
CRISPR-Cas9–Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TIM3 Ligand Expression
2.2. Establishment of TIM3 Knockout NK Cells Using CRISPR-Cas9
2.3. Validating TIM3 Knockout in NK Cells
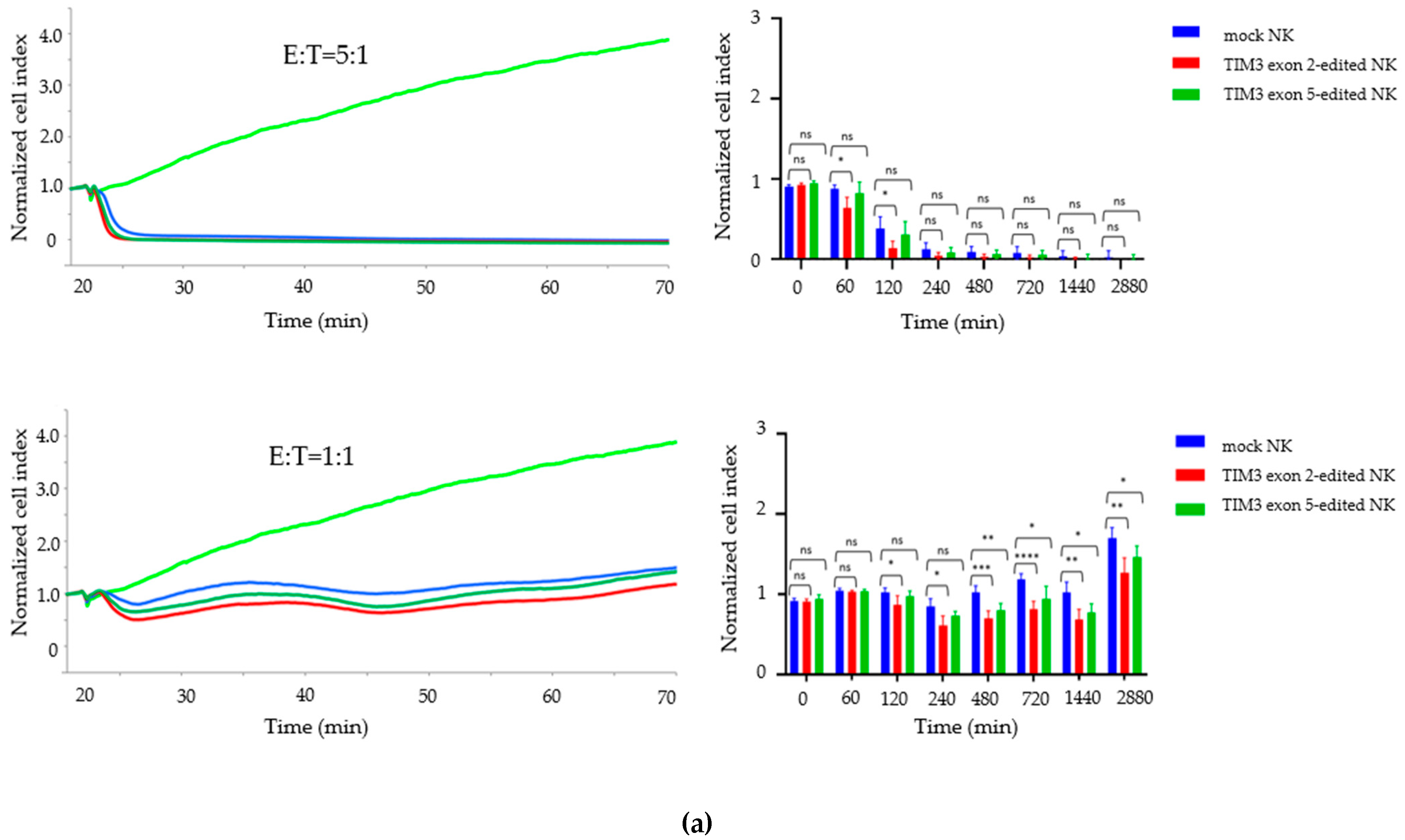
2.4. Growth Inhibition Assay
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Induction of TIM3 Knockout NK Cells
4.3. Antibody Staining and Flow Cytometry
4.4. Designing the sgRNAs
4.5. Gene Deletion Efficacy of CRISPR-Cas9
4.6. Growth Inhibition Assays
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef]
- Li, Y.; Sun, R. Tumor immunotherapy: New aspects of natural killer cells. Chin. J. Cancer Res. 2018, 30, 173–196. [Google Scholar] [CrossRef]
- Rautela, J.; Souza-Fonseca-Guimaraes, F.; Hediyeh-Zadeh, S.; Delconte, R.B.; Davis, M.J.; Huntington, N.D. Molecular insight into targeting the NK cell immune response to cancer. Immunol. Cell Biol. 2018, 96, 477–484. [Google Scholar] [CrossRef]
- Huntington, N.D.; Legrand, N.; Alves, N.L.; Jaron, B.; Weijer, K.; Plet, A.; Corcuff, E.; Mortier, E.; Jacques, Y.; Spits, H.; et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009, 206, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Puthalakath, H.; Gunn, P.; Naik, E.; Michalak, E.M.; Smyth, M.J.; Tabarias, H.; Degli-Esposti, M.A.; Dewson, G.; Willis, S.N.; et al. Interleukin 15–mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 2007, 8, 856–863. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef]
- Richards, J.O.; Chang, X.; Blaser, B.W.; Caligiuri, M.A.; Zheng, P.; Liu, Y. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood 2006, 108, 246–252. [Google Scholar] [CrossRef]
- Mundy-Bosse, B.L.; Scoville, S.D.; Chen, L.; McConnell, K.; Mao, H.C.; Ahmed, E.H.; Zorko, N.; Harvey, S.; Cole, J.; Zhang, X.; et al. MicroRNA-29b mediates altered innate immune development in acute leukemia. J. Clin. Investig. 2016, 126, 4404–4416. [Google Scholar] [CrossRef] [PubMed]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-Dependent Coupling of Tim-3 to T-Cell Receptor Signaling Pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; Dekruyff, R.H. TIMgenes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef]
- Cao, E.; Zang, X.; Ramagopal, U.A.; Mukhopadhaya, A.; Fedorov, A.; Fedorov, E.; Zencheck, W.D.; Lary, J.W.; Cole, J.L.; Deng, H.; et al. T Cell Immunoglobulin Mucin-3 Crystal Structure Reveals a Galectin-9-Independent Ligand-Binding Surface. Immunity 2007, 26, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti–PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Kritikou, J.S.; Dahlberg, C.I.M.; Baptista, M.A.P.; Wagner, A.K.; Banerjee, P.P.; Gwalani, L.A.; Poli, C.; Panda, S.K.; Kärre, K.; Kaech, S.M.; et al. IL-2 in the tumor microenvironment is necessary for Wiskott-Aldrich syndrome protein deficient NK cells to respond to tumors in vivo. Sci. Rep. 2016, 6, 30636. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Cartwright, A.N. Genetic screens to study the immune system in cancer. Curr. Opin. Immunol. 2016, 41, 55–61. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rautela, J.; Surgenor, E.; Huntington, N.D. Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 2020, 108, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237.e6. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.; Stikvoort, A.; Nolan, E.; Kirkham-Mccarthy, L.; Khoruzhenko, S.; Shivakumar, R.; Zweegman, S.; Van De Donk, N.W.C.J.; Mutis, T.; Szegezdi, E.; et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nakazawa, T.; Nakamura, M.; Nishimura, F.; Matsuda, R.; Omoto, K.; Shida, Y.; Murakami, T.; Nakagawa, I.; Motoyama, Y.; et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE 2019, 14, e0212455. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. J. Extracell. Vesicles 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Liu, W.M.; Mei, R.; Di, X.; Ryder, T.B.; Hubbell, E.; Dee, S.; Webster, T.A.; Harrington, C.A.; Ho, M.H.; Baid, J.; et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics 2002, 18, 1593–1599. [Google Scholar] [CrossRef]
- Sutlu, T.; Nyström, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of Intracellular Antiviral Defense Mechanisms Augments Lentiviral Transduction of Human Natural Killer Cells: Implications for Gene Therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef]
- Hu, Y.; Tian, Z.-G.; Zhang, C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol. Sin. 2018, 39, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, I.B.; Pasquinelli, V.; Jurado, J.O.; Abbate, E.; Musella, R.M.; de la Barrera, S.S.; García, V.E. Role Played by the Programmed Death-1–Programmed Death Ligand Pathway during Innate Immunity againstMycobacterium tuberculosis. J. Infect. Dis. 2010, 202, 524–532. [Google Scholar] [CrossRef]
- Norris, S.; Coleman, A.; Kuri-Cervantes, L.; Bower, M.; Nelson, M.; Goodier, M. PD-1 Expression on Natural Killer Cells and CD8 + T Cells During Chronic HIV-1 Infection. Viral Immunol. 2012, 25, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK -cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.R.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; De Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.L.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef]
- Romagné, F.; André, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.; et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Musah-Eroje, A.; Watson, S. A novel 3D in vitro model of glioblastoma reveals resistance to temozolomide which was potentiated by hypoxia. J. Neuro-Oncol. 2019, 142, 231–240. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.-S.; et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted Primary Human Third-Generation CAR-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morimoto, T.; Nakazawa, T.; Matsuda, R.; Nishimura, F.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.-S.; Tsujimura, T.; Nakase, H. CRISPR-Cas9–Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. Int. J. Mol. Sci. 2021, 22, 3489. https://doi.org/10.3390/ijms22073489
Morimoto T, Nakazawa T, Matsuda R, Nishimura F, Nakamura M, Yamada S, Nakagawa I, Park Y-S, Tsujimura T, Nakase H. CRISPR-Cas9–Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. International Journal of Molecular Sciences. 2021; 22(7):3489. https://doi.org/10.3390/ijms22073489
Chicago/Turabian StyleMorimoto, Takayuki, Tsutomu Nakazawa, Ryosuke Matsuda, Fumihiko Nishimura, Mitsutoshi Nakamura, Shuichi Yamada, Ichiro Nakagawa, Young-Soo Park, Takahiro Tsujimura, and Hiroyuki Nakase. 2021. "CRISPR-Cas9–Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells" International Journal of Molecular Sciences 22, no. 7: 3489. https://doi.org/10.3390/ijms22073489
APA StyleMorimoto, T., Nakazawa, T., Matsuda, R., Nishimura, F., Nakamura, M., Yamada, S., Nakagawa, I., Park, Y.-S., Tsujimura, T., & Nakase, H. (2021). CRISPR-Cas9–Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. International Journal of Molecular Sciences, 22(7), 3489. https://doi.org/10.3390/ijms22073489