
Understanding the Molecular Basis of 5-HT4 Receptor Partial Agonists through 3D-QSAR Studies



Abstract
1. Introduction
2. Results and Discussion
2.1. Studied Compounds
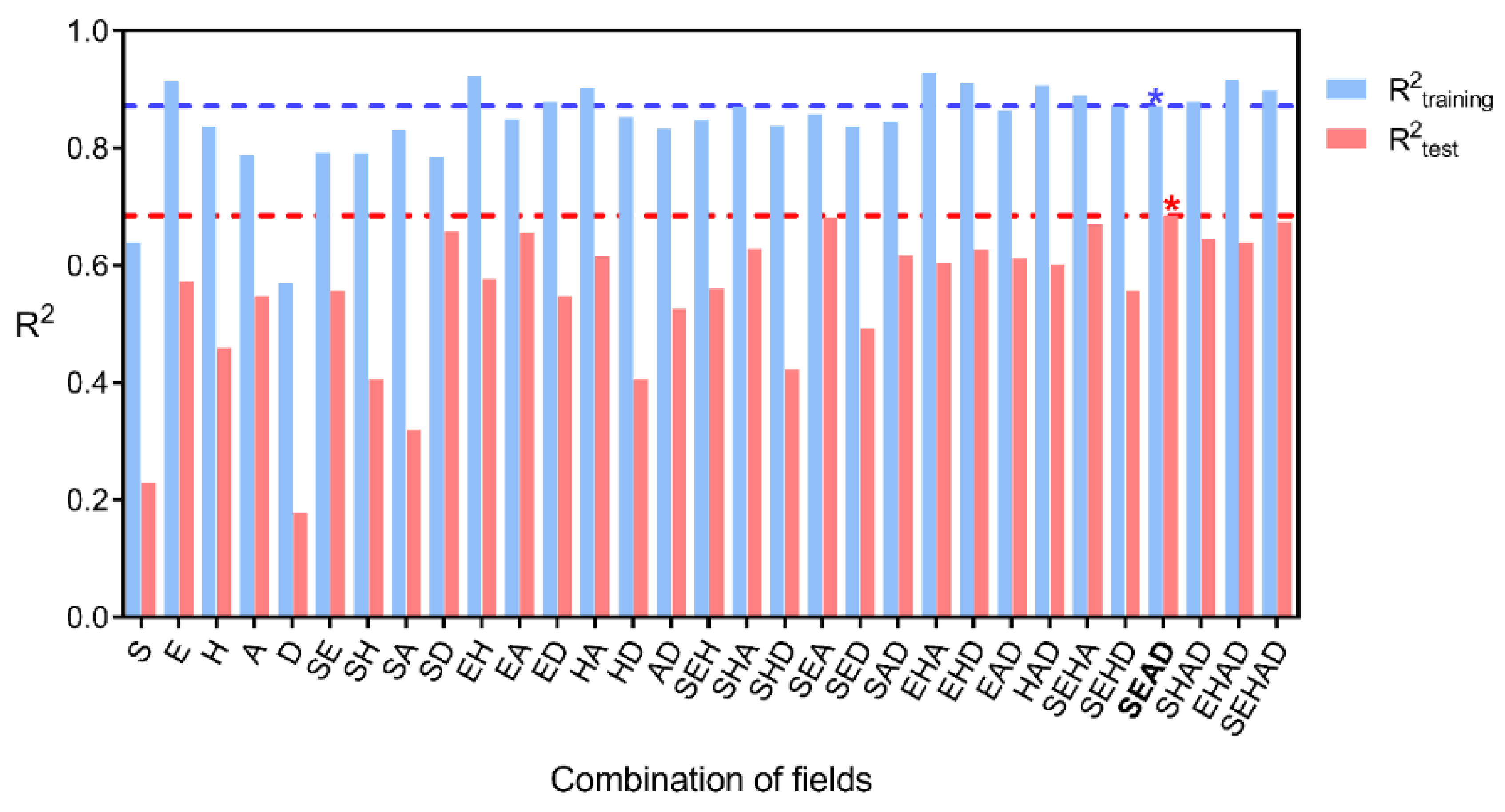
2.2. Statistical Results
2.3. Analysis of the 3D-QSAR Models
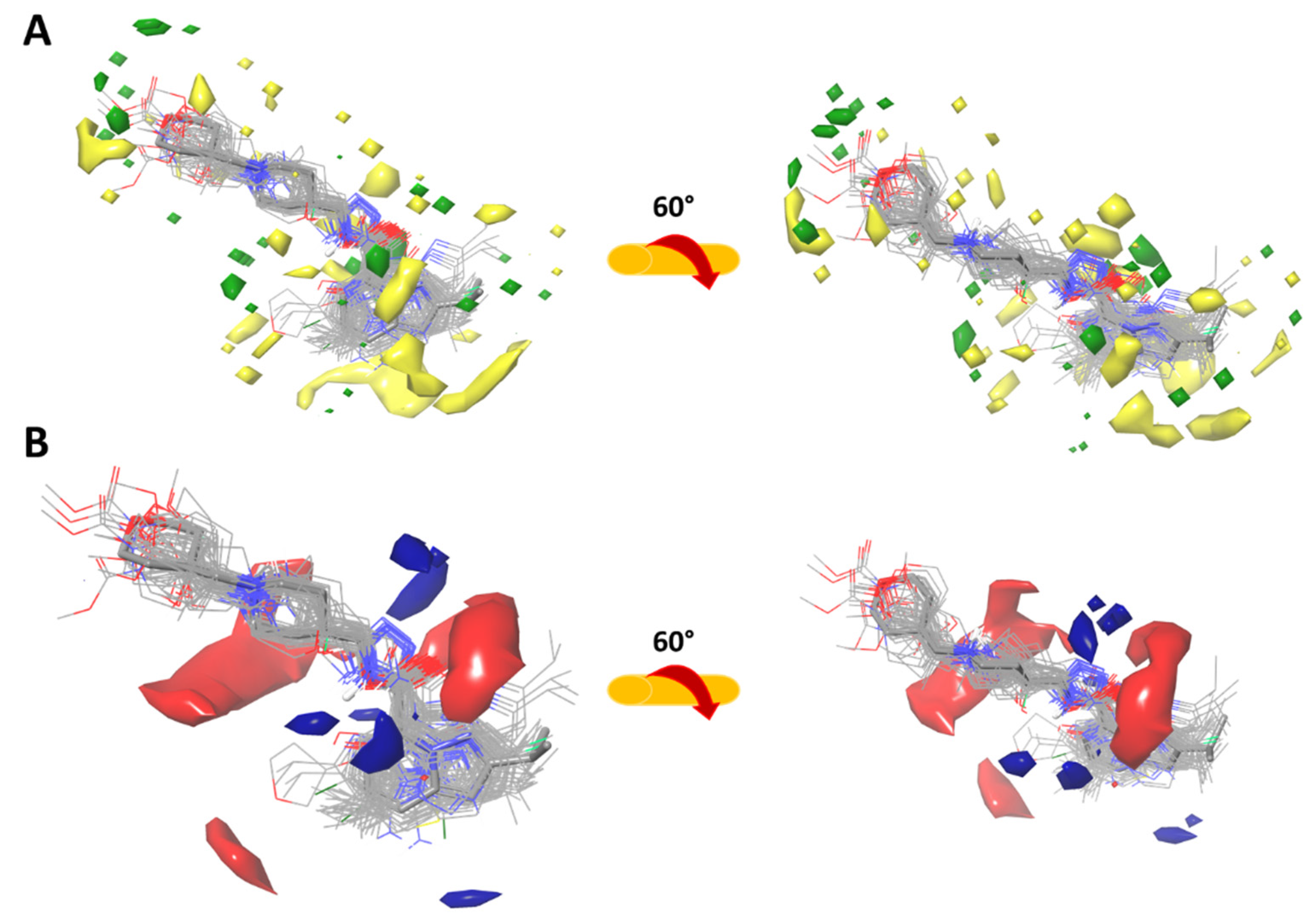
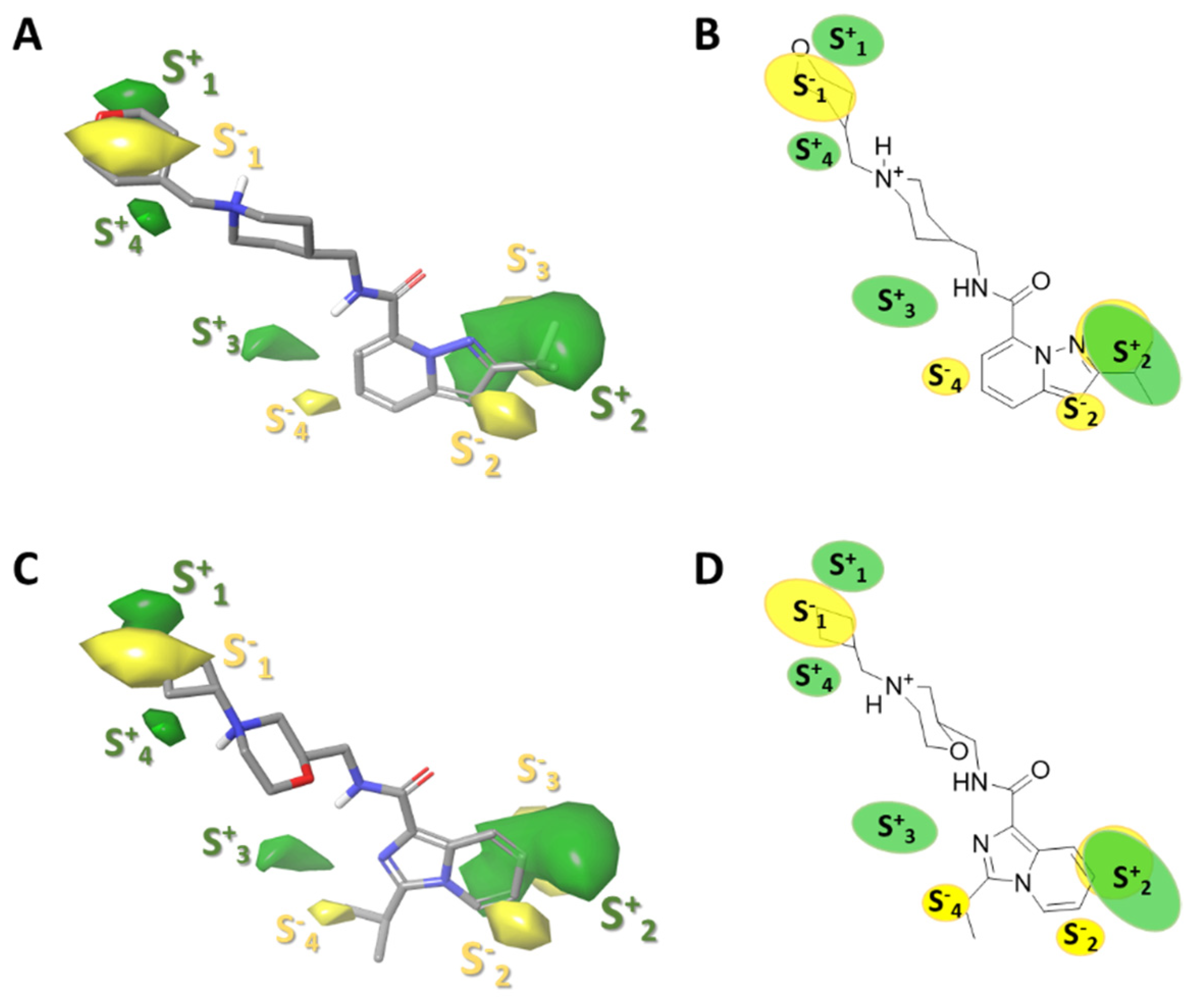
2.3.1. Force-Field Based 3D-QSAR Model—Steric and Electrostatic Contour Map
2.3.2. Gaussian-Field Based 3D-QSAR Model—Steric Contour Map
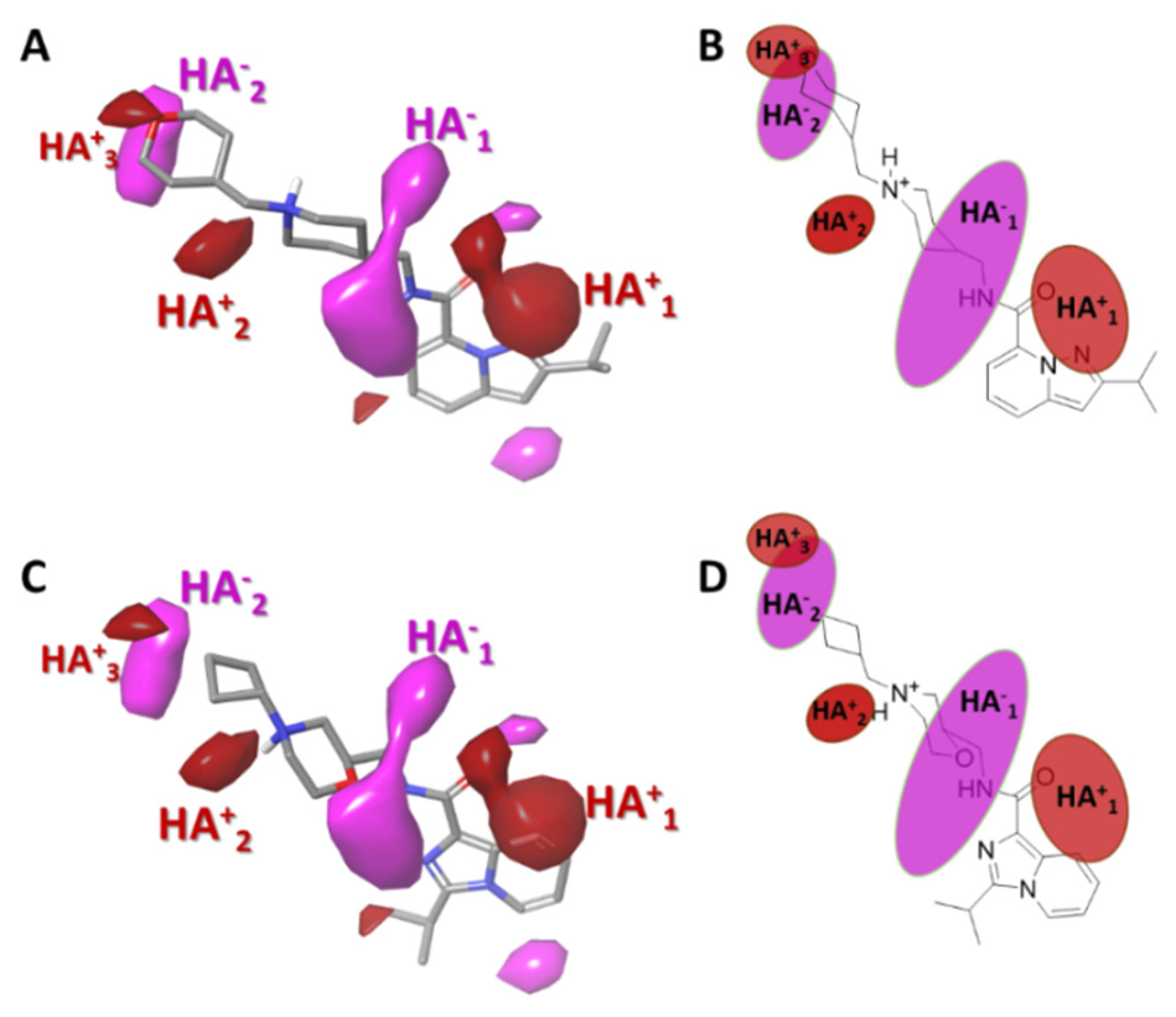
2.3.3. Gaussian-Field Based 3D-QSAR Model—Electrostatic Contour Map
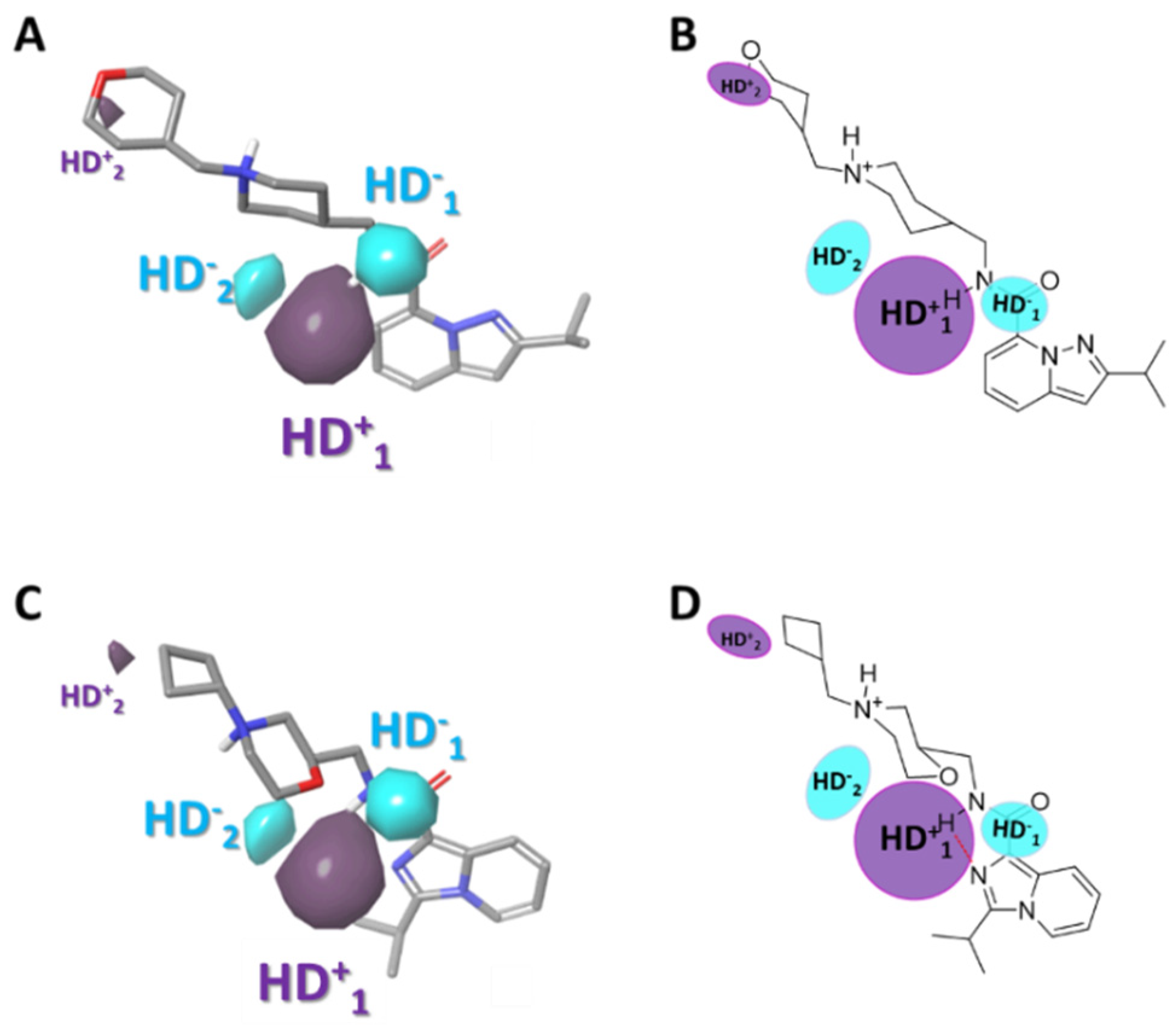
2.3.4. Gaussian-Field Based 3D-QSAR Model—Hydrogen Bond Acceptor Contour Maps
2.3.5. Gaussian-Field Based 3D-QSAR Model—Hydrogen Bond Donor Contour Maps
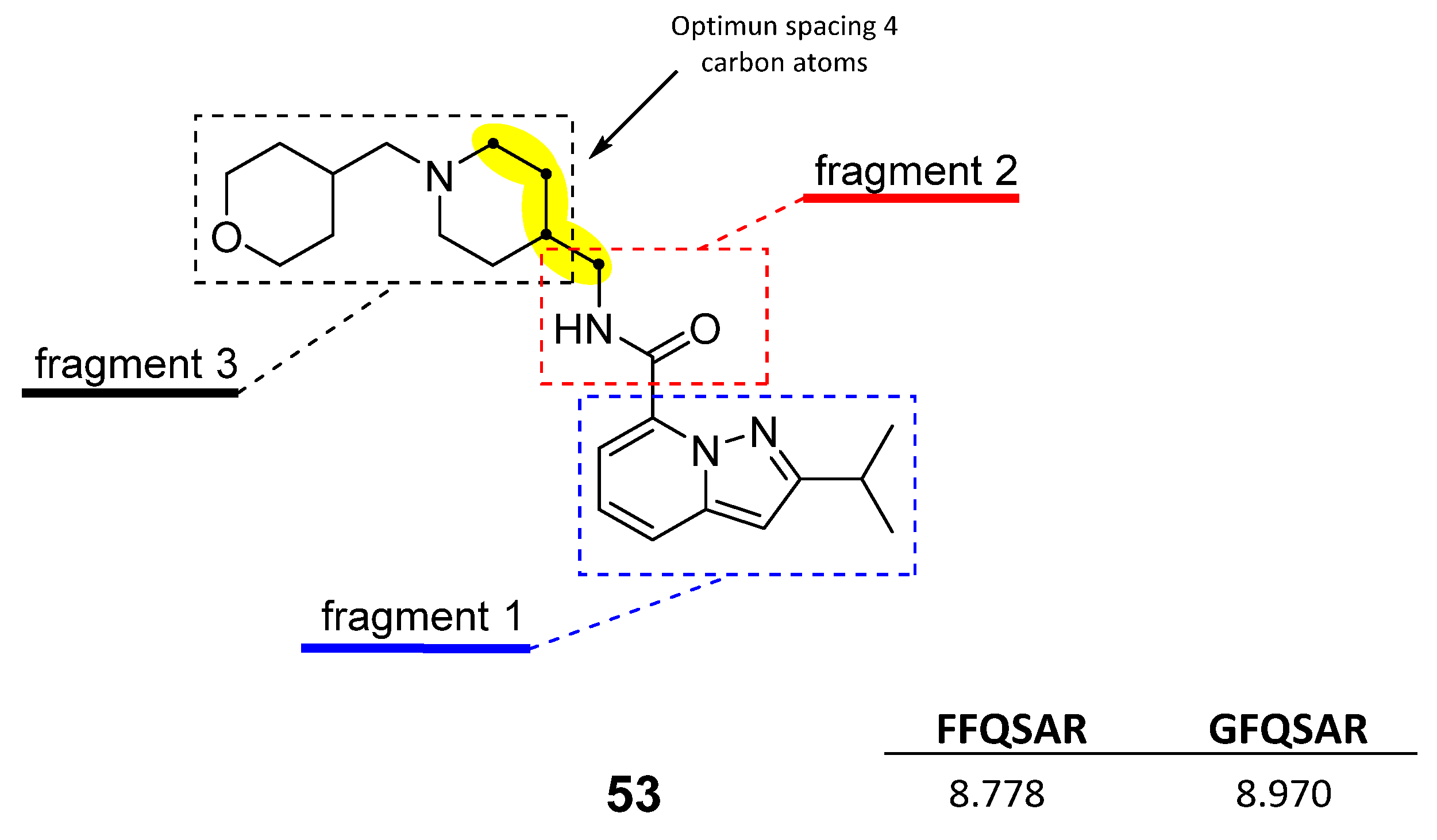
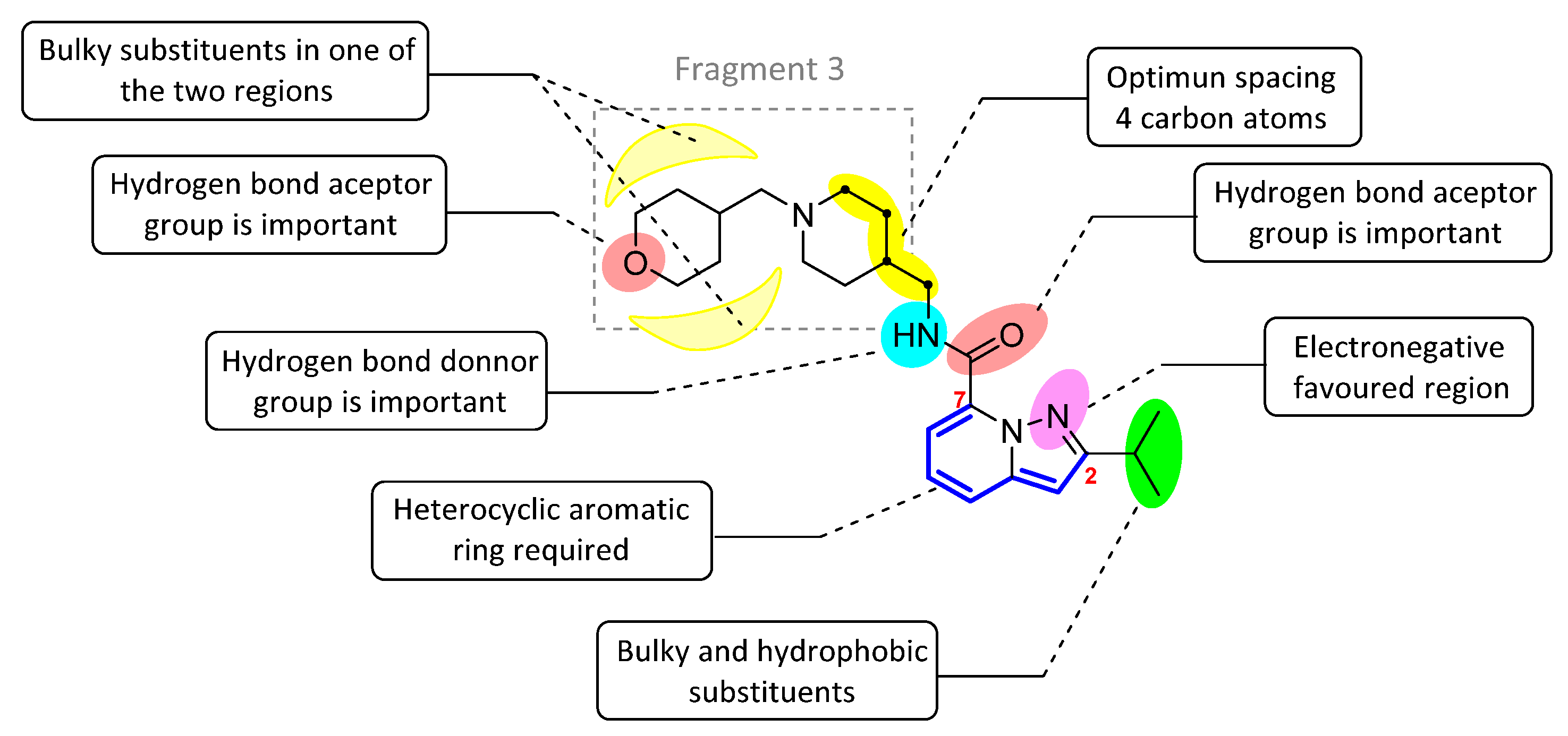
2.4. Design of New Derivatives
2.5. ADMET Predictions
3. Materials and Methods
3.1. Dataset Collection
3.2. Alignment
3.3. Field-Based QSAR Model
3.4. Prediction ADMET Properties
4. Conclusions
- (1)
- The four-carbon atom distance between the amide nitrogen and the aliphatic amine corresponding to fragment 3.
- (2)
- Structural variability in fragment 3 considering aliphatic rings that provide a favourable hydrophobic source for activity
- (3)
- The hydrogen bond acceptor groups in fragment 3 can enhance the activity of compounds.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT4R | Receptor 5-HT4 |
| AD | Alzheimer’s disease |
| S | Steric |
| E | Electrostatic |
| H | Hydrophobic |
| HBA | Hydrogen bond acceptor |
| HBD | Hydrogen bond donor |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| FFQSAR | Force-field based QSAR |
| GFQSAR | Gaussian-field based QSAR |
| CCC | Correlation coefficient of concordance |
| 3D-QSAR | Three-dimensional quantitative structure–activity relationship |
References
- McDowell, S.E. Adverse reactions to drugs used in the treatment of Alzheimer’s disease. Adverse Drug React. Bull. 2011, 1031–1034. [Google Scholar] [CrossRef]
- Gerald, C.; Adham, N.; Kao, H.T.; Olsen, M.A.; Laz, T.M.; Schechter, L.E.; Bard, J.A.; Vaysse, P.J.; Hartig, P.R.; Branchek, T.A.; et al. The 5-HT4 receptor: Molecular cloning and pharmacological characterization of two splice variants. EMBO J. 1995, 14, 2806–2815. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Wong, E.H.F.; Dumuis, A.; Bockaert, J. Central 5-HT4 receptors. Trends Pharmacol. Sci. 1995, 16, 391–398. [Google Scholar] [CrossRef]
- Bockaert, J.; Claeysen, S.; Compan, V.; Dumuis, A. 5-HT4 receptors. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 39–51. [Google Scholar] [CrossRef]
- Bockaert, J.; Claeysen, S.; Compan, V.; Dumuis, A. 5-HT4 receptors, a place in the sun: Act two. Curr. Opin. Pharmacol. 2011, 11, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.; Brown, J.T.; Richardson, J.C.; Medhurst, A.D.; Sehmi, S.S.; Calver, A.R.; Randall, A.D. Modulation of hippocampal excitability by 5-HT 4 receptor agonists persists in a transgenic model of Alzheimer’s disease. Neuroscience 2004, 129, 49–54. [Google Scholar] [CrossRef]
- Maillet, M.; Robert, S.; Lezoualc’h, F. New Insights into Serotonin 5-HT4 Receptors: A Novel Therapeutic Target for Alzheimers Disease? Curr. Alzheimer Res. 2004, 1, 79–85. [Google Scholar] [CrossRef]
- Rebholz, H.; Friedman, E.; Castello, J. Alterations of Expression of the Serotonin 5-HT4 Receptor in Brain Disorders. Int. J. Mol. Sci. 2018, 19, 3581. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Q.; Yao, X.X.; Gao, S.H.; Li, R.; Li, B.J.; Yang, W.; Cui, R.J. Role of 5-HT receptors in neuropathic pain: Potential therapeutic implications. Pharmacol. Res. 2020, 159, 104949. [Google Scholar] [CrossRef] [PubMed]
- Tonini, M.; Pace, F. Drugs Acting on Serotonin Receptors for the Treatment of Functional GI Disorders. Dig. Dis. 2006, 24, 59–69. [Google Scholar] [CrossRef]
- Gershon, M.D. Review article: Serotonin receptors and transporters—Roles in normal and abnormal gastrointestinal motility. In Alimentary Pharmacology and Therapeutics; Supplement; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2004; Volume 20, pp. 3–14. [Google Scholar]
- Nam, Y.; Min, Y.S.; Sohn, U.D. Recent advances in pharmacological research on the management of irritable bowel syndrome. Arch. Pharm. Res. 2018, 41, 955–966. [Google Scholar] [CrossRef]
- Bouras, E.P.; Camilleri, M.; Burton, D.D.; McKinzie, S. Selective stimulation of colonic transit by the benzofuran 5HT4 agonist, prucalopride, in healthy humans. Gut 1999, 44, 682–686. [Google Scholar] [CrossRef]
- Konen, J.R.; Haag, M.M.; Guseva, D.; Hurd, M.; Linton, A.A.; Lavoie, B.; Kerrigan, C.B.; Joyce, E.; Bischoff, S.C.; Swann, S.; et al. Prokinetic actions of luminally acting 5-HT4 receptor agonists. Neurogastroenterol. Motil. 2020, 33, e14026. [Google Scholar] [CrossRef]
- Gwynne, R.M.; Bornstein, J.C. Luminal 5-HT4 receptors—A successful target for prokinetic actions. Neurogastroenterol. Motil. 2019, 31, e13708. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F. The serotonin 5-HT4 receptor and the amyloid precursor protein processing. Exp. Gerontol. 2003, 38, 159–166. [Google Scholar] [CrossRef]
- Lanthier, C.; Dallemagne, P.; Lecoutey, C.; Claeysen, S.; Rochais, C. Therapeutic modulators of the serotonin 5-HT4 receptor: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 495–508. [Google Scholar] [CrossRef]
- Brodney, M.A.; Johnson, D.E.; Sawant-Basak, A.; Coffman, K.J.; Drummond, E.M.; Hudson, E.L.; Fisher, K.E.; Noguchi, H.; Waizumi, N.; McDowell, L.L.; et al. Identification of Multiple 5-HT 4 Partial Agonist Clinical Candidates for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2012, 55, 9240–9254. [Google Scholar] [CrossRef]
- Ahmad, I.; Nirogi, R. 5-HT4 Receptor Agonists for the Treatment of Alzheimer’s Dsease. Neurosci. Med. 2011, 02, 87–92. [Google Scholar] [CrossRef][Green Version]
- Modica, M.; Pittala, V.; Romeo, G.; Salerno, L.; Siracusa, M. Serotonin 5-HT3 and 5-HT4 Ligands: An Update of Medicinal Chemistry Research in the Last Few Years. Curr. Med. Chem. 2010, 17, 334–362. [Google Scholar] [CrossRef]
- Castriconi, F.; Paolino, M.; Giuliani, G.; Anzini, M.; Campiani, G.; Mennuni, L.; Sabatini, C.; Lanza, M.; Caselli, G.; De Rienzo, F.; et al. Synthesis and structure–activity relationship studies in serotonin 5-HT4 receptor ligands based on a benzo[de][2,6]naphthridine scaffold. Eur. J. Med. Chem. 2014, 82, 36–46. [Google Scholar] [CrossRef]
- Nirogi, R.; Mohammed, A.R.; Shinde, A.K.; Bogaraju, N.; Gagginapalli, S.R.; Ravella, S.R.; Kota, L.; Bhyrapuneni, G.; Muddana, N.R.; Benade, V.; et al. Synthesis and SAR of Imidazo[1,5-a]pyridine derivatives as 5-HT4 receptor partial agonists for the treatment of cognitive disorders associated with Alzheimer’s disease. Eur. J. Med. Chem. 2015, 103, 289–301. [Google Scholar] [CrossRef]
- Nirogi, R.; Mohammed, A.R.; Shinde, A.K.; Gagginapally, S.R.; Kancharla, D.M.; Middekadi, V.R.; Bogaraju, N.; Ravella, S.R.; Singh, P.; Birangal, S.R.; et al. Synthesis, Structure–Activity Relationships, and Preclinical Evaluation of Heteroaromatic Amides and 1,3,4-Oxadiazole Derivatives as 5-HT 4 Receptor Partial Agonists. J. Med. Chem. 2018, 61, 4993–5008. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Hassanzadeh, F.; Amanlou, M. Prediction of dual agents as an activator of mutant p53 and inhibitor of Hsp90 by docking, molecular dynamic simulation and virtual screening. J. Mol. Graph. Model. 2015, 61, 186–195. [Google Scholar] [CrossRef]
- Athar, M.; Lone, M.Y.; Khedkar, V.M.; Jha, P.C. Pharmacophore model prediction, 3D-QSAR and molecular docking studies on vinyl sulfones targeting Nrf2-mediated gene transcription intended for anti-Parkinson drug design. J. Biomol. Struct. Dyn. 2016, 34, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Tripuraneni, N.S.; Azam, M.A. A combination of pharmacophore modeling, atom-based 3D-QSAR, molecular docking and molecular dynamics simulation studies on PDE4 enzyme inhibitors. J. Biomol. Struct. Dyn. 2016, 34, 2481–2492. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, L.; Zhong, L.; Dai, D.; Hong, M.; You, R.; Wang, T. Exploration of potential RSK2 inhibitors by pharmacophore modelling, structure-based 3D-QSAR, molecular docking study and molecular dynamics simulation. Mol. Simul. 2017, 43, 534–547. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- pkCSM. Available online: http://biosig.unimelb.edu.au/pkcsm/ (accessed on 19 February 2021).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Molecular Modeling Group. Swiss Institute of Bioinformatics SwissADME. Available online: http://www.swissadme.ch/ (accessed on 19 February 2021).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Paul, C.D.; Hawkins, A.; Geoffrey, S.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2006, 50, 74–82. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]











{kind=link}
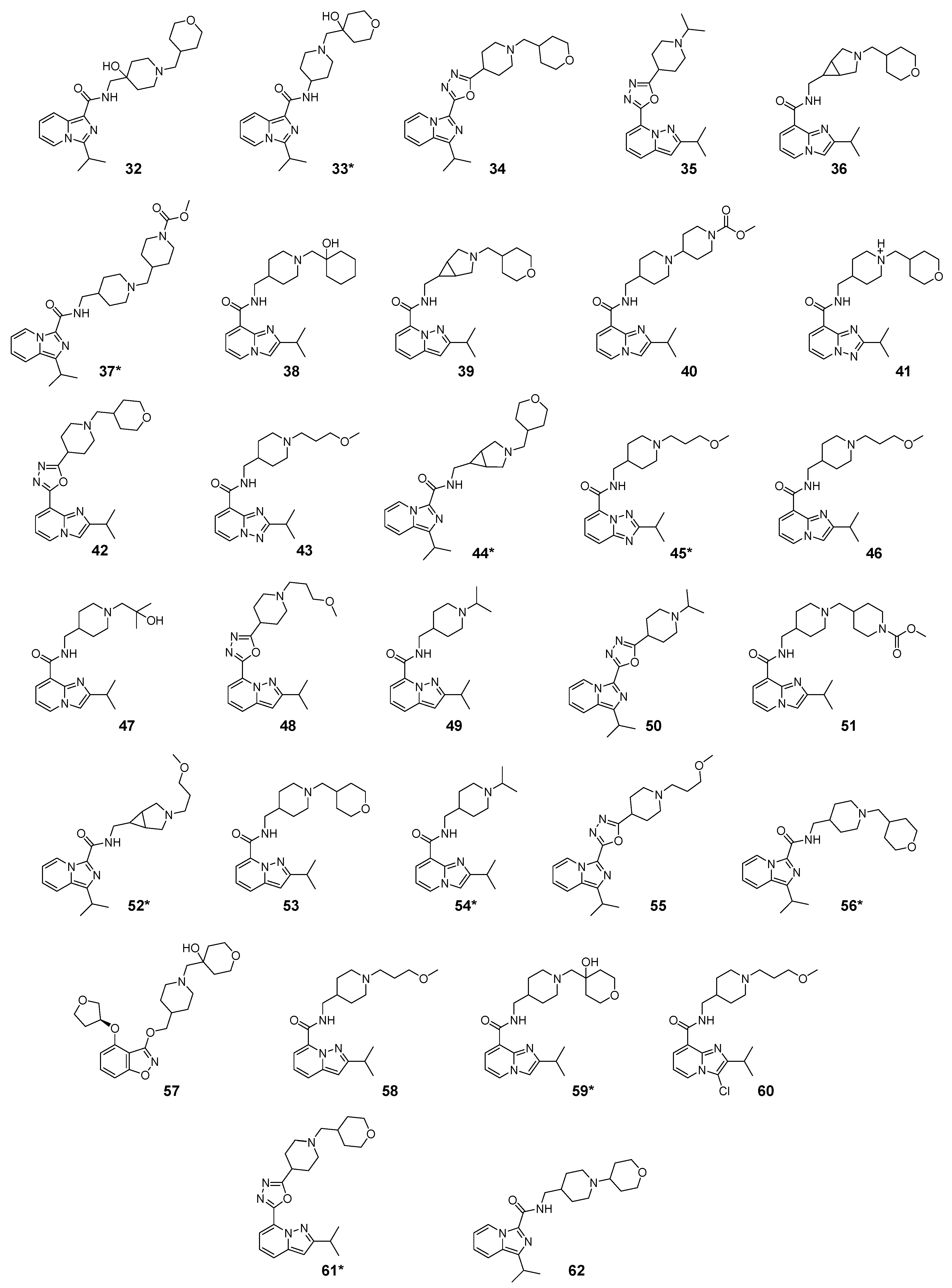{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction of Fields Included in the Model | |||||||
|---|---|---|---|---|---|---|---|
| Fields | SD | R2training | R2Scramble | R2test | Stability | Steric | Electrostatic |
| S | 0.654 | 0.719 | 0.554 | 0.329 | 0.229 | 1 | |
| E | 0.633 | 0.737 | 0.230 | 0.314 | 0.614 | 1 | |
| All | 0.522 | 0.821 | 0.188 | 0.667 | 0.120 | 0.574 | 0.426 |
| Field Contributions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fields | SD | R2training | R2Scramble | R2test | Stability | S | E | H | A | D |
| S | 0.742 | 0.639 | 0.681 | 0.229 | 0.507 | 1.000 | ||||
| E | 0.362 | 0.914 | 0.769 | 0.573 | 0.688 | 1.000 | ||||
| H | 0.498 | 0.837 | 0.804 | 0.460 | 0.341 | 1.000 | ||||
| A | 0.567 | 0.789 | 0.645 | 0.548 | 0.587 | 1.000 | ||||
| D | 0.810 | 0.570 | 0.382 | 0.178 | 0.362 | 1.000 | ||||
| SE | 0.563 | 0.792 | 0.765 | 0.557 | 0.017 | 0.708 | 0.293 | |||
| SH | 0.565 | 0.791 | 0.784 | 0.405 | 0.019 | 0.509 | 0.491 | |||
| SA | 0.507 | 0.831 | 0.790 | 0.319 | 0.253 | 0.534 | 0.466 | |||
| SD | 0.572 | 0.785 | 0.763 | 0.657 | 0.226 | 0.675 | 0.325 | |||
| EH | 0.343 | 0.923 | 0.862 | 0.577 | 0.431 | 0.304 | 0.696 | |||
| EA | 0.479 | 0.849 | 0.743 | 0.656 | 0.593 | 0.345 | 0.655 | |||
| ED | 0.431 | 0.878 | 0.702 | 0.548 | 0.475 | 0.609 | 0.391 | |||
| HA | 0.387 | 0.902 | 0.835 | 0.615 | 0.453 | 0.561 | 0.440 | |||
| HD | 0.473 | 0.853 | 0.824 | 0.405 | 0.217 | 0.734 | 0.266 | |||
| AD | 0.504 | 0.833 | 0.681 | 0.526 | 0.328 | 0.710 | 0.290 | |||
| SEH | 0.483 | 0.847 | 0.817 | 0.560 | 0.141 | 0.417 | 0.170 | 0.413 | ||
| SHA | 0.443 | 0.871 | 0.824 | 0.628 | 0.225 | 0.356 | 0.326 | 0.318 | ||
| SHD | 0.498 | 0.838 | 0.817 | 0.422 | 0.017 | 0.416 | 0.375 | 0.210 | ||
| SEA | 0.467 | 0.857 | 0.807 | 0.681 | 0.250 | 0.466 | 0.147 | 0.387 | ||
| SED | 0.498 | 0.837 | 0.799 | 0.492 | 0.027 | 0.555 | 0.194 | 0.251 | ||
| SAD | 0.486 | 0.845 | 0.812 | 0.617 | 0.148 | 0.470 | 0.356 | 0.175 | ||
| EHA | 0.332 | 0.928 | 0.846 | 0.604 | 0.454 | 0.171 | 0.481 | 0.348 | ||
| EHD | 0.368 | 0.911 | 0.846 | 0.627 | 0.344 | 0.230 | 0.576 | 0.194 | ||
| EAD | 0.455 | 0.864 | 0.739 | 0.611 | 0.447 | 0.275 | 0.504 | 0.221 | ||
| HAD | 0.377 | 0.907 | 0.844 | 0.601 | 0.346 | 0.490 | 0.366 | 0.145 | ||
| SEHA | 0.410 | 0.890 | 0.834 | 0.670 | 0.257 | 0.321 | 0.109 | 0.295 | 0.275 | |
| SEHD | 0.443 | 0.871 | 0.836 | 0.556 | 0.099 | 0.365 | 0.131 | 0.331 | 0.172 | |
| SEAD | 0.442 | 0.898 | 0.826 | 0.695 | 0.172 | 0.420 | 0.125 | 0.304 | 0.151 | |
| SHAD | 0.430 | 0.879 | 0.840 | 0.644 | 0.186 | 0.328 | 0.281 | 0.260 | 0.132 | |
| EHAD | 0.356 | 0.917 | 0.850 | 0.639 | 0.396 | 0.145 | 0.426 | 0.305 | 0.125 | |
| SEHAD | 0.395 | 0.898 | 0.847 | 0.674 | 0.213 | 0.302 | 0.095 | 0.258 | 0.228 | 0.117 |
| FFQSAR | GFQSAR | FFQSAR | GFQSAR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | pEC50_exp | pEC50_calc | Res. | pEC50_calc | Res. | Comp. | pEC50_exp | pEC50_calc | Res. | pEC50_calc | Res. |
| 1 | 8.921 | 8.996 | −0.075 | 8.404 | 0.518 | 32 | 7.246 | 6.825 | 0.421 | 6.993 | 0.253 |
| 2 | 8.585 | 8.836 | −0.251 | 9.127 | −0.542 | 33 | 6.842 | 7.797 | −0.955 | 8.065 | −1.223 |
| 3 | 7.398 | 8.416 | −1.018 | 7.211 | 0.187 | 34 | 6.315 | 6.676 | −0.361 | 6.240 | 0.076 |
| 4 | 7.509 | 7.745 | −0.236 | 7.716 | −0.207 | 35 | 7.268 | 6.893 | 0.375 | 7.441 | −0.173 |
| 5 | 5.648 | 6.033 | −0.385 | 5.894 | −0.246 | 36 | 6.942 | 6.924 | 0.018 | 7.180 | −0.238 |
| 6 | 6.284 | 6.418 | −0.134 | 6.683 | −0.399 | 37 | 8.310 | 8.321 | −0.011 | 8.586 | −0.276 |
| 7 | 6.331 | 5.984 | 0.347 | 6.177 | 0.154 | 38 | 9.523 | 9.551 | −0.028 | 9.631 | −0.108 |
| 8 | 6.133 | 6.173 | −0.040 | 6.098 | 0.035 | 39 | 8.060 | 7.735 | 0.325 | 7.305 | 0.755 |
| 9 | 8.699 | 8.736 | −0.037 | 8.733 | −0.034 | 40 | 8.076 | 8.292 | −0.216 | 7.911 | 0.165 |
| 10 | 7.141 | 8.076 | −0.935 | 7.222 | −0.081 | 41 | 7.102 | 8.080 | −0.978 | 8.083 | −0.981 |
| 11 | 7.703 | 7.675 | 0.028 | 7.967 | −0.264 | 42 | 6.223 | 5.805 | 0.418 | 5.591 | 0.632 |
| 12 | 6.067 | 6.801 | −0.734 | 6.559 | −0.492 | 43 | 5.712 | 6.893 | −1.181 | 6.000 | −0.288 |
| 13 | 8.244 | 7.459 | 0.785 | 8.088 | 0.156 | 44 | 7.983 | 7.459 | 0.524 | 7.512 | 0.471 |
| 14 | 8.000 | 7.932 | 0.068 | 8.196 | −0.196 | 45 | 5.712 | 5.226 | 0.486 | 6.499 | −0.787 |
| 15 | 8.009 | 7.568 | 0.441 | 7.775 | 0.234 | 46 | 8.824 | 9.078 | −0.254 | 9.024 | −0.200 |
| 16 | 8.824 | 8.294 | 0.530 | 8.722 | 0.102 | 47 | 9.155 | 8.822 | 0.333 | 8.597 | 0.558 |
| 17 | 8.469 | 7.331 | 1.138 | 7.541 | 0.928 | 48 | 7.208 | 7.169 | 0.039 | 6.673 | 0.535 |
| 18 | 9.301 | 8.681 | 0.620 | 9.184 | 0.117 | 49 | 7.009 | 6.466 | 0.543 | 7.435 | −0.426 |
| 19 | 6.301 | 6.646 | −0.345 | 5.982 | 0.319 | 50 | 6.120 | 6.400 | −0.280 | 6.303 | −0.183 |
| 20 | 5.867 | 6.333 | −0.466 | 6.292 | −0.425 | 51 | 9.222 | 9.270 | −0.048 | 9.368 | −0.146 |
| 21 | 7.658 | 7.808 | −0.150 | 7.641 | 0.017 | 52 | 7.866 | 7.670 | 0.196 | 7.525 | 0.341 |
| 22 | 7.237 | 7.235 | 0.002 | 7.383 | −0.146 | 53 | 10.000 | 8.778 | 1.222 | 8.970 | 1.030 |
| 23 | 7.469 | 8.233 | −0.764 | 7.944 | −0.475 | 54 | 8.046 | 8.168 | −0.122 | 7.983 | 0.063 |
| 24 | 7.745 | 7.490 | 0.255 | 7.766 | −0.021 | 55 | 6.099 | 6.030 | 0.069 | 5.722 | 0.377 |
| 25 | 7.738 | 7.225 | 0.513 | 7.848 | −0.110 | 56 | 8.056 | 7.701 | 0.355 | 8.100 | −0.044 |
| 26 | 7.409 | 7.349 | 0.060 | 7.366 | 0.043 | 57 | 8.886 | 8.488 | 0.398 | 9.006 | −0.120 |
| 27 | 7.301 | 7.367 | −0.066 | 7.527 | −0.226 | 58 | 9.523 | 8.838 | 0.685 | 8.928 | 0.595 |
| 28 | 7.959 | 8.241 | −0.282 | 7.843 | 0.116 | 59 | 9.398 | 9.487 | −0.089 | 9.815 | −0.417 |
| 29 | 6.076 | 6.727 | −0.651 | 6.162 | −0.086 | 60 | 5.963 | 6.700 | −0.737 | 6.536 | −0.573 |
| 30 | 8.319 | 7.986 | 0.333 | 7.491 | 0.829 | 61 | 7.678 | 6.177 | 1.501 | 6.403 | 1.275 |
| 31 | 8.284 | 8.662 | −0.378 | 8.659 | −0.375 | 62 | 7.377 | 7.497 | −0.120 | 8.061 | −0.684 |
| ID | Structures | FFQSAR | GFQSAR | ID | Structures | FFQSAR | GFQSAR |
|---|---|---|---|---|---|---|---|
| var1 |  | 8.283 | 9.221 | var6 |  | 8.698 | 9.111 |
| var2 |  | 8.209 | 9.375 | var7 |  | 8.690 | 9.547 |
| var3 |  | 8.285 | 9.432 | var8 |  | 9.417 | 9.259 |
| var4 |  | 8.719 | 9.114 | var9 |  | 8.818 | 9.700 |
| var5 |  | 8.775 | 9.513 | var10 |  | 8.641 | 9.849 |
| Absorption | Distribution | Metabolism | Excretion | Toxicity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Inhibitor | ||||||||||||
| ID | IA 1 | VDss 2 | 2D6 | 3A4 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | TC 3 | AMES | Hepatotoxicity | Skin Sensitization |
| 53 | 92.738 | 1.473 | Yes | Yes | No | No | No | Yes | Yes | 0.825 | No | Yes | No |
| var1 | 95.344 | 1.158 | No | Yes | No | No | No | No | Yes | 0.626 | No | Yes | No |
| var2 | 95.054 | 1.152 | No | Yes | No | No | No | No | No | 0.776 | No | Yes | No |
| var3 | 94.483 | 1.084 | No | Yes | No | No | No | No | No | 0.789 | No | Yes | No |
| var4 | 95.045 | 0.941 | No | Yes | No | No | No | No | No | 0.837 | No | Yes | No |
| var5 | 95.572 | 0.826 | No | Yes | No | No | No | No | No | 1.258 | No | Yes | No |
| var6 | 96.306 | 0.84 | No | Yes | No | No | No | No | No | 0.936 | No | Yes | No |
| var7 | 95.844 | 0.952 | No | Yes | No | No | No | No | No | 1.06 | No | Yes | No |
| var8 | 94.815 | 0.584 | No | No | No | No | No | No | No | 1.171 | No | Yes | No |
| var9 | 92.905 | 0.961 | No | No | No | No | No | No | Yes | 0.719 | No | Yes | No |
| var10 | 95.300 | 0.671 | No | No | No | No | No | No | Yes | 0.881 | No | Yes | No |
| ID | Lipinski | Ghose | Veber | Egan | Synthetic Accessibility |
|---|---|---|---|---|---|
| 53 | Yes | Yes | Yes | Yes | 3.26 |
| var1 | Yes | No | Yes | Yes | 4.77 |
| var2 | Yes | Yes | Yes | Yes | 4.55 |
| var3 | Yes | Yes | Yes | Yes | 4.4 |
| var4 | Yes | Yes | Yes | Yes | 3.52 |
| var5 | Yes | Yes | No | Yes | 3.68 |
| var6 | Yes | Yes | Yes | Yes | 4.07 |
| var7 | Yes | Yes | Yes | Yes | 3.97 |
| var8 | Yes | Yes | Yes | Yes | 2.71 |
| var9 | Yes | No | Yes | Yes | 4.89 |
| var10 | Yes | Yes | Yes | Yes | 3.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Alvarez, A.; Chávez-Ángel, E.; Nelson, R. Understanding the Molecular Basis of 5-HT4 Receptor Partial Agonists through 3D-QSAR Studies. Int. J. Mol. Sci. 2021, 22, 3602. https://doi.org/10.3390/ijms22073602
Castro-Alvarez A, Chávez-Ángel E, Nelson R. Understanding the Molecular Basis of 5-HT4 Receptor Partial Agonists through 3D-QSAR Studies. International Journal of Molecular Sciences. 2021; 22(7):3602. https://doi.org/10.3390/ijms22073602
Chicago/Turabian StyleCastro-Alvarez, Alejandro, Emigdio Chávez-Ángel, and Ronald Nelson. 2021. "Understanding the Molecular Basis of 5-HT4 Receptor Partial Agonists through 3D-QSAR Studies" International Journal of Molecular Sciences 22, no. 7: 3602. https://doi.org/10.3390/ijms22073602
APA StyleCastro-Alvarez, A., Chávez-Ángel, E., & Nelson, R. (2021). Understanding the Molecular Basis of 5-HT4 Receptor Partial Agonists through 3D-QSAR Studies. International Journal of Molecular Sciences, 22(7), 3602. https://doi.org/10.3390/ijms22073602