
Is Chelation Therapy a Potential Treatment for Parkinson’s Disease?




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Perspective and Introduction
2. Parkinson’s Disease
2.1. Iron Loading in PD
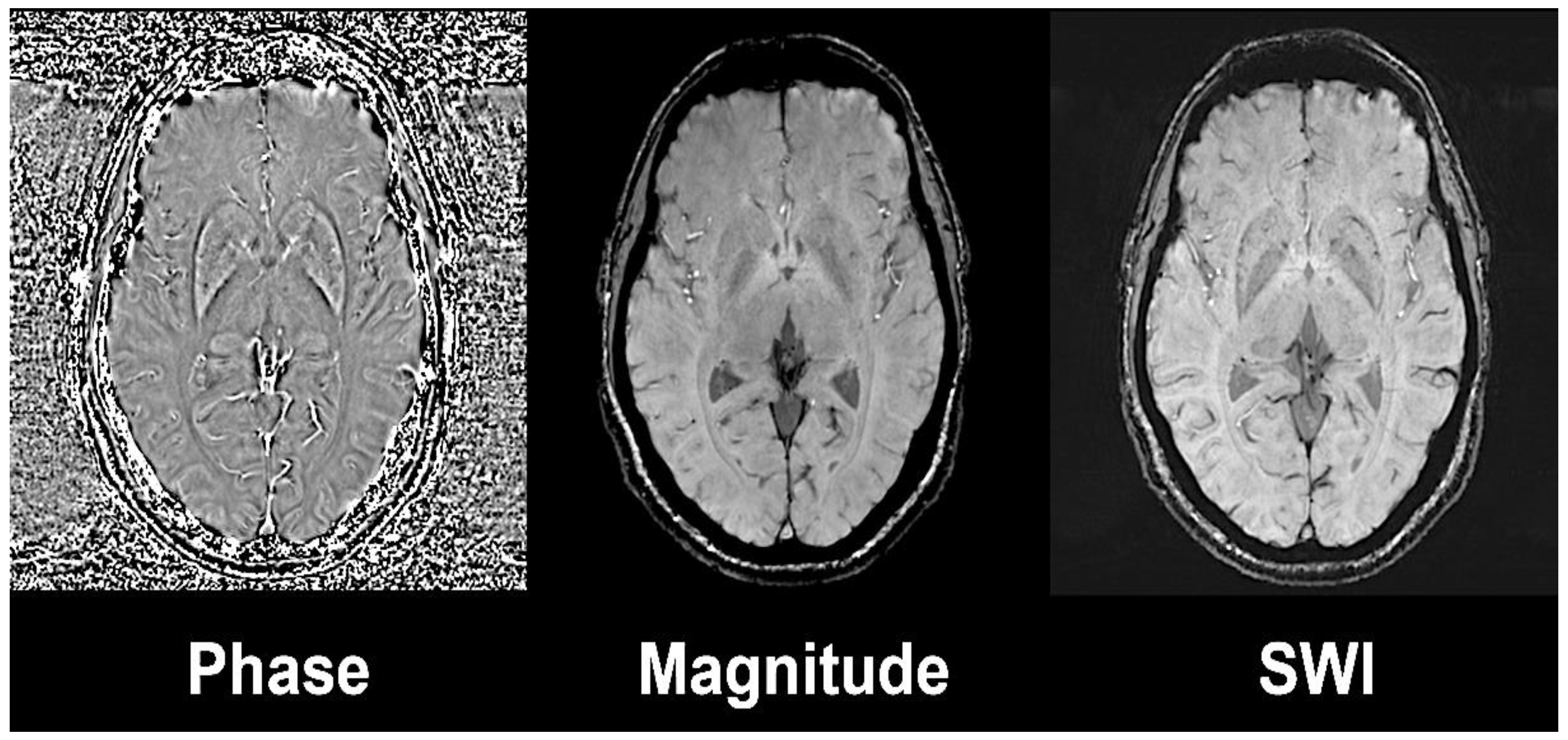
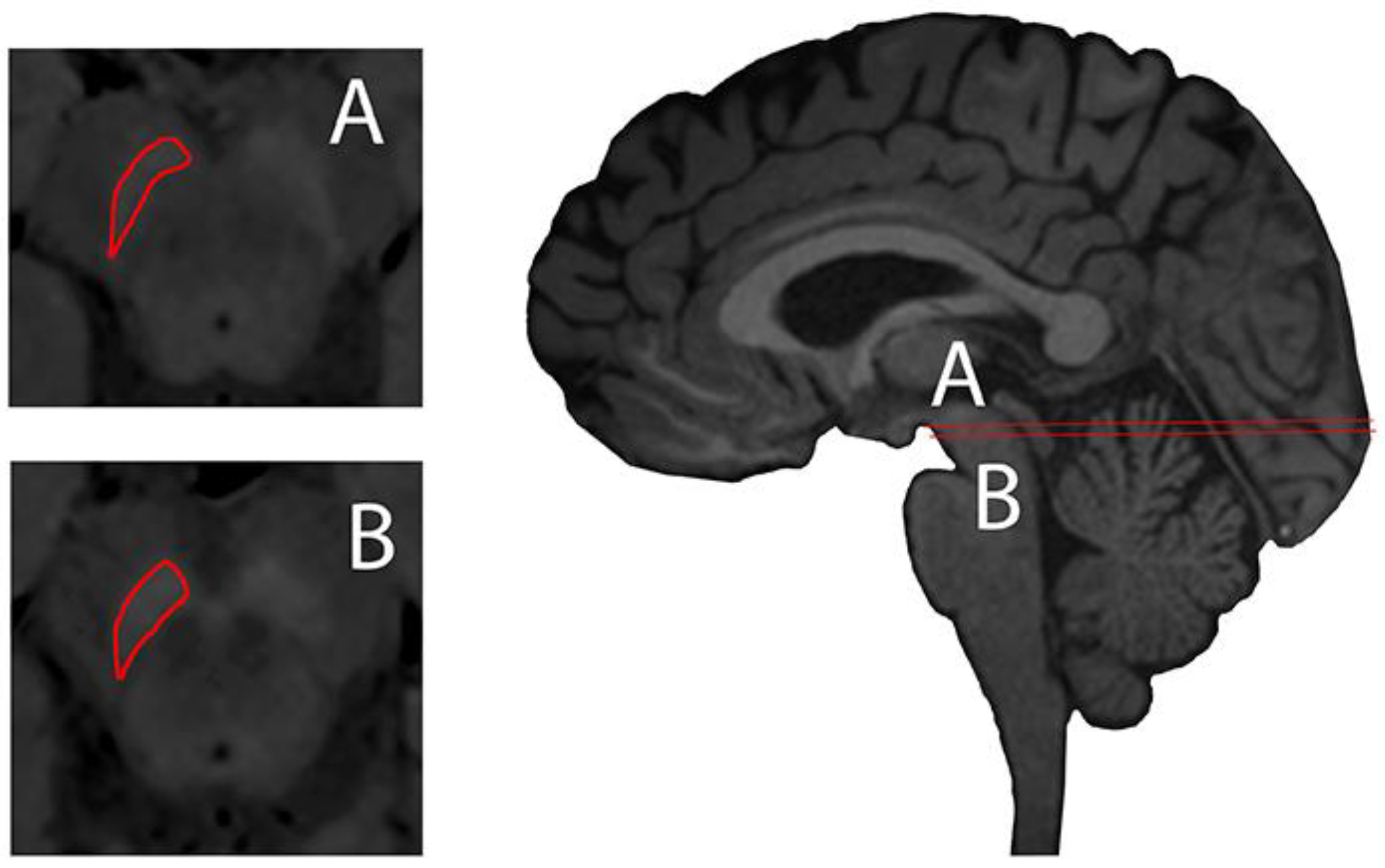
2.2. Non-Invasive Evaluation of Iron Loading
2.3. Neuroinflammation in PD
2.4. Non-Invasive Evaluation of Neuroinflammation
2.5. Anti-Inflammatory Drugs in PD
3. Chelation Therapy
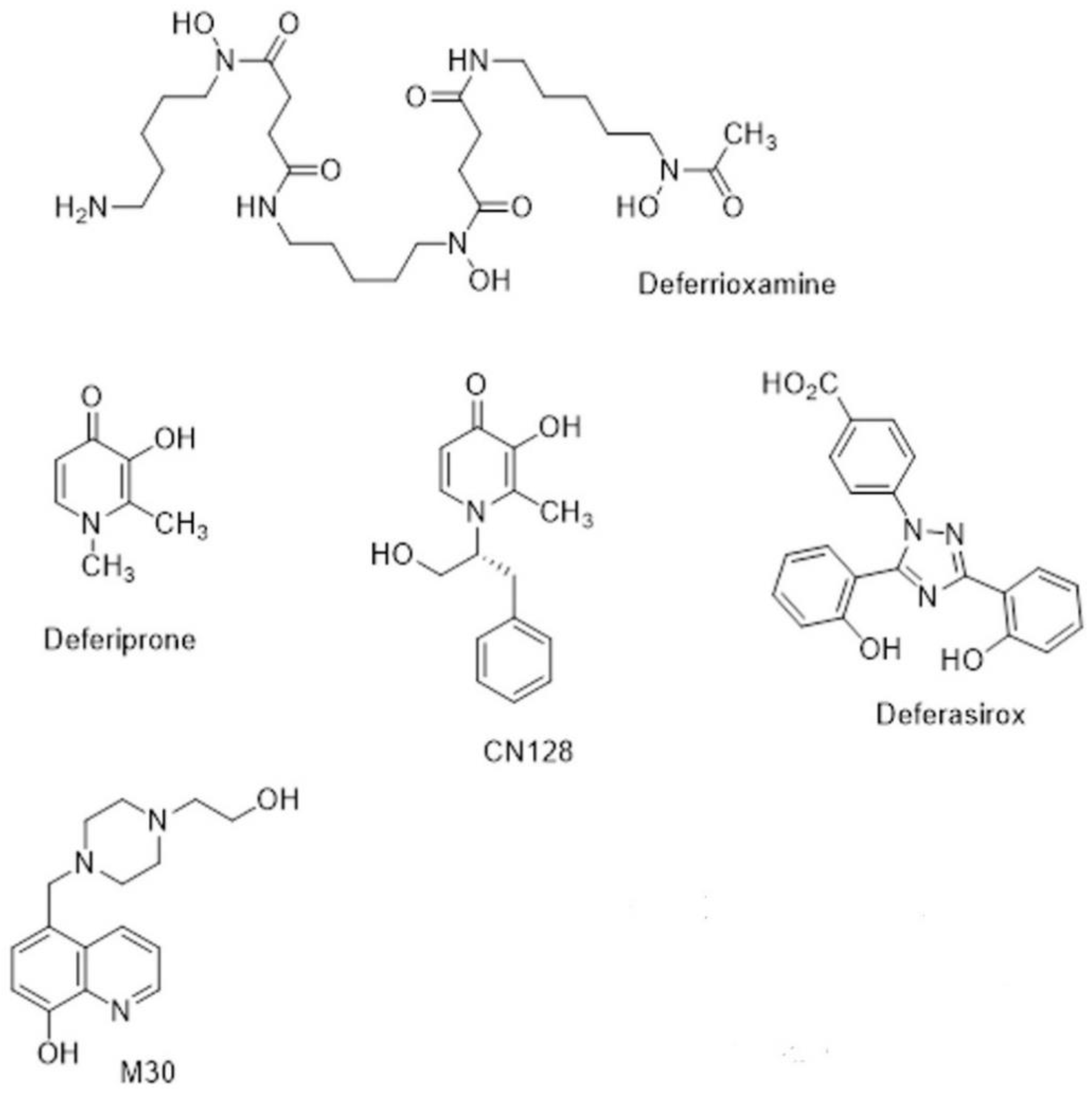
3.1. Iron Chelators in Current Clinical Use
3.2. Appication of Currently Available Chelators in Clinical Studies of PD
3.3. Non-Invasive Evaluation of Brain Iron Chelation Therapy
3.4. New Chelators for Brain Iron Chelation
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Life Expectancy. Available online: https://ourworldindata.org/grapher/life-expectancy1770.2019 (accessed on 3 December 2020).
- Decade of Healthy Ageing 2020–2030. World Health Organization. Available online: https://cdn.who.int/media/docs/default-source/decade-of-healthy-ageing/final-decade-proposal/decade-proposal-final-apr2020-en.pdf (accessed on 3 December 2020).
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Ward, R.J.; Crichton, R.R. Ironing out the Brain. In Essential Metals in Medicine, 19th ed.; Siegel, A., Freisinger, E., Siegel, R., Eds.; Walter de Gruyter: Berlin, Germany, 2019; pp. 87–122. [Google Scholar]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain of Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.; Ferrer, I.; Fernando, G.; Hilfiker, S. Biomonitorization of iron accumulation in the substantia nigra from Lewy body disease patients. Toxicol. Rep. 2017, 4, 188–193. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Tilley, B.; Bansal, S.; Gentleman, S.; Dexter, D.T.; Ward, R.J. Parkinson’s Disease, Iron and Inflammation. J. Neural Transm. 2020, 128, 15–25. [Google Scholar] [CrossRef]
- Jellinger, K.; Paulus, W.; Grundke-Iqbal, I.; Riederer, P.; Youdim, M.B. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J. Neural. Transm. Park. Dis. Dement. Sect. 1990, 2, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for Parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef]
- Hadzhieva, M.; Kirches, E.; Mawrin, C. Review: Iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2014, 40, 240–257. [Google Scholar] [CrossRef]
- Dexter, D.T.; Agid, F.; Agid, Y.; Wells, F.R.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- Haacke, E.M.; Xu, Y.; Cheng, Y.C.N.; Reichenbach, J.R. Susceptibility weighted imaging (SWI). Magn. Reason. Med. 2004, 52, 612–618. [Google Scholar] [CrossRef]
- Martin-Bastida, A. Nigral iron and neuromelanin imaging studies in Parkinson´s disease. Ph.D. Thesis, Medicine. Imperial College London, London, UK, 2019. [Google Scholar]
- Deistung, A.; Schweser, F.; Reichenbach, J.R. Overview of quantitative susceptibility mapping. NMR Biomed. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Schenck, J.F. Magnetic resonance imaging of brain iron. J. Neurolog. Sci. 2003, 207, 99–102. [Google Scholar] [CrossRef]
- Hallgren, B.; Sourander, P. The effect of age on the non-haemin iron in thr human brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef]
- Haacke, E.M.; Cheng, N.Y.C.; House, M.J.; Liu, Q.; Neelavalli, J.; Ogg, R.J.; Khan, A.; Ayaz, M.; Kirsch, W.; Obenaus, A. Imaging iron stores in the brain using magnetic resonance imaging. Mag. Res. Imaging 2005, 23, 1–25. [Google Scholar] [CrossRef]
- Antonini, A.; Leenders, K.L.; Meier, D.; Oertel, W.H.; Boesiger, P.; Anliker, M. T2 relaxation time in patients with parkinson’s disease. Neurology 1993, 43, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.W.; Wieler, M.; Gee, M. Midbrain iron content in early Parkinson disease: A potential biomarker of disease status. Neurology 2008, 70, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.H.O.; Santos, A.C.; Tumas, V.; Liu, M.; Zheng, W.; Haacke, C.E.G. Quantifying brain iron deposition in patients with Parkinson’s disease using quantitative susceptibility mapping, R2 and R2*. Mag. Res. Imaging 2015, 33, 559–565. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Lao-Kaim, N.P.; Loane, C.; Politis, M.; Roussakis, A.A.; Valle-Guzman, N.; Kefalopoulou, Z.; Paul-Visse, G.; Widner, H.; Xing, Y.; et al. Motor associations of iron accumulation in deep grey matter nuclei in Parkinson’s disease: A cross-sectional study of iron-related magnetic resonance imaging susceptibility. Eur. J. Neurol. 2017, 24, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.M.; Du, G.; Kidacki, M.; Patel, N.; Shaffer, M.L.; Mailman, R.B.; Huang, X. Higher iron in the red nucleus marks Parkinson’s dyskinesia. Neurobiol. Aging. 2013, 34, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Ryvlin, P.; Broussoll, E.; Piollet, H.; Viallet, F.; Khalfallah, Y.; Chazot, G. Magnetic Resonance Imaging Evidence of Decreased Putamenal Iron Content in Idiopathic Parkinson’s Disease. Arch. Neurol. 1995, 52, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulze, R.D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pig. Cell Res. 2004, 17, 610–617. [Google Scholar] [CrossRef]
- Zecca, L.; Casella, L.; Albertini, A.; Bellei, C.; Zucca, F.A.; Engelen, M.; Zadlo, A.; Szewczy, G.; Zareba, M.; Sarna, T. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J. Neurochem. 2008, 106, 1866–1875. [Google Scholar]
- Double, K.L.; Gerlach, M.; Schünemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.H.; Riederer, P.; Ben-Shachar, D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Pietracupa, S.; Piccini, P. Neuromelanin in parkinsonian disorders: An update. Int. J. Neurosci. 2017, 127, 1116–1123. [Google Scholar] [CrossRef]
- Sasaki, M.; Shibata, E.; Tohyama, K.; Takahashi, J.; Otsuka, K.; Tsuchiy, K.; Takahash, S.; Ehara, S.; Terayama, Y.; Sakai, A. Neuromelanin magnetic resonance imaging of locus ceruleus and substantia nigra in Parkinson’s disease. NeuroReport 2006, 19, 1649–1654. [Google Scholar] [CrossRef]
- Schwarz, S.T.; Rittman, T.; Gontu, V.; Morgan, P.S.; Bajaj, N.; Auer, D.P. T1-Weighted MRI shows stage-dependent substantia nigra signal loss in Parkinson’s disease. Mov. Disord. 2011, 26, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, G.; Fernández-Seara, M.A.; Lorenzo-Betancor, O.; Ortega-Cubero, S.; Puigvert, M.; Uranga, J.; Vidorreta, M.; Irigoyen, J.; Lorenzo, E.; Munoz-Barrutia, A.; et al. Automated Neuromelanin Imaging as a Diagnostic Biomarker for Parkinson’s Disease. Mov. Disord. 2015, 30, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Martín-Bastida, A.; Lao-Kaim, N.P.; Roussakis, A.A.; Searle, G.E.; Xing, Y.; Gunn, R.N.; Schwarz, S.T.; Barker, R.A.; Auer, D.P.; Piccini, P. Relationship between neuromelanin and dopamine terminals within the Parkinson’s nigrostriatal system. Brain 2019, 142, 2023–2036. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itajaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Federoff, H.J.; Maguire-Zeiss, K.A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 2009, 16, 238–254. [Google Scholar] [CrossRef]
- Zecca, L.; Wilms, H.; Geick, S.; Claasen, J.-H.; Brandenburg, L.-O.; Holzknecht, C.; Panizza, M.L.; Zucca, F.A.; Deuschl, G.; Sievers, J.; et al. Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: Implications for Parkinson’s disease. Acta Neuropathol. 2008, 116, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Banati, R.B.; Myers, R.; Kreutzberg, G.W. PK (‘peripheral benzodiazepine’)—Binding sites in the CNS indicate early and discrete brain lesions: Microautoradiographic detection of [3H]PK 11195 binding to activated microglia. J. Neurocytol. 1998, 26, 77–82. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.J.; Lucassen, P.J.; van Dam, A.-M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Cytokines in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 58, 143–151. [Google Scholar]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New progress on the role of glia in iron metabolism and iron-induced degeneration of dopamine neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Redensek, A.; David, S. Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-alpha and TGF-beta1. Glia 2012, 60, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef]
- Dringen, R.; Bishop, G.M.; Koeppe, M.; Dang, T.N.; Robinson, S.R. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 2007, 32, 1884–1890. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Cage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dang, T.N.; Dringen, R. Accumulation of non-transferrin bound iron by neurons, astrocytes and microglia. Neurotox. Res. 2007, 19, 443–451. [Google Scholar] [CrossRef]
- Pelizzoni, I.; Zacchetti, D.; Campanella, A.; Grohovaz, F.; Codazzi, F. Iron uptake in quiescent and inflammation-activated astrocytes: A potentially neuroprotective control of iron burden. Biochim. Biophys. Acta 2013, 1832, 1326–1333. [Google Scholar] [CrossRef]
- Yu, L.H.; Yan, C.Z.; Zheng, B.J.; Ci, Y.-H.; Chang, S.-Y.; Yu, P.; Gao, G.-F.; Li, H.-Y.; Dong, T.-Y.; Chang, Y.-Z. Hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death 2017, e2676. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Guilarte, T.R. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol. Ther. 2008, 118, 1–17. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brook, D.J. In vivo imaging of microglial activation with [11C] (R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.; Doorduin, J.; de Vries, E.F.J.; Dierckx, R.A.; Leenders, K.L. [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 2010, 16, 57–59. [Google Scholar] [CrossRef]
- Dodel, R.; Spottke, A.; Gerhard, A.; Reuss, A.; Reinecker, S.; Schimke, N.; Trenkwalder, C.; Sixel-Döring, F.; Herting, B.; Kamm, C.; et al. Minocycline 1-year therapy in multiple-system-atrophy: Effect on clinical symptoms and [11C] (R)-PK11195 PET (MEMSA-trial). Mov. Disord. 2010, 25, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cerebr. Blood Flow Metabol. 2012, 32, 1–5. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C. Increased microglial activation in patients with Parkinson disease using [18F]-DPA714 TSPO PET imaging. Parkinson Relat. Diord. 2020, 82, 29–36. [Google Scholar] [CrossRef]
- Olmos, G.; Alemany, R.; Escriba, P.V.; Garcia-Sevilla, J.A. The effects of chronic imidazoline drug treatment on glial fibrillary acidic protein concentrations in rat brain. Br. J. Pharmacol. 1994, 111, 997–1002. [Google Scholar] [CrossRef]
- Siemian, J.N.; LaMacchia, Z.M.; Spreuer, V.; Tian, J.; Ignatowski, T.A.; Paez, P.M.; Zhang, Y.; Li, J.-X. The imidazoline I2 receptor agonist 2-BFI attenuates hypersensitivity and spinal neuroinflammation in a rat model of neuropathic pain. Biochem. Pharmacol. 2018, 153, 260–268. [Google Scholar] [CrossRef]
- Fowler, J.S.; MacGregor, R.R.; Wolf, A.P.; Arnett, C.D.; Dewey, S.L.; Schlyer, D.; Christman, D.; Logan, J.; Smith, M.; Sachs, H.; et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 1987, 235, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Tyacke, R.J.; Fisher, A.; Robinson, E.S.; Grundt, P.; Turner, E.M.; Husbands, S.M.; Hudson, A.L.; Parker, C.A.; Nutt, D.J. Evaluation and initial in vitro and ex vivo characterization of the potential positron emission tomography ligand, BU99008 (2–(4,5-dihydro-1H-imidazol-2-yl)-1- methyl-1H-indole). for the imidazoline2 binding site. Synapse 2012, 66, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Tyacke, R.J.; Myers, J.F.M.; Venkataraman, A.; Mick, I.; Turton, S.; Passchier, J.; Husbands, S.M.; Rabiner, E.A.; Gunn, R.N.; Murphy, P.S.; et al. Evaluation of 11C-BU99008, a PET Ligand for the Imidazoline2 binding site in human brain. J. Nucl. Med. 2018, 59, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Tyacke, R.J.; Polychronis, S.; Myers, J.; Gunn, R.N.; Rabiner, E.A.; Nutt, D.; Politis, M. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’ disease: An in vivo11C-BU99008 PET study. Brain 2019, 142, 3116–3128. [Google Scholar] [CrossRef]
- Carrera, I.; Cacabelos, R. Current Drugs and Potential Future Neuroprotective Compounds for Parkinson’s Disease. Curr. Neuropharmacol. 2019, 17, 295–306. [Google Scholar] [CrossRef] [PubMed]
- English, J.M.; Cobb, M.H. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci. 2002, 23, 40–45. [Google Scholar] [CrossRef]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed]
- Schiess, M. Nonsteroidal anti-inflammatory drugs protect against Parkinson neurodegeneration: Can an NSAID a day keep Parkinson disease away? Arch. Neurol. 2003, 60, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.H.; Maraganore, D.M.; Peterson, B.J.; Ahlskog, J.E.; Rocca, W.A. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: A case-control study. Neurology 2006, 67, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal anti-inflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willet, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 6, 1059–1064. [Google Scholar] [CrossRef]
- Driver, J.A.; Logroscino, G.; Lu, L.; Gaziano, J.M.; Kurth, T. Use of non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease: Nested case-control study. BMJ 2011, 342, 198. [Google Scholar] [CrossRef] [PubMed]
- Gagne, J.J.; Power, M.C. Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology 2010, 74, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; de Lima Barros, P.; Levy, D.; Bydlowski, S.P. Ferroptosis mechanisms involved in neurological diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’ disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.G. Thalassemia: The continued challenge. Ann. N. Y. Acad. Sci. 2005, 1054, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.G. Thalassemia: A look to the future. Ann. N. Y. Acad. Sci. 2016, 1368, 11–15. [Google Scholar] [CrossRef]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Crichton, R.R.; Ward, R.J.; Hider, R.C. The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the pituitary—Ironing out the Brain. Pharmaceuticals 2019, 12, 138. [Google Scholar] [CrossRef]
- Propper, R.D.; Cooper, B.; Rufo, R.R.; Nienhuis, A.W.; Anderson, W.F.; Bunn, H.F.; Rosenthal, A.; Nathan, D.G. Continuous subcutaneous administration of deferoxamine in patients with iron overload. N. Engl. J. Med. 1977, 297, 418–423. [Google Scholar] [CrossRef]
- Pippard, M.J.; Callender, S.T.; Weatherall, D.J. Chelation regimens with desferrioxamine. Lancet 1977, 1, 1101. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar] [PubMed]
- Olivieri, N.F.; Nathan, D.G.; MacMillan, J.H.; Wayne, A.S.; Liu, P.P.; McGee, A.; Martin, M.; Koren, G.; Cohen, A.R. Survival in medically treated patients with homozygous beta thalassemia. N. Engl. J. Med. 1994, 331, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Khan, M.; Darlison, M. Survival in beta-thalassaemia major in the UK: Data from the UK Thalassaemia Register. Lancet 2000, 355, 2051–2052. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Aldouri, M.A.; Hoffbrand, A.V.; Barr, J.; Wonke, B.; Kourouclaris, T.; Sheppard, L. Effective chelation of iron in beta thalassaemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one. BMJ 1987, 295, 1509–1512. [Google Scholar] [CrossRef]
- Olivieri, N.F.; Koren, G.; Hermann, C.; Bentur, Y.; Chung, D.; Klein, J.; St Louis, P.; Freedman, M.H.; McClelland, R.A.; Templeton, D.M. Comparison of oral iron chelator L1 and desferrioxamine in iron-loaded patients. Lancet 1990, 336, 1275–1279. [Google Scholar] [CrossRef]
- Hoyes, K.P.; Porter, J.B. Subcellular distribution of desferrioxamine and hydroxypyridin-4-one chelators in K562 cells affects chelation of intracellular iron pools. Br. J. Haematol. 1993, 85, 393–400. [Google Scholar] [CrossRef]
- Huang, X.P.; Spino, M.; Thiessen, J.J. Transport kinetics of iron chelators and their chelates in Caco-2 cells. Pharm. Res. 2006, 23, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Botzenhardt, S.; Li, N.; Chan, E.W. Safety profiles of iron chelators in young patients with haemoglobinopathies. Eur. J. Haematol. 2017, 98, 198–217. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Link, G.; Pinson, A.; Peter, H.H.; Dobbin, P.; Hider, R.C. Iron mobilization from myocardial cells by 3-hydroxypyridin-4-one chelators: Studies in rat heart cells in culture. Blood 1991, 77, 2049–2053. [Google Scholar] [CrossRef]
- Anderson, L.J.; Wonke, B.; Prescott, E.; Holden, S.; Walker, J.M.; Pennell, D.J. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet 2002, 9332, 516–520. [Google Scholar] [CrossRef]
- Pepe, A.; Meloni, A.; Rossi, G.; Cuccia, L.; D’Ascola, G.D.; Santodirocco, M.; Cianciulli, P.; Caruso, V.; Romeo, M.A.; Filosa, A.; et al. Cardiac and hepatic iron and ejection fraction in thalassemia major: Multicentre prospective comparison of combined deferiprone and deferoxamine therapy against deferiprone or deferoxamine monotherapy. J. Cardiovasc. Magn. Reson. 2013, 15. [Google Scholar] [CrossRef]
- Nick, H.; Acklin, P.; Lattmann, R.; Buehlmayer, P.; Hauffe, S.; Schupp, J.; Alberti, D. Development of tridentate iron chelators: From desferrithiocin to ICL670. Curr. Med. Chem. 2003, 10, 1065–1076. [Google Scholar] [CrossRef]
- Longueville, A.; Crichton, R.R. An animal model of iron overload and its application to study hepatic ferritin iron mobilization by chelators. Biochem. Pharmacol. 1986, 35, 3669–3678. [Google Scholar] [CrossRef]
- Wolfe, L.C.; Nicolosi, R.J.; Renaud, M.M.; Finger, J.; Hegsted, M.; Peter, H.; Nathan, D.G. A non-human primate model for the study of oral iron chelators. Br. J. Haematol. 1989, 72, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.J.; Streiff, R.R.; Wiegand, J.; Vinson, J.R.; Luchetta, G.; Evans, K.M.; Peter, H.; Jenny, H.B. A comparative evaluation of iron clearance models. Ann. N. Y. Acad. Sci. 1990, 612, 378–393. [Google Scholar] [CrossRef]
- Baker, E.; Wong, A.; Peter, H.; Jacobs, A. Desferrithiocin is an effective iron chelator in vivo and in vitro but ferrithiocin is toxic. Br. J. Haematol. 1992, 81, 424–431. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Wiegand, J.; McManis, J.S.; Bharti, N. A search for clinically effective iron chelators. J. Med. Chem. 2014, 57, 9259–9291. [Google Scholar] [CrossRef]
- Daar, S.; Pathare, A.; Nick, H.; Kriemler-Krahn, U.; Hmissi, A.; Habr, D.; Taher, A. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily loaded patients with beta-thalassaemia. Eur. J. Haematol. 2009, 82, 454–457. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator in patients with best-thalassemia. Blood 2006, 207, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Porter, J.D.; El-Beshlawy, A.; Li, C.K.; Seymour, J.F.; Elalfy, M.; Gattermann, N.; Giraudier, S.; Lee, J.W.; Chan, L.L.; et al. Tailoring iron chelation by iron intake and serum ferritin; the perspective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 2010, 95, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Boddaert, N.; Le Quan Sang, K.H.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.; Florence, A.; Aouad, F.; Hider, R.; Jenner, P.; Crichton, R.R. Brain iron in the ferrocene-loaded rat: Its chelation and influence on dopamine metabolism. Biochem. Pharmacol. 1995, 49, 1821–1826. [Google Scholar] [CrossRef]
- Dexter, D.T.; Ward, R.J.; Florence, A.; Jenner, P.; Crichton, R.R. Effects of desferrithiocin and its derivatives on peripheral iron and striatal dopamine and 5-hydroxytryptamine metabolism in the ferrocene-loaded rat. Biochem. Pharmacol. 1999, 58, 151–155. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Connor, J.R.; Menzies, S.L. Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, X.; Li, Z.; Lu, Z.; Kong, S.; Jiang, H.; Du, H.; Pan, X.; Nandi, M.; Kong, X.; et al. CN128: A New Orally Active Hydroxypyridinone Iron Chelator. J. Med. Chem. 2020, 63, 4215–4226. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pham, A.N.; Hider, R.C. Effectiveness of the Iron Chelator CN128 in Mitigating the Formation of Dopamine Oxidation Products Associated with the Progression of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 3646–3657. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Kupershmidt, L.; Amit, T.; Weinreb, O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S132–S136. [Google Scholar] [CrossRef]
- Bar-Am, O.; Amit, T.; Youdim, M.B.; Weinreb, O. Neuroprotective and neurorestorative potential of propargylamine derivatives in ageing: Focus on mitochondrial targets. J. Neural Transm. 2016, 123, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Orvig, C. Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem. Rev. 2009, 109, 4885–4910. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, D.S.; Lázaro, D.F.; Sacco, P.; Ferreira, P.R.; Diniz, R.; Fernández, C.O.; Outeiro, T.F.; Rey, N.A. X1INH, an improved next-generation affinity-optimized hydrazonic ligand, attenuates abnormal copper(I)/copper (II)-α-Syn interactions and affects protein aggregation in a cellular model of synucleinopathy. Dalton Trans. 2020, 49, 16252–16267. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Kim, S.R. Beneficial Effects of Flavonoids against Parkinson’s Disease. J. Med. Food 2018, 21, 421–432. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Amit, T.; Youdim, M.B. Neurological mechanisms of green tea polyphenols in Alzheimer’s and Parkinson’s diseases. J. Nutr. Biochem. 2004, 15, 506–516. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef]
- Zhang, H.; Bai, L.; He, J.; Zhong, L.; Duan, X.; Ouyang, L.; Zhu, Y.; Wang, T.; Zhang, Y.; Shi, J. Recent advances in discovery and development of natural products as source for anti-Parkinson’s disease lead compounds. Eur. J. Med. Chem. 2017, 141, 257–272. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, R.J.; Dexter, D.T.; Martin-Bastida, A.; Crichton, R.R. Is Chelation Therapy a Potential Treatment for Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3338. https://doi.org/10.3390/ijms22073338
Ward RJ, Dexter DT, Martin-Bastida A, Crichton RR. Is Chelation Therapy a Potential Treatment for Parkinson’s Disease? International Journal of Molecular Sciences. 2021; 22(7):3338. https://doi.org/10.3390/ijms22073338
Chicago/Turabian StyleWard, Roberta J., David T. Dexter, Antonio Martin-Bastida, and Robert R. Crichton. 2021. "Is Chelation Therapy a Potential Treatment for Parkinson’s Disease?" International Journal of Molecular Sciences 22, no. 7: 3338. https://doi.org/10.3390/ijms22073338
APA StyleWard, R. J., Dexter, D. T., Martin-Bastida, A., & Crichton, R. R. (2021). Is Chelation Therapy a Potential Treatment for Parkinson’s Disease? International Journal of Molecular Sciences, 22(7), 3338. https://doi.org/10.3390/ijms22073338