
Cubic Octa-Carbon: Quantum-Chemical Design of Molecular Structure and Potential Way of Its Synthesis from Cubane



Abstract
:1. Introduction
2. Calculation Method
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kharisov, B.I.; Kharissova, O.V. Carbon Allotropes: Metal-Complex Chemistry, Properties and Applications; Springer Nature AG: Cham, Switzerland, 2019; ISBN 978-3-030-03504-4. [Google Scholar] [CrossRef]
- Belenkov, E.A.; Mavrinsky, V.V. Crystal structure of a perfect carbyne. Crystallogr. Rep. 2008, 53, 83–87. [Google Scholar] [CrossRef]
- Pan, B.; Xiao, J.; Li, J.; Liu, P.; Wang, C.; Yang, G. Carbyne with finite length: The one-dimensional sp carbon. Sci. Adv. 2015, 1, e1500857. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Rohringer, P.; Suenaga, K.; Niimi, K.S.Y.; Kotakoski, J.; Meyer, J.; Peterlik, H.; Wanko, M.; Jahangirov, S.; Rubio, A.; et al. Confined linear carbon chains as a route to bulk carbyne. Nat. Mater. 2016, 15, 634–639. [Google Scholar] [CrossRef]
- Schueller, O.J.A.; Brittain, S.T.; Whitesides, G.M. Fabrication of glassy carbon microstructures by pyrolysis of microfabricated polymeric precursors. Adv. Mater. 1997, 9, 477–480. [Google Scholar] [CrossRef]
- Jorio, A.; Dresselhaus, M.S.; Dresselhaus, G.; Gogotsi, Y. Carbon Nanotubes: Advanced Topics in the Synthesis, Structure, Properties and Applications, 1st ed; Springer: Berlin, Germany, 2008; ISBN 978-3-540-72864-1. [Google Scholar] [CrossRef]
- Tibbetts, G.; Lake, M.; Strong, K.; Rice, B. A review of the fabrication and properties of vapor-grown carbon nanofiber/polymer composites. Compos. Sci. Technol. 2007, 67, 1709–1718. [Google Scholar] [CrossRef]
- Khamatgalimov, A.; Kovalenko, V. Substructural Approach for Assessing the Stability of Higher Fullerenes. Int. J. Mol. Sci. 2021, 22, 3760. [Google Scholar] [CrossRef]
- McCulloch, D.G.; McKenzie, D.R.; Goringe, C.M. Ab initio simulations of the structure of amorphous carbon. Phys. Rev. 2000, B61, 2349. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R Rep. 2002, 37, 129–281. [Google Scholar] [CrossRef] [Green Version]
- Katsnelson, M.; Novoselov, K. Graphene: New bridge between condensed matter physics and quantum electrodynamics. Solid State Commun. 2007, 143, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Novoselov, K. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ghuge, A.D.; Shirode, A.R.; Kadam, V.J. Graphene: A comprehensive review. Curr. Drug Targets 2017, 18, 724–733. [Google Scholar] [CrossRef]
- Wu, J.; Pisula, W.; Mullen, K. Graphenes as potential material for electronics. Chem. Rev. 2007, 107, 718–747. [Google Scholar] [CrossRef]
- Yu, X.; Cheng, H.; Zhang, M.; Zhao, Y.; Qu, L.; Shi, G. Graphene-based smart materials. Nature Rev. Mater. 2017, 2, 17046. [Google Scholar] [CrossRef]
- Novoselov, K.S. Technology: Rapid progress in producing graphene. Nature 2014, 505, 291. [Google Scholar] [CrossRef] [Green Version]
- Van den Brink, J. Graphene: From strength to strength. Nat. Nanotechnol. 2007, 2, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S. Structure, stability, and fragmentation of small carbon clusters. J. Chem. Phys. 1987, 87, 2191–2197. [Google Scholar]
- Martin, J.M.L.; Francois, J.P.; Gijbels, R. Ab initio study of the infrared spectra of linear Cn clusters (n = 6–9). J. Chem. Phys. 1990, 93, 8850–8861. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Francois, J.P.; Gijbels, R. A critical comparison of MINDO/3, MNDO, AM1, and PM3 for a model problem: Carbon clusters C2-C10. An ad hoc reparametrization of MNDO well suited for the accurate prediction of their spectroscopic constants. J. Comput. Chem. 1991, 12, 52–70. [Google Scholar] [CrossRef]
- Parasuk, V.; Almlöf, J. The electronic and molecular structure of carbon clusters: C8 and C10. Theor. Chim. Acta 1992, 83, 227–237. [Google Scholar] [CrossRef]
- Hutter, J.; Luethi, H.P.; Diederich, F. Structures and vibrational frequencies of the carbon molecules C2-C18 calculated by density functional theory. J. Am. Chem. Soc. 1994, 116, 750–756. [Google Scholar] [CrossRef]
- Tseng, S.; Shen, M.; Yu, C. A MNDO study of carbon clusters with specifically fitted parameters. Theor. Chim. Acta 1995, 92, 269–280. [Google Scholar] [CrossRef]
- Martin, J.M.L.; El-Yazal, J.; Francois, J.-P. Structure and vibrational spectra of carbon clusters Cn (n = 2–10, 12, 14, 16, 18) using density functional theory including exact exchange. Chem. Phys. Lett. 1995, 242, 570–579. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Taylor, P.R. Structure and Vibrations of Small Carbon Clusters from Coupled-Cluster Calculations. J. Phys. Chem. 1996, 100, 6047–6056. [Google Scholar] [CrossRef]
- Wang, S.L.; Rittby, C.M.L.; Graham, W.R.M. Detection of cyclic carbon clusters. II. Isotopic study of the 𝜈12(e𝑢) mode of cyclic C8 in solid Ar. J. Chem. Phys. 1997, 107, 7025–7033. [Google Scholar] [CrossRef]
- Nyrönen, T.H.; Reijo Suontamo, R. An MO study of neutral C high-symmetry clusters. Chem. Phys. Lett. 1997, 280, 227–232. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional study of carbon clusters C2n (2 ≤ n ≤ 16). I. Structure and bonding in the neutral clusters. J. Chem. Phys. 1999, 110, 5189–5200. [Google Scholar] [CrossRef] [Green Version]
- Sharapa, D.; Hirsch, A.; Meyer, B.; Clark, T. Cubic C8: An Observable Allotrope of Carbon? ChemPhysChem 2015, 16, 2165–2171. [Google Scholar] [CrossRef] [Green Version]
- Varandas, A.J.C. Even numbered carbon clusters: Cost-effective wavefunction-based method for calculation and automated location of most structural isomers. Eur. Phys. J. D 2018, 72, 134. [Google Scholar] [CrossRef]
- Rocha, C.M.R.; Li, J.; Varandas, A.J.C. Difficulties and Virtues in Assessing the Potential Energy Surfaces of Carbon Clusters via DMBE Theory: Stationary Points of Cκ (κ = 2−10) at the Focal Point. J. Phys. Chem. A 2019, 123, 3121–3130. [Google Scholar] [CrossRef]
- Chaglayan, B.; Huran, A.W.; Amor, N.B.; Brumas, V.; Evangelisti, S.; Leininger, T. Spherical aromaticity and electron delocalization in C8 and B4N4 cubic systems. Theor. Chem. Acc. 2019, 138, 5. [Google Scholar] [CrossRef]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pople, A.; Krishnan, R.; Schlegel, H.B.; Binkley, J.S. Electron Correlation Theories and Their Application to the Study of Simple Reaction Potential Surfaces. Int. J. Quantum Chem. 1978, 14, 545–560. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D., III. Many-body perturbation-theory, coupled-pair many-electron theory, and importance of quadruple excitations for correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model—The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Krishnan, R. Quadratic configuration interaction—A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Revs. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Revs. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian Inc.: Wallingford, UK, 2000. [Google Scholar]
- Eaton, P.E. Cubanes: Starting materials for the chemistry of the 1990s and the new century. Angew. Chem. Int. Ed. 1992, 31, 1421–1436. [Google Scholar] [CrossRef]
- Biegasiewicz, K.F.; Griffiths, J.R.; Savage, G.P.; Tsanaktsidis, J.; Priefer, R. Cubane: 50 Years Later. Chem. Rev. 2015, 115, 6719–6745. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, L.; Hedberg, K.; Eaton, P.E.; Nodari, N.; Robiette, A.G. Bond lengths and quadratic force field for cubane. J. Am. Chem. Soc. 1991, 113, 1514–1517. [Google Scholar] [CrossRef]
- Schubert, W.; Yoshimine, M.; Pacansky, J. Ab initio self-consistent field calculations on the structure of cubane, cubene, and the cubyl radical. J. Phys. Chem. 1981, 85, 1340–1342. [Google Scholar] [CrossRef]
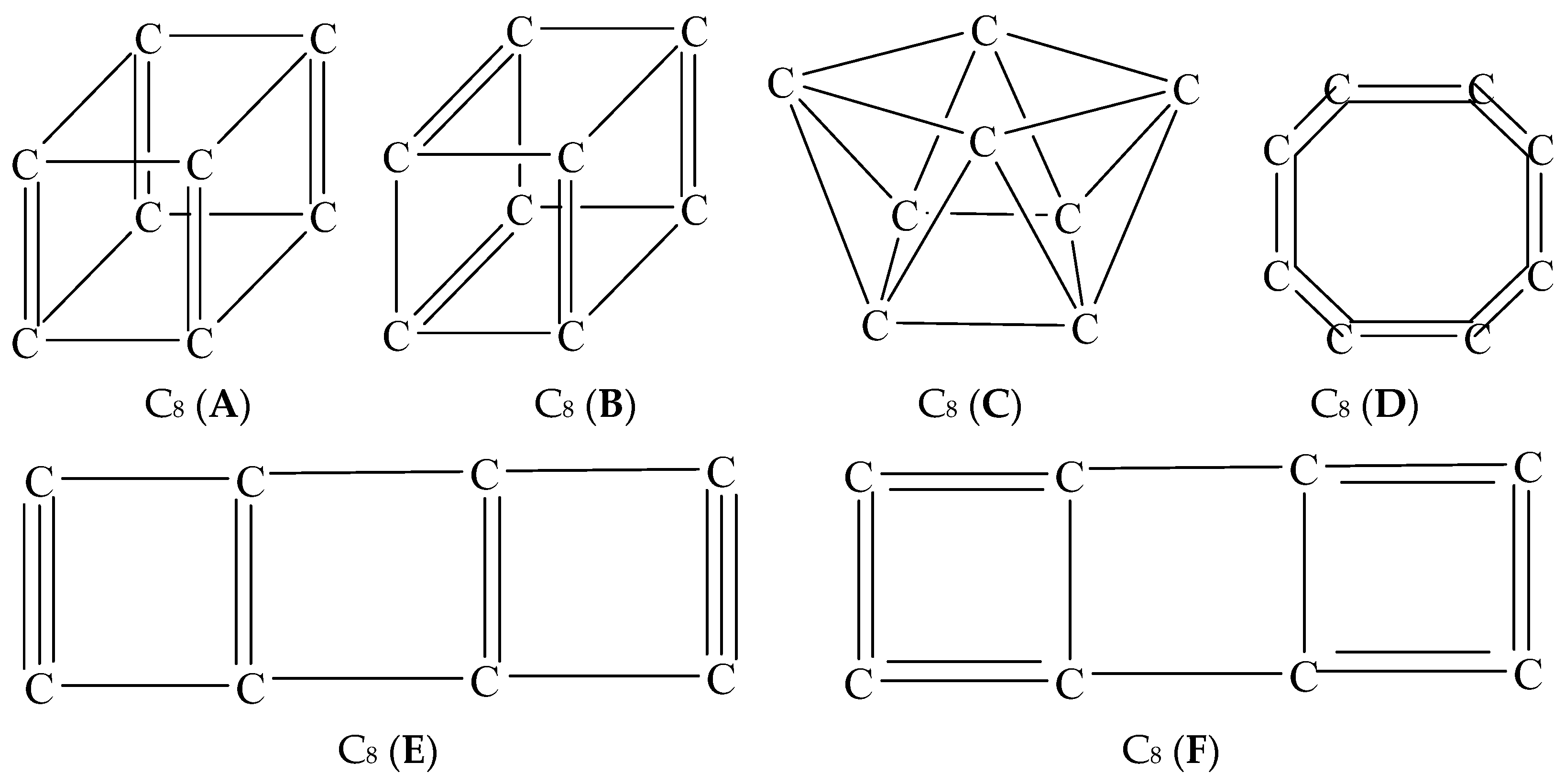
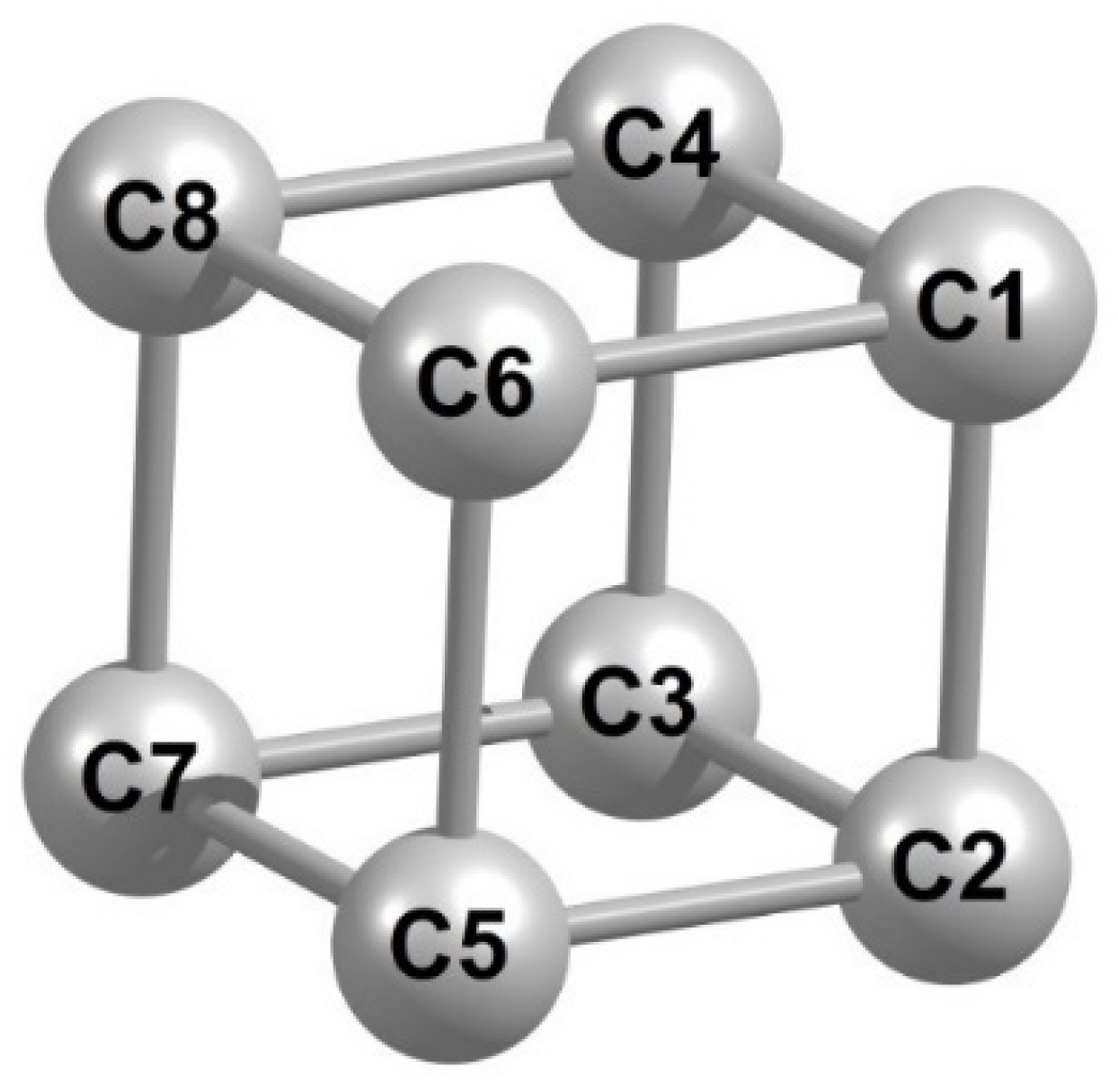

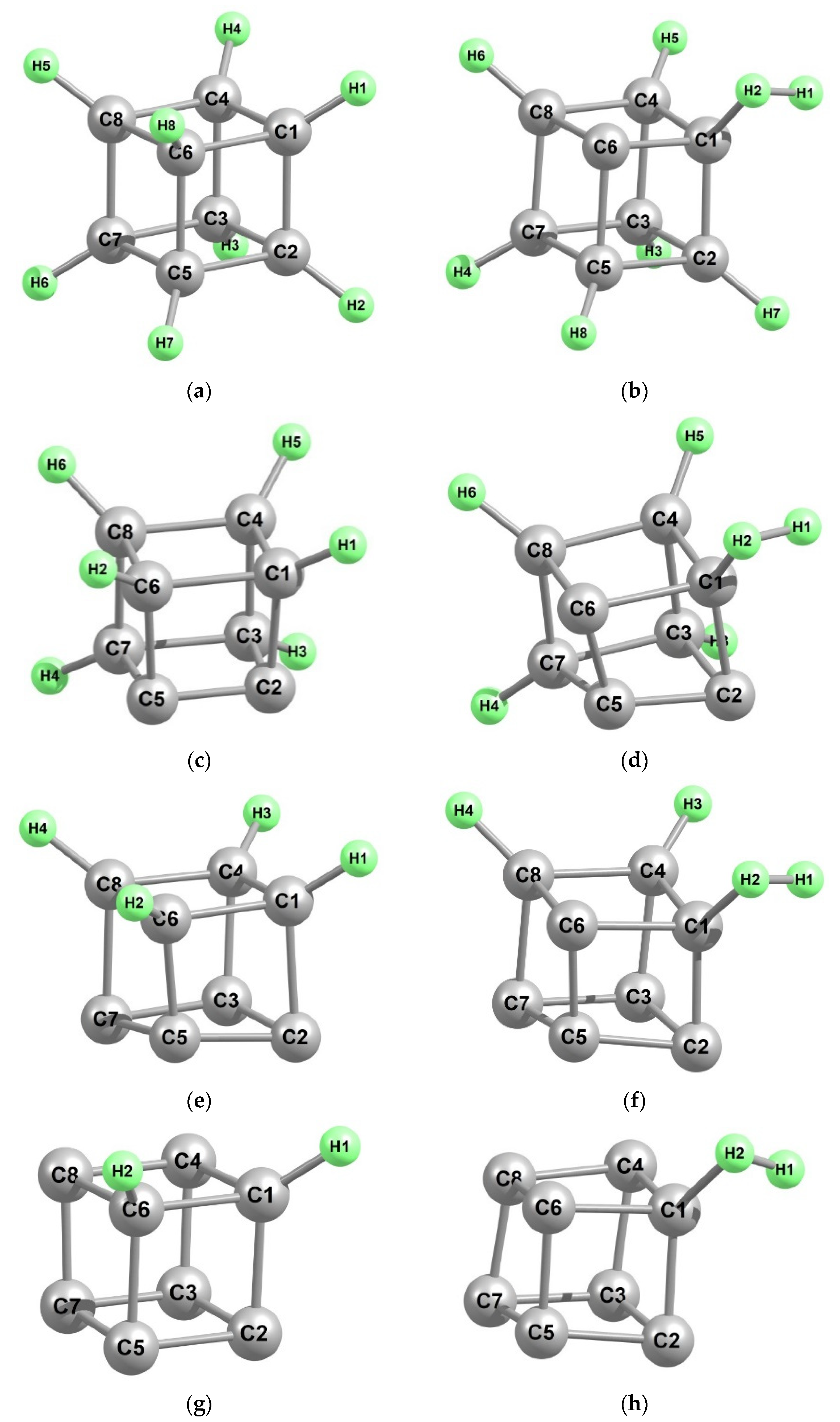

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Calculated by | Parameter | Calculated by | ||
|---|---|---|---|---|---|
| CCSD(T)/QZVP | B3PW91/QZVP | CCSD(T)/QZVP | B3PW91/QZVP | ||
| Carbon–Carbon Bond Lengths, pm | |||||
| (C1C4) | 148.3 | 146.7 | (C7C5) | 148.0 | 146.7 |
| (C4C8) | 148.0 | 146.7 | (C5C2) | 148.6 | 146.7 |
| (C8C6) | 148.2 | 146.7 | (C1C2) | 148.3 | 146.7 |
| (C6C1) | 148.1 | 146.7 | (C3C4) | 148.3 | 146.7 |
| (C2C3) | 148.3 | 146.7 | (C5C6) | 148.5 | 146.7 |
| (C3C7) | 148.5 | 146.7 | (C7C8) | 148.5 | 146.7 |
| Bond Angles, deg | |||||
| (C1C4C8) | 90.4 | 90.0 | (C5C6C1) | 90.4 | 90.0 |
| (C4C8C6) | 89.6 | 90.0 | (C6C1C2) | 89.8 | 90.0 |
| (C8C6C1) | 90.3 | 90.0 | (C6C5C7) | 89.9 | 90.0 |
| (C6C1C4) | 89.7 | 90.0 | (C5C7C8) | 90.2 | 90.0 |
| (C2C5C7) | 89.9 | 90.0 | (C7C8C6) | 89.8 | 90.0 |
| (C5C7C3) | 90.3 | 90.0 | (C8C6C5) | 90.1 | 90.0 |
| (C1C2C3) | 90.2 | 90.0 | (C7C8C4) | 89.8 | 90.0 |
| (C2C3C4) | 89.8 | 90.0 | (C8C4C3) | 90.4 | 90.0 |
| (C3C4C1) | 90.2 | 90.0 | (C4C3C7) | 89.7 | 90.0 |
| (C6C1C4) | 89.7 | 90.0 | (C3C7C8) | 90.1 | 90.0 |
| (C1C2C5) | 90.3 | 90.0 | (C7C8C4) | 89.8 | 90.0 |
| (C2C5C6) | 89.9 | 90.0 | (C8C4C3) | 90.4 | 90.0 |
| Selected Torsion (Dihedral) Angles, deg | |||||
| (C1C4C8C6) | –0.2 | 0.0 | (C1C2C7C8) | 0.0 | 0.0 |
| (C1C4C3C2) | 0.4 | 0.0 | (C4C6C5C3) | 0.2 | 0.0 |
| (C1C2C5C6) | 0.0 | 0.0 | (C1C2C3C7) | –90.1 | –90.0 |
| (C2C3C7C5) | –0.2 | 0.0 | (C2C5C6C8) | 90.3 | 90.0 |
| (C5C6C8C7) | –0.4 | 0.0 | (C5C6C8C4) | –90.2 | –90.0 |
| (C3C4C8C7) | 0.2 | 0.0 | (C7C8C4C1) | –90.0 | –90.0 |
| Calculation Method | Effective Charge on Carbon Atoms, in Units of Electron Charge (ē) | |||||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | |
| CCSD(T)/QZVP | −0.0104 | +0.0099 | −0.0098 | +0.0102 | −0.0097 | +0.0102 | +0.0099 | −0.0103 |
| B3PW91/QZVP | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
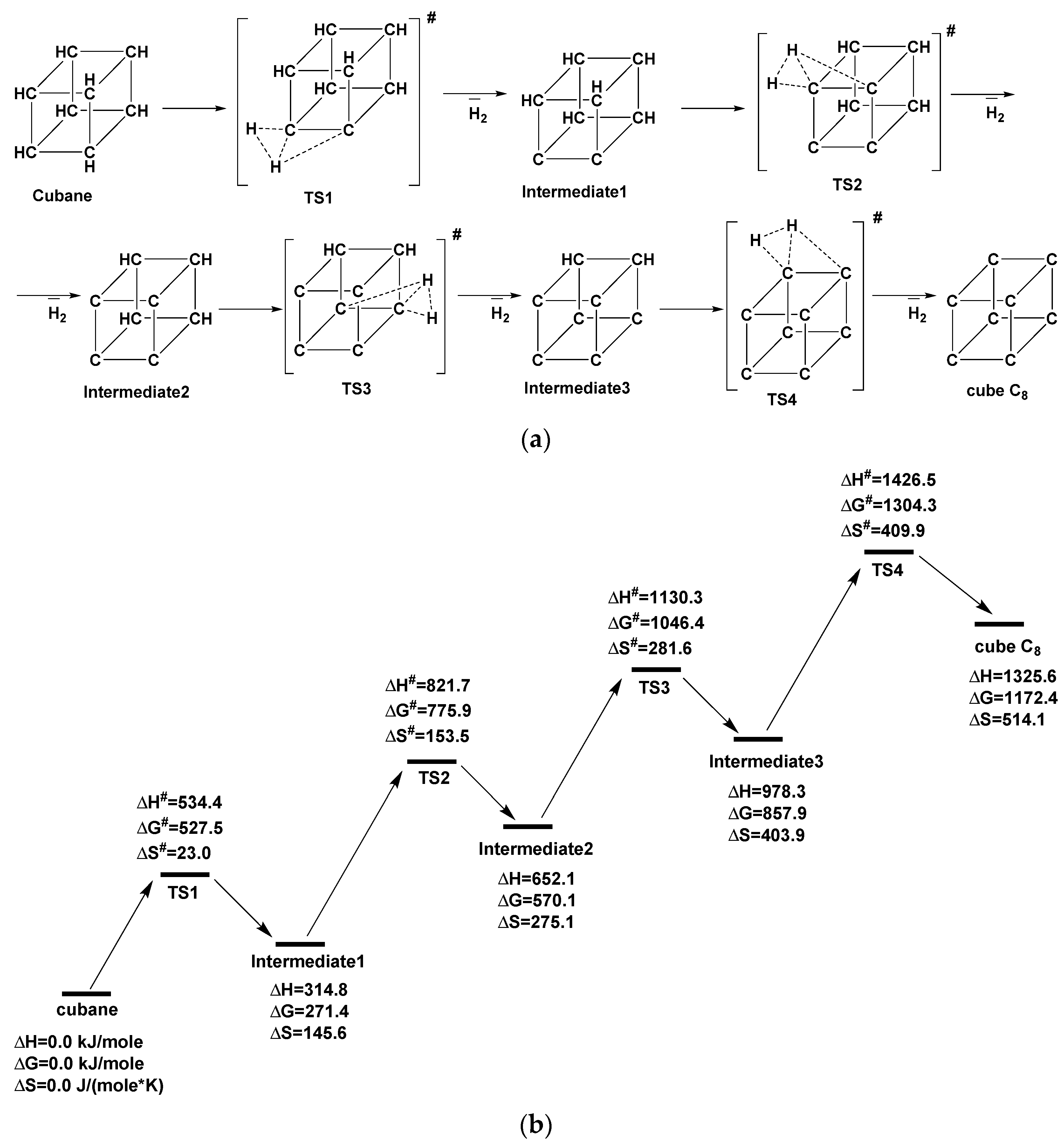
| Transition Stage (TSn) | TS1 | TS2 | TS3 | TS4 |
|---|---|---|---|---|
| ΔH# ΔG# ΔS# | 534.4 527.5 23.0 | 506.9 504.5 8.0 | 478.2 476.3 6.5 | 448.2 446.5 5.9 |
| ΔHr ΔGr ΔSr | 314.8 271.4 145.6 | 337.3 298.6 129.6 | 326.2 287.8 128.8 | 347.3 314.5 110.1 |
| r(H1H2), pm | 90.9 | 90.4 | 85.2 | 84.3 |
| r(C1H1), pm | 140.2 | 134.3 | 144.4 | 137.6 |
| r(C1H2), pm | 125.5 | 124.4 | 132.8 | 134.5 |
| r(C2H2), pm | 197.2 | 209.4 | 218.9 | 219.2 |
| r(C1C2), pm | 155.0 | 160.7 | 157.7 | 154.4 |
| ∠H1C1H2, deg | 39.6 | 40.7 | 35.5 | 36.1 |
| ∠H2C1C2, deg | 88.7 | 93.7 | 97.4 | 98.5 |
| ν1, cm−1 | −1569 | −1258 | −1144 | −1022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chachkov, D.V.; Mikhailov, O.V. Cubic Octa-Carbon: Quantum-Chemical Design of Molecular Structure and Potential Way of Its Synthesis from Cubane. Int. J. Mol. Sci. 2021, 22, 12067. https://doi.org/10.3390/ijms222112067
Chachkov DV, Mikhailov OV. Cubic Octa-Carbon: Quantum-Chemical Design of Molecular Structure and Potential Way of Its Synthesis from Cubane. International Journal of Molecular Sciences. 2021; 22(21):12067. https://doi.org/10.3390/ijms222112067
Chicago/Turabian StyleChachkov, Denis V., and Oleg V. Mikhailov. 2021. "Cubic Octa-Carbon: Quantum-Chemical Design of Molecular Structure and Potential Way of Its Synthesis from Cubane" International Journal of Molecular Sciences 22, no. 21: 12067. https://doi.org/10.3390/ijms222112067
APA StyleChachkov, D. V., & Mikhailov, O. V. (2021). Cubic Octa-Carbon: Quantum-Chemical Design of Molecular Structure and Potential Way of Its Synthesis from Cubane. International Journal of Molecular Sciences, 22(21), 12067. https://doi.org/10.3390/ijms222112067