.R.; Kovalenko, V.I. Electronic and geometric structure of some isomers of C90 fullerene and the structure of their chlorine and perfluoroalkyl polyadducts. Butl. Comm. 2014, 37, 1–12. (In Russian) [Google Scholar]
- Tuktamysheva, R.A.; Khamatgalimov, A.R.; Kovalenko, V.I. Theoretical analysis of the electronic state of [C84CF3] monoradicals: IPR isomers 22 (D2), 23 (D2d) and 4 (D2d). Bull. Technol. Univ. 2015, 11, 62–66. (In Russian) [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. Molecular structures of the open-shell IPR isomers of fullerene C90. Fuller. Nanotub. Carbon Nanostruct. 2017, 25, 179–184. [Google Scholar] [CrossRef]
- Mitroshkina, M.V.; Khamatgalimov, A.R.; Kovalenko, V.I. Molecule structure of two isomers 234 (Cs) and 258 (C1) of higher fullerene C104. Butl. Comm. 2017, 49, 28–33. (In Russian) [Google Scholar]
- Gainullina, A.A.; Khamatgalimov, A.R.; Kovalenko, V.I. Structure and stability of fullerene C104: IPR isomers 812 (D2) and 822 (D3d). Butl. Comm. 2017, 49, 75–83. (In Russian) [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I.; Gimranova, G.G. The structure of fullerene C66 and its endohedral analog Sc2@C66. In Proceedings of the Structure and Dynamics of Molecular Systems, Yalchik, Russia, 25 June–1 July 2006; pp. 346–350. (In Russian). [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. The structure of fullerene C66, which does not obey the rule of isolated pentagons, and endohedral metallofullerene Sc2@C66: Quantum-chemical calculations. Russ. J. Phys. Chem. A 2008, 82, 1164–1169. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Korolev, S.S.; Arkhipov, A.A.; Arkhipov, A.A.; Kovalenko, V.I. Stability of the non-IPR isomers 6140 (D3) and 6275 (D3) of fullerene C68. Fuller. Nanotub. Carbon Nanostruct. 2008, 16, 542–545. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Kovalenko, V.I. The structure of non-IPR isomer 4169 (Cs) of fullerene C66 and the reasons of its stabilization in the form of derivatives. Butl. Comm. 2012, 32, 141–148. (In Russian) [Google Scholar]
- Khamatgalimov, A.R.; Kovalenko, V.I. The structure of a linear combination of three pentagons in non-IPR isomer 4169 (Cs) of fullerene C66. Russ. J. Phys. Chem. A 2013, 87, 1884–1888. [Google Scholar]
- Khamatgalimov, A.R.; Idrisov, R.I.; Kamaletdinov, I.I.; Kovalenko, V.I. The key feature of instability of small non-IPR closed-shell fullerenes: Three isomers of C40. Mendeleev Commun. 2020, 30, 725–727. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Yakupova, L.I.; Kovalenko, V.I. Features of molecular structure of small non-IPR fullerenes: The two isomers of C50. Theor. Chem. Acc. 2020, 139, 1–8. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Idrisov, R.I.; Kamaletdinov, I.I.; Kovalenko, V.I. Open-shell nature of non-IPR fullerene C40: Isomers 29 (C2) and 40 (Td). J. Mol. Model. 2021, 27, 22. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, N.F. Quantum Mechanics and Quantum Chemistry; Mir: Moscow, Russia, 2001. (In Russian) [Google Scholar]
- Minkin, V.I.; Minyaev, R.M.; Hoffmann, R. Nonclassical structures of organic compounds: Non-standard stereochemistry and hypercoordination. Usp. Khim. 2002, 71, 989–1015. [Google Scholar] [CrossRef]
- Stepanov, N.F.; Novakovskaya, Y.F. Quantum chemistry today. Ross. Khimicheskij Zhurnal (Zhurnal Ross. Khimicheskogo Obs. Im. D.I. Mendeleeva) 2007, 51, 5–17. [Google Scholar]
- Burshtein, K.Y.; Sharygin, P.P. Quantum-Chemical Calculations in Organic Chemistry and Molecular Spectros-Copy; Nauka: Moscow, Russia, 1989. (In Russian) [Google Scholar]
- Clark, T. Computer Chemistry; Mir: Moscow, Russia, 1990. (In Russian) [Google Scholar]
- Foresman, J.B.; Frish, A. Exploring Chemistry with Electronic Structure Methods; Gaussian Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computation Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 1999. [Google Scholar]
- Rogers, D.W. Computational Chemistry Using the PC; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Slanina, Z.; Zhao, X.; Deota, P.; Osawa, E. Calculations of Higher Fullerenes and Quasi-Fullerenes. In Fullerenes: Chemistry, Physics, and Technology; John Wiley & Sons: New York, NY, USA, 2000; pp. 283–330. [Google Scholar]
- Straitvizer, E. Molecular Orbital Theory; Mir: Moscow, Russia, 1965. (In Russian) [Google Scholar]
- Schwerdtfeger, P.; Wirz, L.N.; Avery, J. The topology of fullerenes. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 96–145. [Google Scholar] [CrossRef] [PubMed]
- Murry, R.L.; Colt, J.R.; Scuseria, G.E. How accurate are molecular mechanics predictions for fullerenes? A benchmark comparison with Hartree-Fock self-consistent field results. J. Phys. Chem. 1993, 97, 4954–4959. [Google Scholar] [CrossRef]
- Chen, Z.; Thiel, W. Performance of semiempirical methods in fullerene chemistry: Relative energies and nucleus-independent chemical shifts. Chem. Phys. Lett. 2003, 367, 15–25. [Google Scholar] [CrossRef]
- Zheng, G.; Irle, S.; Morokuma, K. Performance of the DFTB method in comparison to DFT and semiempirical methods for geometries and energies of C20-C86 fullerene isomers. Chem. Phys. Lett. 2005, 412, 210–216. [Google Scholar] [CrossRef]
- Rostami, Z.; Hosseinian, A.; Monfared, A. DFT results against experimental data for electronic properties of C60 and C70 fullerene derivatives. J. Mol. Graph. Model. 2018, 81, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Shao, N.; Gao, Y.; Zeng, X.C. Search for lowest-energy fullerenes 2: C38 to C80 and C112 to C120. J. Phys. Chem. C 2007, 111, 17671–17677. [Google Scholar] [CrossRef]
- Shao, N.; Gao, Y.; Yoo, S.; An, W.; Zeng, X.C. Search for lowest-energy fullerenes: C98 to C110. J. Phys. Chem. A 2006, 110, 7672–7676. [Google Scholar] [CrossRef] [PubMed]
- Paulus, B. Electronic and structural properties of the cage-like molecules C20 to C36. Phys. Chem. Chem. Phys. 2003, 5, 3364–3367. [Google Scholar] [CrossRef]
- Kareev, I.E.; Kuvychko, I.V.; Shustova, N.B.; Lebedkin, S.F.; Bubnov, V.P.; Anderson, O.P.; Popov, A.A.; Boltalina, O.V.; Strauss, S.H. C1-(C84-C2(11))(CF3)12: Trifluoromethylation yields structural proof of a minor C84 cage and reveals a principle of higher fullerene reactivity. Angew. Chem. Int. Ed. 2008, 47, 6204–6207. [Google Scholar] [CrossRef]
- Kareev, I.E.; Popov, A.A.; Kuvychko, I.V.; Shustova, N.B.; Lebedkin, S.F.; Bubnov, V.P.; Anderson, O.P.; Seppelt, K.; Strauss, S.H.; Boltalina, O.V. Synthesis and X-ray or NMR/DFT structure elucidation of twenty-one new trifluoromethyl derivatives of soluble cage isomers of C76, C78, C84, and C90. J. Am. Chem. Soc. 2008, 130, 13471–13489. [Google Scholar] [CrossRef]
- Amsharov, K.; Jansen, M. Synthesis of a higher fullerene precursor-An “unrolled” C84 fullerene. Chem. Commun. 2009, 2009, 2691–2693. [Google Scholar] [CrossRef]
- Rehaman, A.; Gagliardi, L.; Pyykko, P. Pocket and antipocket conformations for the CH4@C84 endohedral fullerene. Int. J. Quantum Chem. 2007, 107, 1162–1169. [Google Scholar] [CrossRef][Green Version]
- Slanina, Z.; Uhlík, F.; Bao, L.; Akasaka, T.; Lu, X.; Adamowicz, L. Calculated relative populations for the Eu@C84 isomers. Fuller. Nanotub. Carbon Nanostruct. 2021, 29, 144–148. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Bao, L.; Akasaka, T.; Lu, X.; Adamowicz, L. Eu@C86 isomers: Calculated relative populations. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 565–570. [Google Scholar] [CrossRef]
- Slanina, Z.; Adamowicz, L.; Bakowies, D.; Thiel, W. Fullerene C50 isomers: Temperature-induced interchange of relative stabilities. Thermochim. Acta 1992, 202, 249–254. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlik, F.; Lee, S.-L.; Osawa, E. Geometrical and thermodynamic approaches to the relative stabilities of fullerene isomers. MATCH Commun. Math. Comput. Chem. 2001, 44, 335–348. [Google Scholar]
- Slanina, Z.; Kobayashi, K.; Nagase, S. Ca@C72 IPR and non-IPR structures: Computed temperature development of their relative concentrations. Chem. Phys. Lett. 2003, 372, 810–814. [Google Scholar] [CrossRef]
- Slanina, Z.; Kobayashi, K.; Nagase, S. Ca@C74 isomers: Relative concentrations at higher temperatures. Chem. Phys. 2004, 301, 153–157. [Google Scholar] [CrossRef]
- Slanina, Z.; Kobayashi, K.; Nagase, S. Computed temperature development of the relative stabilities of La@C82 isomers. Chem. Phys. Lett. 2004, 388, 74–78. [Google Scholar] [CrossRef]
- Khamatgalimov, A.R.; Luzhetskii, A.V.; Kovalenko, V.I. Unusual pentagon and hexagon geometry of three isomers (No 1, 20, and 23) of fullerene C84. Int. J. Quantum Chem. 2008, 108, 1334–1339. [Google Scholar] [CrossRef]
- Sun, G.; Kertesz, M. Isomer identification for fullerene C84 by 13C NMR spectrum: A density-functional theory study. J. Phys. Chem. A 2001, 105, 5212–5220. [Google Scholar] [CrossRef]
- Chen, Z.; Cioslowski, J.; Rao, N.; Moncrieff, D.; Bühl, M.; Hirsch, A.; Thiel, W. Endohedral chemical shifts in higher fullerenes with 72-86 carbon atoms. Theor. Chem. Acc. 2001, 106, 364–368. [Google Scholar] [CrossRef]
- Cioslowski, J.; Rao, N.; Moncrieff, D. Standard enthalpies of formation of fullerenes and their dependence on structural motifs. J. Am. Chem. Soc. 2000, 122, 8265–8270. [Google Scholar] [CrossRef]
- Epple, L.; Amsharov, K.; Simeonov, K.; Dix, I.; Jansen, M. Crystallographic characterization and identification of a minor isomer of C84 fullerene. Chem. Commun. 2008, 5610–5612. [Google Scholar] [CrossRef]
- Tamm, N.B.; Sidorov, L.N.; Kemnitz, E.; Troyanov, S.I. Isolation and structural X-ray investigation of perfluoroalkyl derivatives of six cage isomers of C84. Chem. A Eur. J. 2009, 15, 10486–10492. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fullerene | HOMO−LUMO | T | BREmin |
|---|---|---|---|
| C60 (Ih) | 0.7566 | 45.40 | 0.082 |
| C70 (D5h) | 0.5293 | 37.05 | 0.052 |
| C72 (D6d) | 0.7023 | 50.57 | 0.0845 |
| C74 (D3h) | 0.1031 | 7.63 | −0.0053 |
| C82 isomer 1 (C2) | 0.1495 | 12.26 | −0.060 |
| isomer 2 (Cs) | 0.3313 | 27.17 | 0.009 |
| isomer 3 (C2) | 0.256 | 21.06 | −0.040 |
| isomer 4 (Cs) | 0.2450 | 20.09 | −0.031 |
| isomer 5 (C2) | 0.1300 | 10.66 | −0.117 |
| isomer 6 (Cs) | 0.0683 | 5.60 | −0.162 |
| isomer 7 (C3v) | 0.0000 | 0.00 | −0.241 |
| isomer 8 (C3v) | 0.0467 | 3.83 | −0.188 |
| isomer 9 (C2v) | 0.0160 | 1.31 | −0.227 |
| C84 isomer 1 (D2) | 0.6143 | 51.60 | 0.0819 |
| isomer 20 (Td) | 0.6962 | 58.48 | 0.0771 |
| isomer 24 (D6h) | 0.5293 | 44.46 | 0.0351 |
 | a 1 | b | c | d |
|---|---|---|---|---|
| (D6h) | (C5v) | (C4v) | (C3v) | |
| Bonds lengths, Å | ||||
| CCcentral 2 | 1.43 | 1.42 | 1.45 | 1.45 |
| CCradial | 1.42 | 1.38 | 1.37 | 1.37 |
| CCside | 1.42 | 1.45 | 1.46 | 1.48 |
| CCborder | 1.37 | 1.39 | 1.38 | 1.38 |
| CH | 1.09 | 1.09 | 1.09 | 1.09 |
| Sum of valence angles Σ, grad | 360.00 | 354.07 | 332.17 | 298.88 |
| Dihedral angles, grad. | ||||
| I3 | 180.00 | 159.09 | 134.03 | 105.64 |
| II | 180.00 | 153.52 | 127.08 | 109.00 |
 | Neutral | Anion | Dianion |
|---|---|---|---|
| C13H9—phenalenyl | |||
| a 1 | 1.43 | 1.45 | 1.47 |
| b | 1.39 | 1.39 | 1.42 |
| c | 1.42 | 1.43 | 1.42 |
| Electron affinity | 0.84 | 5.97 | – |
| HOMO–LUMO gap | 1.94 | 3.58 | 1.41 |
| C13H6(CH3)3—2,5,8–tri–methyl–phenalenyl | |||
| a | 1.44 | 1.45 | 1.44–1.45 |
| b | 1.41 | 1.41 | 1.40–1.44 |
| c | 1.43–1.44 | 1.44 | 1.43–1.48 |
| Electron affinity | 0.59 | 5.18 | – |
| HOMO–LUMO gap | 1.80 | 3.29 | 1.35 |
| C13H6(C4H9)3—2,5,8–tri–tert–butyl–phenalenyl | |||
| a | 1.43 | 1.44 | 1.44–1.48 |
| b | 1.40–1.41 | 1.40 | 1.40–1.45 |
| c | 42 | 1.42 | 1.41–1.44 |
| Electron affinity | 0.91 | 5.29 | – |
| HOMO–LUMO gap | 1.92 | 3.56 | 1.06 |
| C13H6(C4H9)3—2,5,8–tri–tert–butyl–phenalenyl (experimental values) [56] | |||
| a | 1.41–1.42 | ||
| b | 1.37–1.39 | ||
| c | 1.41–1.42 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamatgalimov, A.R.; Kovalenko, V.I. Substructural Approach for Assessing the Stability of Higher Fullerenes. Int. J. Mol. Sci. 2021, 22, 3760. https://doi.org/10.3390/ijms22073760
Khamatgalimov AR, Kovalenko VI. Substructural Approach for Assessing the Stability of Higher Fullerenes. International Journal of Molecular Sciences. 2021; 22(7):3760. https://doi.org/10.3390/ijms22073760
Chicago/Turabian StyleKhamatgalimov, Ayrat R., and Valeri I. Kovalenko. 2021. "Substructural Approach for Assessing the Stability of Higher Fullerenes" International Journal of Molecular Sciences 22, no. 7: 3760. https://doi.org/10.3390/ijms22073760
APA StyleKhamatgalimov, A. R., & Kovalenko, V. I. (2021). Substructural Approach for Assessing the Stability of Higher Fullerenes. International Journal of Molecular Sciences, 22(7), 3760. https://doi.org/10.3390/ijms22073760