
EnvIRONmental Aspects in Myelodysplastic Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Aspects on Iron, ROS and Implications for MDS
2.1. Effects of Iron/ROS in the Bone Marrow Niche and Hematopoietic Output
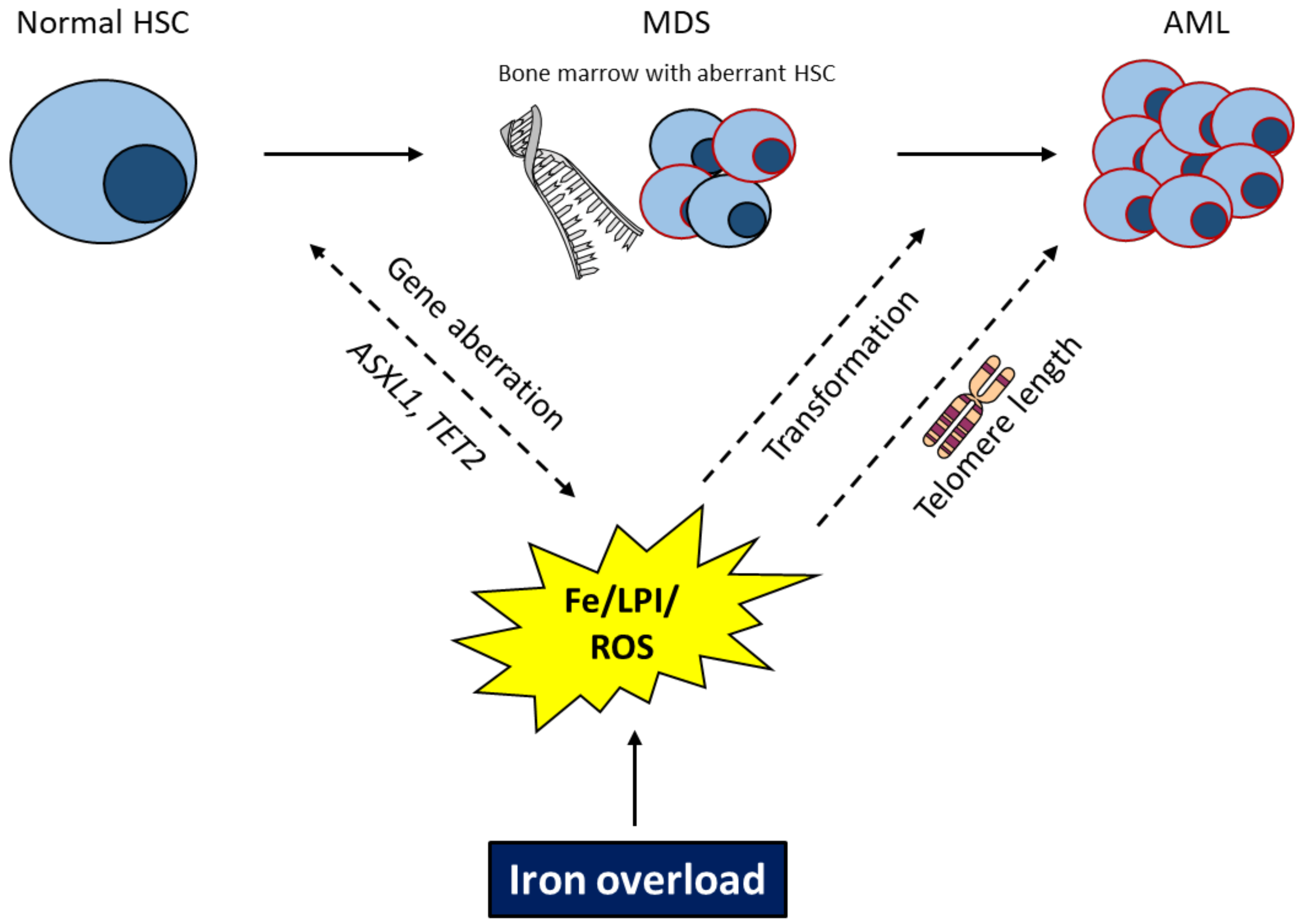
2.2. Effects of Iron/ROS for Genetic Instability and Leukemic Transformation
3. Aspects on Iron, Hepcidin, Inflammation and MDS
4. Aspects on TET2, Inflammation, Iron Metabolism and MDS
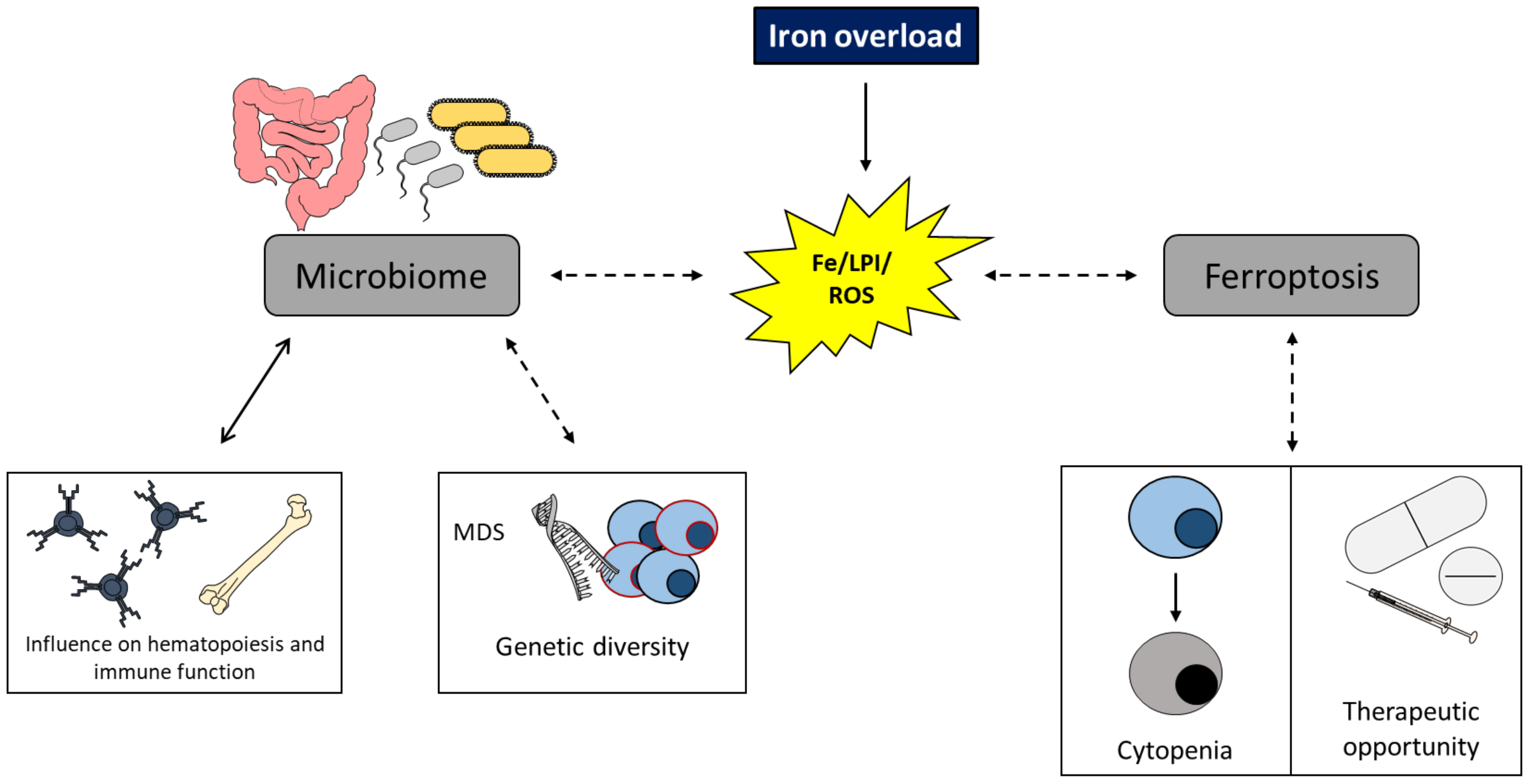
5. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Franke, G.N.; Kubasch, A.S.; Cross, M.; Vucinic, V.; Platzbecker, U. Iron overload and its impact on outcome of patients with hematological diseases. Mol. Aspects Med. 2020, 75, 100868. [Google Scholar] [CrossRef]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Sinha, S.; Pereira-Reis, J.; Guerra, A.; Rivella, S.; Duarte, D. The Role of Iron in Benign and Malignant Hematopoiesis. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Ito, H.; Kurokawa, H.; Matsui, H. Mitochondrial reactive oxygen species and heme, non-heme iron metabolism. Arch. Biochem. Biophys. 2021, 700, 108695. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Petzer, V.; Theurl, I.; Weiss, G. Established and emerging concepts to treat imbalances of iron homeostasis in inflammatory diseases. Pharmaceuticals 2018, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Troadec, M.B.; Loréal, O.; Brissot, E. Pathophysiology and classification of iron overload diseases; update 2018. Transfus. Clin. Biol. 2019, 26, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Viveiros, A.; Panzer, M.; Baumgartner, N.; Schaefer, B.; Finkenstedt, A.; Henninger, B.; Theurl, I.; Nachbaur, K.; Weiss, G.; Haubner, R.; et al. Reduced iron export associated with hepcidin resistance can explain the iron overload spectrum in ferroportin disease. Liver Int. 2020, 40, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; De Franceschi, L.; Muckenthaler, M.; Taher, A.; Rees, D.; De Montalembert, M.; Rivella, S.; Eleftheriou, A.; Cappellini, M.D. EHA research roadmap on hemoglobinopathies and thalassemia: An update. HemaSphere 2019, 3, e208. [Google Scholar] [CrossRef]
- Ikawa, Y.; Miccio, A.; Magrin, E.; Kwiatkowski, J.L.; Rivella, S.; Cavazzana, M. Gene therapy of hemoglobinopathies: Progress and future challenges. Hum. Mol. Genet. 2019, 28, R24–R30. [Google Scholar] [CrossRef] [PubMed]
- Gardenghi, S.; Marongiu, M.F.; Ramos, P.; Guy, E.; Breda, L.; Chadburn, A.; Liu, Y.F.; Amariglio, N.; Rechavi, G.; Rachmilewitz, E.A.; et al. Ineffective erythropoiesis in β-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007, 109, 5027–5035. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective Erythropoiesis: Anemia and Iron Overload. Hematol. Oncol. Clin. N. Am. 2018, 32, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.; Rivella, S. β-thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118, 4321–4330. [Google Scholar] [CrossRef]
- Rivella, S. Ineffective erythropoiesis and thalassemias. Curr. Opin. Hematol. 2009, 16, 187–194. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 2016, 38, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Viprakasit, V.; Kattamis, A.; Rivella, S.; Taher, A.T. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am. J. Hematol. 2021, 96, E54–E56. [Google Scholar] [CrossRef]
- Casu, C.; Oikonomidou, P.R.; Chen, H.; Nandi, V.; Ginzburg, Y.; Prasad, P.; Fleming, R.E.; Shah, Y.M.; Valore, E.V.; Nemeth, E.; et al. Minihepcidin peptides as disease modifiers in mice affected by-thalassemia and polycythemia vera. Blood 2016, 128, 265–276. [Google Scholar] [CrossRef]
- Casu, C.; Chessa, R.; Liu, A.; Gupta, R.; Drakesmith, H.; Fleming, R.; Ginzburg, Y.Z.; MacDonald, B.; Rivella, S. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult β-thalassemia major. Haematologica 2020, 105, 1835–1844. [Google Scholar] [CrossRef]
- Moreno Berggren, D.; Folkvaljon, Y.; Engvall, M.; Sundberg, J.; Lambe, M.; Antunovic, P.; Garelius, H.; Lorenz, F.; Nilsson, L.; Rasmussen, B.; et al. Prognostic scoring systems for myelodysplastic syndromes (MDS) in a population-based setting: A report from the Swedish MDS register. Br. J. Haematol. 2018, 181, 614–627. [Google Scholar] [CrossRef]
- Malcovati, L.; Hellström-Lindberg, E.; Bowen, D.; Adès, L.; Cermak, J.; Del Cañizo, C.; Della Porta, M.G.; Fenaux, P.; Gattermann, N.; Germing, U.; et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European LeukemiaNet. Blood 2013, 122, 2943–2964. [Google Scholar] [CrossRef]
- Alessandrino, E.P.; Della Porta, M.G.; Bacigalupo, A.; Malcovati, L.; Angelucci, E.; Van Lint, M.T.; Falda, M.; Onida, F.; Bernardi, M.; Guidi, S.; et al. Prognostic impact of pre-transplantation transfusion history and secondary iron overload in patients with myelodysplastic syndrome undergoing allogeneic stem cell transplantation: A GITMO study. Haematologica 2010, 95, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Porwit, A.; Saft, L. The AML-MDS interface-leukemic transformation in myelodysplastic syndromes. J. Hematop. 2011, 4, 69–79. [Google Scholar] [CrossRef]
- Malcovati, L.; Della Porta, M.G.; Cazzola, M. Predicting survival and leukemic evolution in patients with myelodysplastic syndrome. Haematologica 2006, 91, 1588–1590. [Google Scholar]
- Jaeger, M.; Aul, C.; Söhngen, D.; Germing, U.; Schneider, W. Secondary hemochromatosis in polytransfused patients with myelodysplastic syndromes. Beitr. Infusionsther. 1992, 30, 464–468. [Google Scholar]
- Cazzola, M.; Barosi, G.; Gobbi, P.G.; Invernizzi, R.; Riccardi, A.; Ascari, E. Natural history of idiopathic refractory sideroblastic anemia. Blood 1988, 71, 305–312. [Google Scholar] [CrossRef]
- Wermke, M.; Eckoldt, J.; Götze, K.S.; Klein, S.A.; Bug, G.; de Wreede, L.C.; Kramer, M.; Stölzel, F.; von Bonin, M.; Schetelig, J.; et al. Enhanced labile plasma iron and outcome in acute myeloid leukaemia and myelodysplastic syndrome after allogeneic haemopoietic cell transplantation (ALLIVE): A prospective, multicentre, observational trial. Lancet Haematol. 2018, 5, e201–e210. [Google Scholar] [CrossRef]
- De Swart, L.; Smith, A.; Johnston, T.W.; Haase, D.; Droste, J.; Fenaux, P.; Symeonidis, A.; Sanz, G.; Hellström-Lindberg, E.; Cermák, J.; et al. Validation of the revised international prognostic scoring system (IPSS-R) in patients with lower-risk myelodysplastic syndromes: A report from the prospective European LeukaemiaNet MDS (EUMDS) registry. Br. J. Haematol. 2015, 170, 372–383. [Google Scholar] [CrossRef]
- Neukirchen, J.; Fox, F.; Kündgen, A.; Nachtkamp, K.; Strupp, C.; Haas, R.; Germing, U.; Gattermann, N. Improved survival in MDS patients receiving iron chelation therapy-A matched pair analysis of 188 patients from the Düsseldorf MDS registry. Leuk. Res. 2012, 36, 1067–1070. [Google Scholar] [CrossRef]
- Gattermann, N. Iron overload in myelodysplastic syndromes (MDS). Int. J. Hematol. 2018, 107, 55–63. [Google Scholar] [CrossRef]
- Goldberg, S.L.; Chen, E.; Corral, M.; Guo, A.; Mody-Patel, N.; Pecora, A.L.; Laouri, M. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J. Clin. Oncol. 2010, 28, 2847–2852. [Google Scholar] [CrossRef]
- Malcovati, L.; Germing, U.; Kuendgen, A.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Giagounidis, A.; Hildebrandt, B.; Bernasconi, P.; Knipp, S.; et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J. Clin. Oncol. 2007, 25, 3503–3510. [Google Scholar] [CrossRef]
- Malcovati, L.; Porta, M.G.D.; Strupp, C.; Ambaglio, I.; Kuendgen, A.; Nachtkamp, K.; Travaglino, E.; Invernizzi, R.; Pascutto, C.; Lazzarino, M.; et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based prognostic scoring system (WPSS). Haematologica 2011, 96, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Boni, M.; Travaglino, E.; Passamonti, F.; Arcaini, L.; Maffioli, M.; Bernasconi, P.; et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: A basis for clinical decision making. J. Clin. Oncol. 2005, 23, 7594–7603. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Parmon, A.; Kurrle, N.; Schnütgen, F.; Serve, H. The Clinical Significance of Iron Overload and Iron Metabolism in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 627662. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Hell, S.; Platzbecker, U. Controversies on the consequences of iron overload and chelation in MDS. HemaSphere 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Lobão, M.J.; Pereira, A.; Nascimento Costa, J.M.; et al. Oxidative stress and mitochondrial dysfunction play a role in myelodysplastic syndrome development, diagnosis, and prognosis: A pilot study. Free Radic. Res. 2015, 49, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Saigo, K.; Takenokuchi, M.; Hiramatsu, Y.; Tada, H.; Hishita, T.; Takata, M.; Misawa, M.; Imoto, S.; Imashuku, S. Oxidative stress levels in myelodysplastic syndrome patients: Their relationship to serum ferritin and haemoglobin values. J. Int. Med. Res. 2011, 39, 1941–1945. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.F.; Barbosa, M.C.; De Jesus Santos, T.E.; De Jesus Ponte Carvalho, T.M.; De Freitas, R.M.; Martins, M.R.A.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhães, S.M.M. Increased parameters of oxidative stress and its relation to transfusion iron overload in patients with myelodysplastic syndromes. J. Clin. Pathol. 2013, 66, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Ivars, D.; Orero, M.T.; Javier, K.; Díaz-Vico, L.; García-Giménez, J.L.; Mena, S.; Tormos, C.; Egea, M.; Pérez, P.L.; Arrizabalaga, B.; et al. Oxidative imbalance in low/intermediate-1-risk myelodysplastic syndrome patients: The influence of iron overload. Clin. Biochem. 2017, 50, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, M.; Bagguley, T.; van Marrewijk, C.; Smith, A.; Bowen, D.; Culligan, D.; Kolade, S.; Symeonidis, A.; Garelius, H.; Spanoudakis, M.; et al. Toxic iron species in lower-risk myelodysplastic syndrome patients: Course of disease and effects on outcome. Leukemia 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.; Paragh, G.; Jeney, V. Influence of iron on bone homeostasis. Pharmaceuticals 2018, 11, 107. [Google Scholar] [CrossRef]
- Samimi, A.; Khodayar, M.J.; Alidadi, H.; Khodadi, E. The Dual Role of ROS in Hematological Malignancies: Stem Cell Protection and Cancer Cell Metastasis. Stem Cell Rev. Rep. 2020, 16, 262–275. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Jin, X.; He, X.; Cao, X.; Xu, P.; Xing, Y.; Sui, S.; Wang, L.; Meng, J.; Lu, W.; Cui, R.; et al. Iron overload impairs normal hematopoietic stem and progenitor cells through reactive oxygen species and shortens survival in myelodysplastic syndrome mice. Haematologica 2018, 103, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Braulke, F.; Sinzig, U.; Wulf, G.; Maas, J.H.; Konietschke, F.; Haase, D. Iron overload impairs proliferation of erythroid progenitors cells (BFU-E) from patients with myelodysplastic syndromes. Leuk. Res. 2013, 37, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Gale, R.P.; Zhu, G.; Xu, Z.; Qin, T.; Zhang, Y.; Huang, G.; Li, B.; Fang, L.; Zhang, H.; et al. Serum iron metabolism and erythropoiesis in patients with myelodysplastic syndrome not receiving RBC transfusions. Leuk. Res. 2014, 38, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Petzer, V.; Tymoszuk, P.; Asshoff, M.; Carvalho, J.; Papworth, J.; Deantonio, C.; Bayliss, L.; Wake, M.S.; Seifert, M.; Brigo, N.; et al. A fully human anti-BMP6 antibody reduces the need for erythropoietin in rodent models of the anemia of chronic disease. Blood 2020, 136, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.-Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Q.; Zhao, Y.S.; Guo, J.; Zhao, S.D.; Song, L.X.; Fei, C.M.; Zhang, Z.; Li, X.; Chang, C. kang Iron overload promotes erythroid apoptosis through regulating HIF-1a/ROS signaling pathway in patients with myelodysplastic syndrome. Leuk. Res. 2017, 58, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Meunier, M.; Ancelet, S.; Lefebvre, C.; Arnaud, J.; Garrel, C.; Pezet, M.; Wang, Y.; Faure, P.; Szymanski, G.; Duployez, N.; et al. Reactive oxygen species levels control NF-κB activation by low dose deferasirox in erythroid progenitors of low risk myelodysplastic syndromes. Oncotarget 2017, 8, 105510–105524. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)–“2021 update on diagnosis, risk-stratification, and management”. Am. J. Hematol. 2021, 96, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Invernizzi, R. Ring sideroblasts and sideroblastic anemias. Haematologica 2011, 96, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Hamel, J.F.; Toma, A.; Kelaidi, C.; Thépot, S.; Campelo, M.D.; Santini, V.; Sekeres, M.A.; Balleari, E.; Kaivers, J.; et al. Outcome of lower-risk patients with myelodysplastic syndromes without 5q deletion after failure of erythropoiesis-stimulating agents. J. Clin. Oncol. 2017, 35, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Fenaux, P.; Platzbecker, U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021, 5, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Oikonomidou, P.R.; Sinha, S.; Zhang, J.; Presti, V.L.; Hamilton, C.R.; Breda, L.; Casu, C.; La, P.; Martins, A.C.; et al. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of b-thalassemia. Blood 2019, 134, 568–572. [Google Scholar] [CrossRef]
- Martinez, P.A.; Li, R.; Ramanathan, H.N.; Bhasin, M.; Pearsall, R.S.; Kumar, R.; Suragani, R.N.V.S. Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine β-thalassaemia by increasing GATA-1 availability. J. Cell. Mol. Med. 2020, 24, 6162–6177. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhao, M.; Rajbhandary, S.; Xie, F.; Chai, X.; Mu, J.; Meng, J.; Liu, Y.; Jiang, Y.; Xu, X.; et al. Free iron catalyzes oxidative damage to hematopoietic cells/mesenchymal stem cells in vitro and suppresses hematopoiesis in iron overload patients. Eur. J. Haematol. 2013, 91, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Fei, C.; Xiao, C.; Wu, D.; Wu, L.; Li, X.; Chang, C. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhai, W.; Zhao, M.; Li, D.; Chai, X.; Cao, X.; Meng, J.; Chen, J.; Xiao, X.; Li, Q.; et al. Effects of iron overload on the bone marrow microenvironment in mice. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Suzuki, T.; Uehara, E.; Ueda, M.; Nagai, T.; Ozawa, K. The bone marrow hematopoietic microenvironment is impaired in iron-overloaded mice. Eur. J. Haematol. 2014, 93, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Meng, F.; Hu, X.; Huang, L.; Liu, H.; Liu, Z.; Li, L. Iron overload regulate the cytokine of mesenchymal stromal cells through ROS/HIF-1α pathway in Myelodysplastic syndromes. Leuk. Res. 2020, 93. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Z.; Liu, H.; Ding, K.; Mi, F.; Xiang, C.; Wang, G.; Guo, Y.; Fu, R. Iron overload impairs bone marrow mesenchymal stromal cells from higher-risk MDS patients by regulating the ROS-related Wnt/β-catenin pathway. Stem. Cells Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Wulfert, M.; Küpper, A.C.; Tapprich, C.; Bottomley, S.S.; Bowen, D.; Germing, U.; Haas, R.; Gattermann, N. Analysis of mitochondrial DNA in 104 patients with myelodysplastic syndromes. Exp. Hematol. 2008, 36, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Ravera, S.; Calabrese, C.; Gaidano, V.; Niscola, P.; Balleari, E.; Gallo, D.; Petiti, J.; Signorino, E.; Rosso, V.; et al. Iron overload alters the energy metabolism in patients with myelodysplastic syndromes: Results from the multicenter FISM BIOFER study. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Volani, C.; Paglia, G.; Smarason, S.; Pramstaller, P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef] [PubMed]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Giri, S.; DeVeaux, M.; Ballas, S.K.; Duong, V.H. Systematic review and meta-analysis of the effect of iron chelation therapy on overall survival and disease progression in patients with lower-risk myelodysplastic syndromes. Ann. Hematol. 2019, 98, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Marek, B.J.; Paley, C.; Esposito, J.; McNamara, K.; Richards, P.D.; DiBella, N.; Garcia-Manero, G. Relation between chelation and clinical outcomes in lower-risk patients with myelodysplastic syndromes: Registry analysis at 5 years. Leuk. Res. 2017, 56, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sapena, R.; Kelaidi, C.; Vassilieff, D.; Bordessoule, D.; Stamatoullas, A.; Cheze, S.; Beyne-Rauzy, O.; Vey, N.; Rose, C.; et al. Ferritin level at diagnosis is not correlated with poorer survival in non RBC transfusion dependent lower risk de novo MDS. Leuk. Res. 2011, 35, 1530–1533. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, Z.; An, T.; Zhao, L. The impact of iron chelation therapy on patients with lower/intermediate IPSS MDS and the prognostic role of elevated serum ferritin in patients with MDS and AML: A meta-analysis. Medicine 2019, 98. [Google Scholar] [CrossRef] [PubMed]
- Leitch, H.A.; Buckstein, R.; Zhu, N.; Nevill, T.J.; Yee, K.W.L.; Leber, B.; Keating, M.M.; Hilaire, E.; Kumar, R.; Delage, R.; et al. Iron overload in myelodysplastic syndromes: Evidence based guidelines from the Canadian consortium on MDS. Leuk. Res. 2018, 74, 21–41. [Google Scholar] [CrossRef]
- Gimferrer, E.; Nomdedeu, J.; Gich, I.; Barcelo, M.J.; Baiget, M. Prevalence of hemochromatosis related HFE gene mutations in patients with acute myeloid leukemia. Leuk. Res. 1999, 23, 597–598. [Google Scholar] [CrossRef]
- Veneri, D.; Franchini, M.; Zanetti, F.; Krampera, M.; de Matteis, G.; Pizzolo, G. Iron overload in acute myeloid leukemia patients is not related to HFE and TFR2 gene mutations. Haematologica 2003, 88, 1069–1070. [Google Scholar]
- Viola, A.; Pagano, L.; Laudati, D.; D’Elia, R.; D’Amico, M.R.; Ammirabile, M.; Palmieri, S.; Prossomariti, L.; Ferrara, F. HFE gene mutations in patients with acute leukemia. Leuk. Lymphoma 2006, 47, 2331–2334. [Google Scholar] [CrossRef]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; De Graaff, B.; McLaren, C.E.; Loreál, O. Haemochromatosis. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. Epigenetic changes in the myelodysplastic syndrome. Hematol. Oncol. Clin. N. Am. 2010, 24, 317–330. [Google Scholar] [CrossRef]
- Bejar, R.; Levine, R.; Ebert, B.L. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Mainous, A.G.; Wright, R.U.; Hulihan, M.M.; Twal, W.O.; McLaren, C.E.; Diaz, V.A.; McLaren, G.D.; Argraves, W.S.; Grant, A.M. Telomere length and elevated iron: The influence of phenotype and HFE genotype. Am. J. Hematol. 2013, 88, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Colla, S.; Ong, D.S.T.; Ogoti, Y.; Marchesini, M.; Mistry, N.A.; Clise-Dwyer, K.; Ang, S.A.; Storti, P.; Viale, A.; Giuliani, N.; et al. Telomere Dysfunction Drives Aberrant Hematopoietic Differentiation and Myelodysplastic Syndrome. Cancer Cell 2015, 27, 644–657. [Google Scholar] [CrossRef]
- Lange, K.; Holm, L.; Nielsen, K.V.; Hahn, A.; Hofmann, W.; Kreipe, H.; Schlegelberger, B.; Göhring, G. Telomere shortening and chromosomal instability in myelodysplastic syndromes. Genes Chromosom. Cancer 2010, 49, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Rollison, D.E.; Epling-Burnette, P.K.; Park, J.Y.; Lee, J.H.; Park, H.; Jonathan, K.; Cole, A.L.; Painter, J.S.; Guerrier, M.; Meléndez-Santiago, J.; et al. Telomere length in myelodysplastic syndromes. Leuk. Lymphoma 2011, 52, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.M.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. New insights into iron deficiency and iron deficiency anemia. Blood Rev. 2017, 31, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Grants, J.M.; Wegrzyn, J.; Hui, T.; O’Neill, K.; Shadbolt, M.; Knapp, D.J.H.F.; Parker, J.; Deng, Y.; Gopal, A.; Roderick Docking, T.; et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood 2020, 135, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Calvi, L.M.; Becker, M.W.; Liesveld, J.L. Flaming and fanning: The Spectrum of inflammatory influences in myelodysplastic syndromes. Blood Rev. 2019, 36, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kawakami, T.; Yamamoto, N.; Tomizawa, M.; Fujiwara, T.; Ishii, T.; Harigae, H.; Ogasawara, K. Activation of the NLRP3 inflammasome by cellular labile iron. Exp. Hematol. 2016, 44, 116–124. [Google Scholar] [CrossRef]
- Lee, P.; Peng, H.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef]
- Santini, V.; Girelli, D.; Sanna, A.; Martinelli, N.; Duca, L.; Campostrini, N.; Cortelezzi, A.; Corbella, M.; Bosi, A.; Reda, G.; et al. Hepcidin levels and their determinants in different types of myelodysplastic syndromes. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Gu, S.; Xv, Y.; Fei, C.; Xiao, C.; Guo, J.; Zhao, Y.; Xv, F.; Li, X.; Chang, C. Labile plasma iron, more practical and more sensitive to iron overload in myelodysplastic syndromes. Hematology 2017, 22, 9–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambaglio, I.; Malcovati, L.; Papaemmanuil, E.; Laarakkers, C.M.; Della Porta, M.G.; Gallì, A.; Da Vià, M.C.; Bono, E.; Ubezio, M.; Travaglino, E.; et al. Inappropriately low hepcidin levels in patients with myelodysplastic syndrome carrying a somatic mutation of SF3B1. Haematologica 2013, 98, 420–423. [Google Scholar] [CrossRef]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef]
- Bondu, S.; Alary, A.S.; Lefèvre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Mossner, M.; Stöhr, A.; Jann, J.C.; Streuer, A.; Schmitt, N.; Knaflic, A.; Nowak, V.; Weimer, N.; Obländer, J.; et al. High erythroferrone expression in CD71+ erythroid progenitors predicts superior survival in myelodysplastic syndromes. Br. J. Haematol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kosmider, O.; Maloisel, F.; Drenou, B.; Chapuis, N.; Lefebvre, T.; Karim, Z.; Puy, H.; Alary, A.S.; Ducamp, S.; et al. Dyserythropoiesis evaluated by the RED score and hepcidin:ferritin ratio predicts response to erythropoietin in lower-risk myelodysplastic syndromes. Haematologica 2019, 104, 497–504. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-associated TET2 mutations: Common drivers of myeloid dysfunction, cancer and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 626. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56.e13–70.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833.e5–849.e5. [Google Scholar] [CrossRef]
- Zhao, L.-P.; Boy, M.; Azoulay, C.; Clappier, E.; Sébert, M.; Amable, L.; Klibi, J.; Benlagha, K.; Espéli, M.; Balabanian, K.; et al. Genomic landscape of MDS/CMML associated with systemic inflammatory and autoimmune disease. Leukemia 2021, 1–5. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The Methylcytosine Dioxygenase Tet2 Promotes DNA Demethylation and Activation of Cytokine Gene Expression in T Cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef]
- Yue, X.; Trifari, S.; Äijö, T.; Tsagaratou, A.; Pastor, W.A.; Zepeda-Martínez, J.A.; Lio, C.W.J.; Li, X.; Huang, Y.; Vijayanand, P.; et al. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 2016, 213, 377–397. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef]
- Shearstone, J.R.; Pop, R.; Bock, C.; Boyle, P.; Meissner, A.; Socolovsky, M. Global DNA demethylation during mouse erythropoiesis in vivo. Science 2011, 334, 799–802. [Google Scholar] [CrossRef]
- Madzo, J.; Liu, H.; Rodriguez, A.; Vasanthakumar, A.; Sundaravel, S.; Caces, D.B.D.; Looney, T.J.; Zhang, L.; Lepore, J.B.; Macrae, T.; et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. 2014, 6, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zhang, S.; Wang, S.; Wang, Y.; Li, W.; Huang, Y.; Zhao, H.; Wu, X.; An, C.; Guo, X.; et al. TET2 deficiency leads to stem cell factor–dependent clonal expansion of dysfunctional erythroid progenitors. Blood 2018, 132, 2406. [Google Scholar] [CrossRef]
- Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013, 14, 17643–17663. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef]
- Guo, S.; Jiang, X.; Wang, Y.; Chen, L.; Li, H.; Li, X.; Jia, Y. The protective role of TET2 in erythroid iron homeostasis against oxidative stress and erythropoiesis. Cell. Signal. 2017, 38, 106–115. [Google Scholar] [CrossRef]
- Koury, M.J.; Haase, V.H. Anaemia in kidney disease: Harnessing hypoxia responses for therapy. Nat. Rev. Nephrol. 2015, 11, 394–410. [Google Scholar] [CrossRef]
- Sonnweber, T.; Nachbaur, D.; Schroll, A.; Nairz, M.; Seifert, M.; Demetz, E.; Haschka, D.; Mitterstiller, A.M.; Kleinsasser, A.; Burtscher, M.; et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 2014, 63, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.C.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Inokura, K.; Fujiwara, T.; Saito, K.; Iino, T.; Hatta, S.; Okitsu, Y.; Fukuhara, N.; Onishi, Y.; Ishizawa, K.; Shimoda, K.; et al. Impact of TET2 deficiency on iron metabolism in erythroblasts. Exp. Hematol. 2017, 49, 56.e5–67.e5. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Büsche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Füreder, W.; Bettelheim, P.; Pfeilstöcker, M.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis with Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Demetz, E.; Tymoszuk, P.; Hilbe, R.; Volani, C.; Haschka, D.; Heim, C.; Auer, K.; Lener, D.; Zeiger, L.B.; Pfeifhofer-Obermair, C.; et al. The haemochromatosis gene Hfe and Kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur. Heart J. 2020, 41, 3949–3959B. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Dubois-deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Yusuf, R.Z.; Saez, B.; Sharda, A.; van Gastel, N.; Yu, V.W.C.; Baryawno, N.; Scadden, E.W.; Acharya, S.; Chattophadhyay, S.; Huang, C.; et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood 2020, 136, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Niu, H.; Yue, L.; Liu, J.; Yang, L.; Liu, C.; Jiang, H.; Dong, S.; Shao, Z.; Xing, L.; et al. Abnormal Ferroptosis in Myelodysplastic Syndrome. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Jaeggi, T.; Kortman, G.A.M.; Moretti, D.; Chassard, C.; Holding, P.; Dostal, A.; Boekhorst, J.; Timmerman, H.M.; Swinkels, D.W.; Tjalsma, H.; et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in Kenyan infants. Gut 2015, 64, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Clavel, T.; Smirnov, K.; Schmidt, A.; Lagkouvardos, I.; Walker, A.; Lucio, M.; Michalke, B.; Schmitt-Kopplin, P.; Fedorak, R.; et al. Oral versus intravenous iron replacement therapy distinctly alters the gut microbiota and metabolome in patients with IBD. Gut 2017, 66, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Li, X.Y.; Shen, L.; Ji, H.F. Regulatory effects of transition metals supplementation/deficiency on the gut microbiota. Appl. Microbiol. Biotechnol. 2021, 105, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Visitchanakun, P.; Saisorn, W.; Wongphoom, J.; Chatthanathon, P.; Somboonna, N.; Svasti, S.; Fucharoen, S.; Leelahavanichkul, A. Gut leakage enhances sepsis susceptibility in iron-overloaded β-thalassemia mice through macrophage hyperinflammatory responses. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G966–G979. [Google Scholar] [CrossRef] [PubMed]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115.e6–130.e6. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petzer, V.; Theurl, I.; Weiss, G.; Wolf, D. EnvIRONmental Aspects in Myelodysplastic Syndrome. Int. J. Mol. Sci. 2021, 22, 5202. https://doi.org/10.3390/ijms22105202
Petzer V, Theurl I, Weiss G, Wolf D. EnvIRONmental Aspects in Myelodysplastic Syndrome. International Journal of Molecular Sciences. 2021; 22(10):5202. https://doi.org/10.3390/ijms22105202
Chicago/Turabian StylePetzer, Verena, Igor Theurl, Günter Weiss, and Dominik Wolf. 2021. "EnvIRONmental Aspects in Myelodysplastic Syndrome" International Journal of Molecular Sciences 22, no. 10: 5202. https://doi.org/10.3390/ijms22105202
APA StylePetzer, V., Theurl, I., Weiss, G., & Wolf, D. (2021). EnvIRONmental Aspects in Myelodysplastic Syndrome. International Journal of Molecular Sciences, 22(10), 5202. https://doi.org/10.3390/ijms22105202