
Biological and Physico-Chemical Characteristics of Arginine-Rich Peptide Gemini Surfactants with Lysine and Cystine Spacers

 , , and
, , and 
Abstract
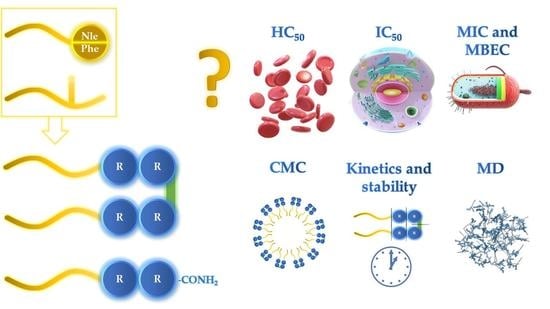
1. Introduction
2. Results and Discussion
2.1. Determination of Peptide Hydrophobicity and Critical Aggregation Concentration
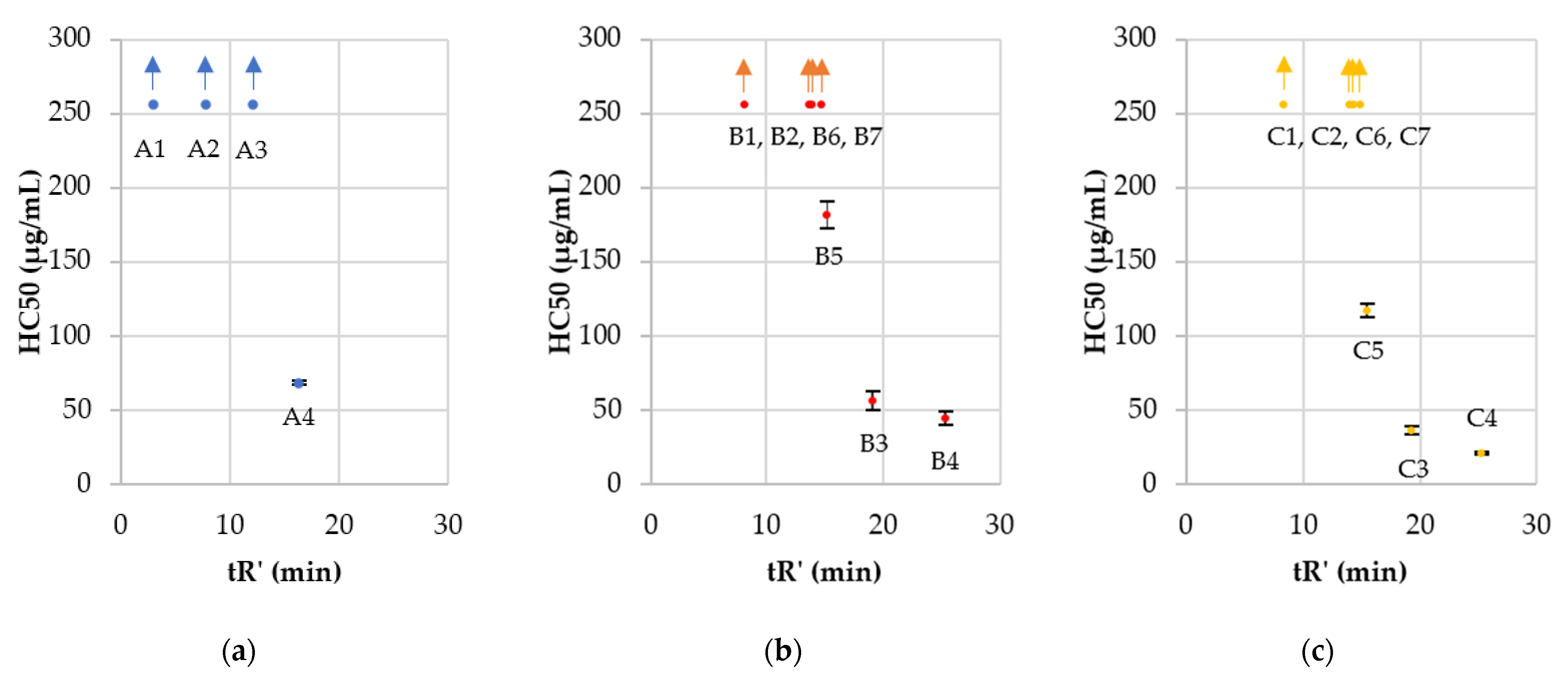
2.2. Antimicrobial Activity, Cytotoxicity, and Hemolysis of Lipopeptides
2.3. Antimicrobial Activity in the Presence of Serum
2.4. Antibiofilm Activity
2.5. Membrane Permeabilization
2.6. Serum Stability
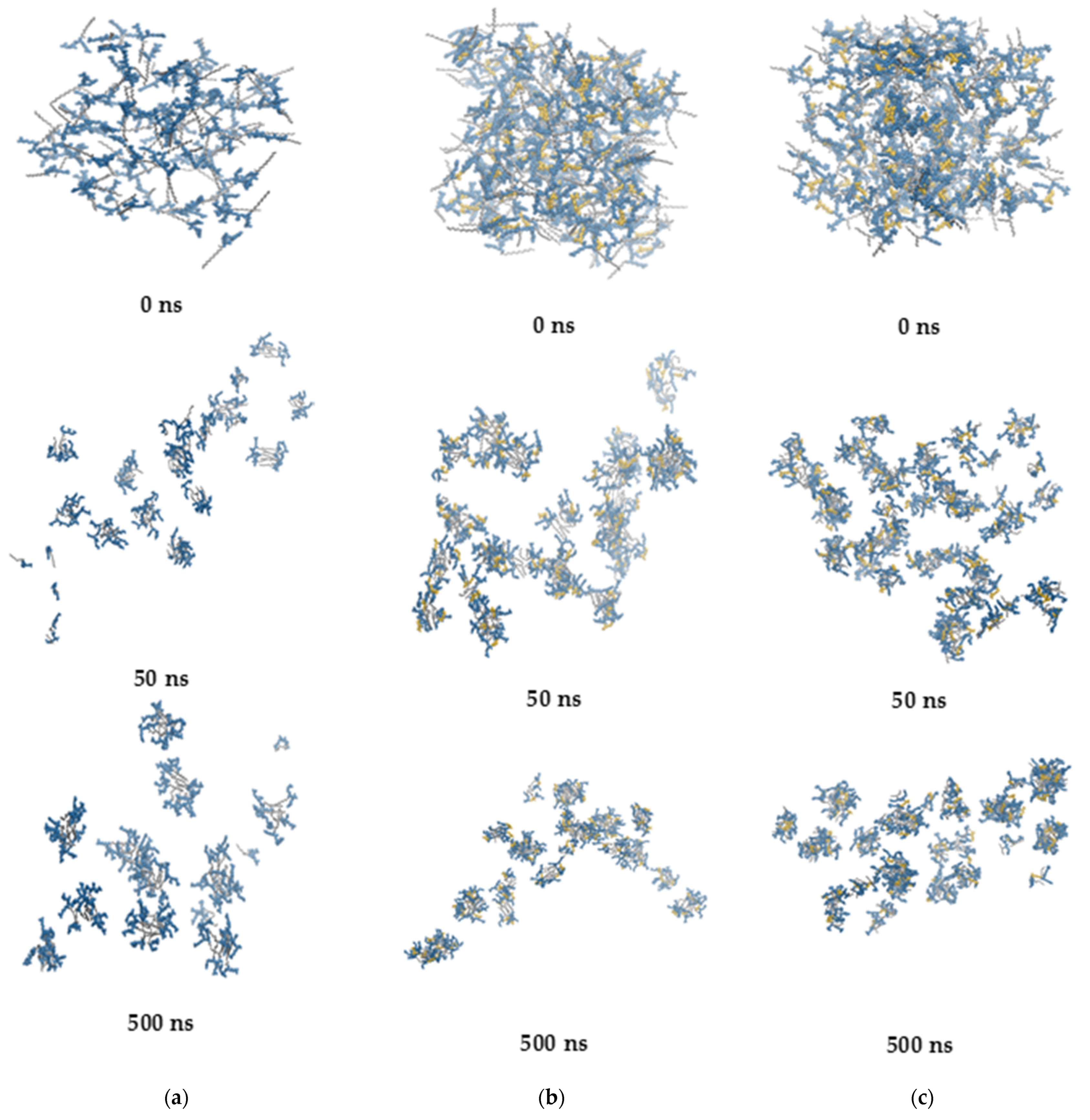
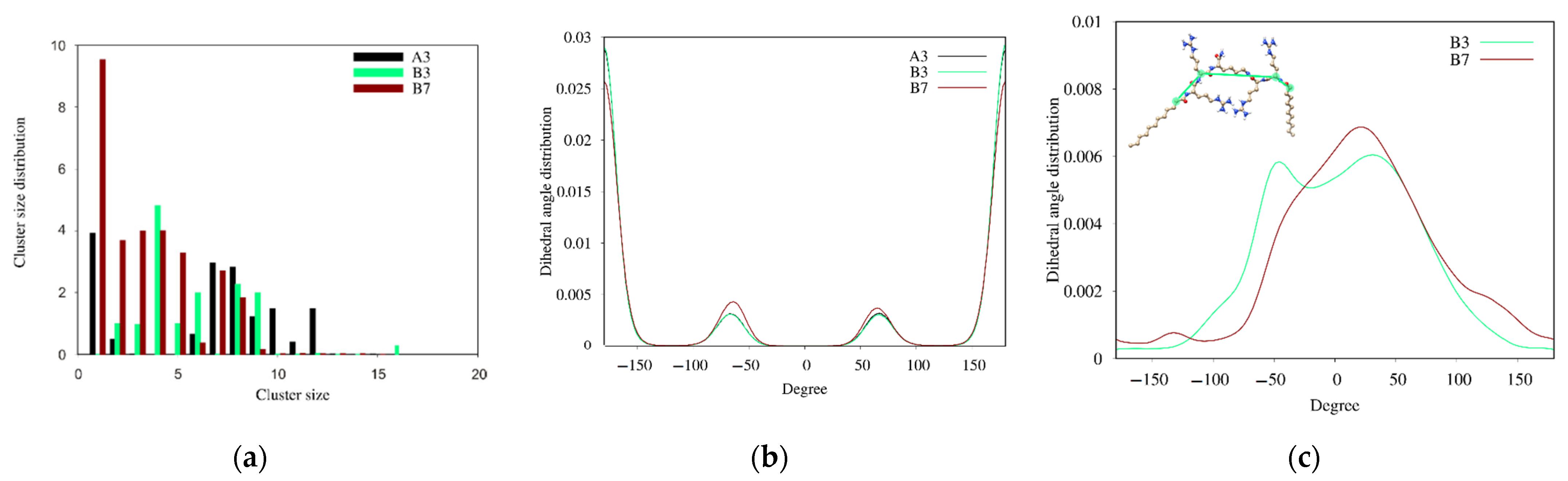
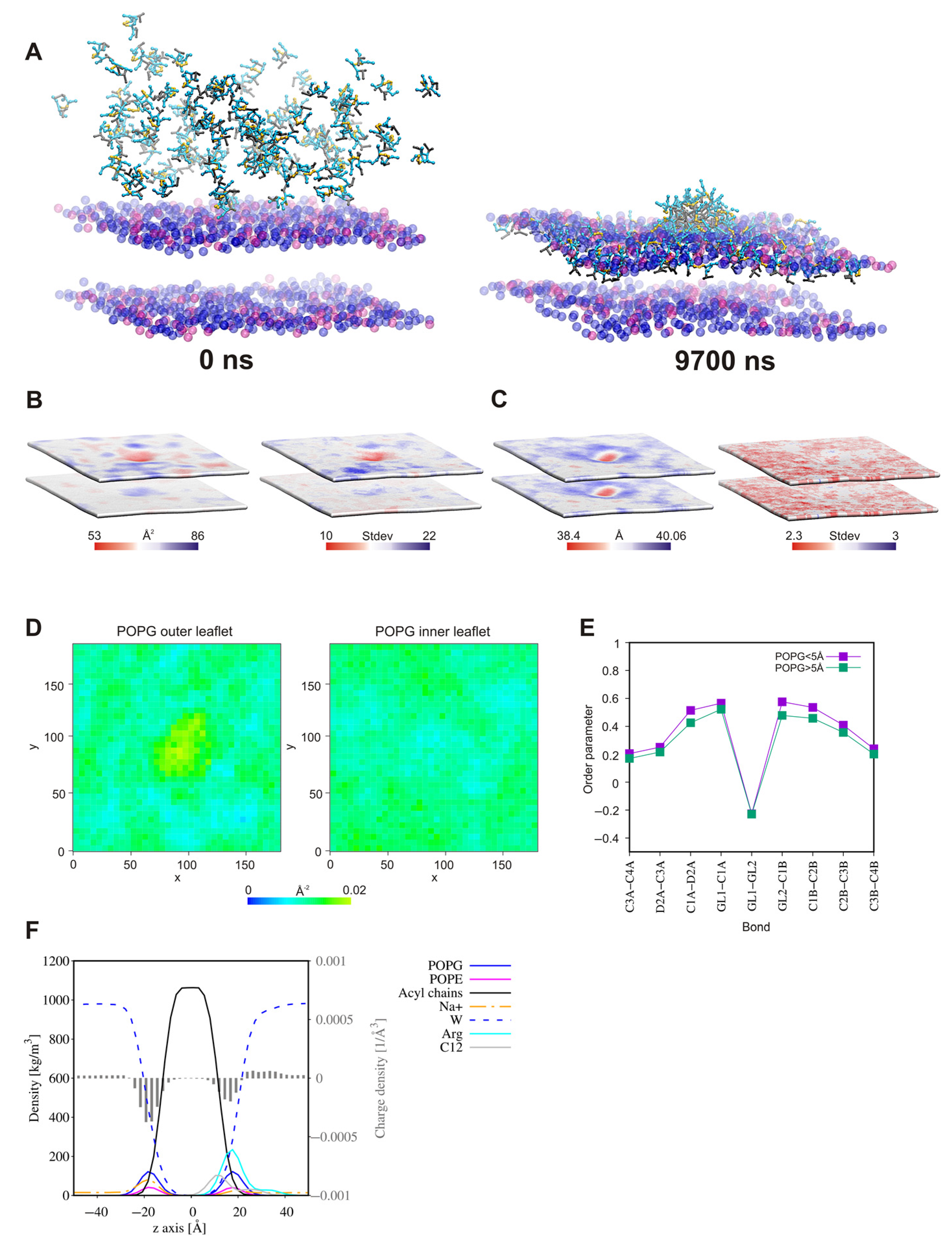
2.7. Self-Assembly Tendency via Molecular Dynamic Simulations
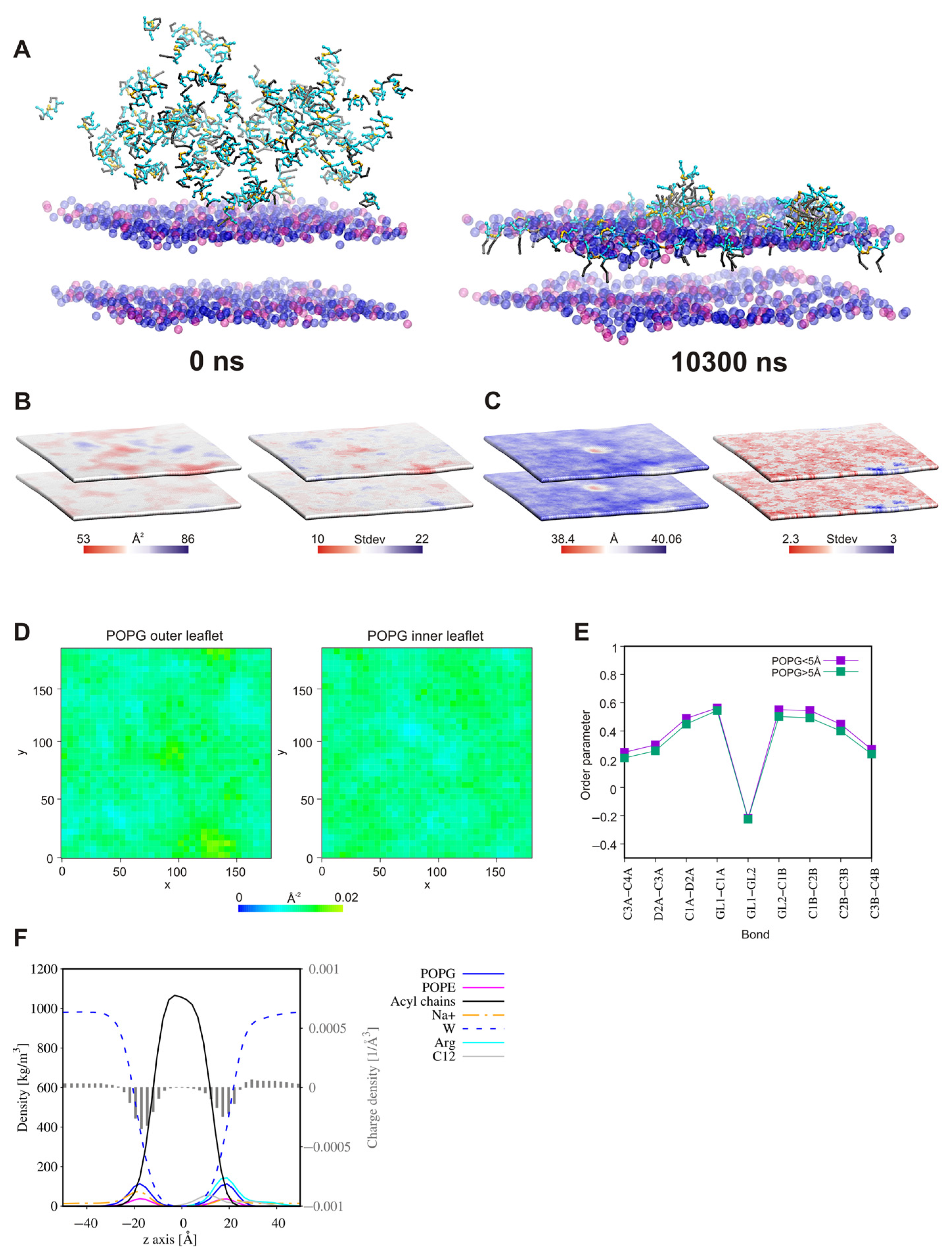
2.8. Surfactant—Membrane Interactions via Molecular Dynamic Simulations
3. Materials and Methods
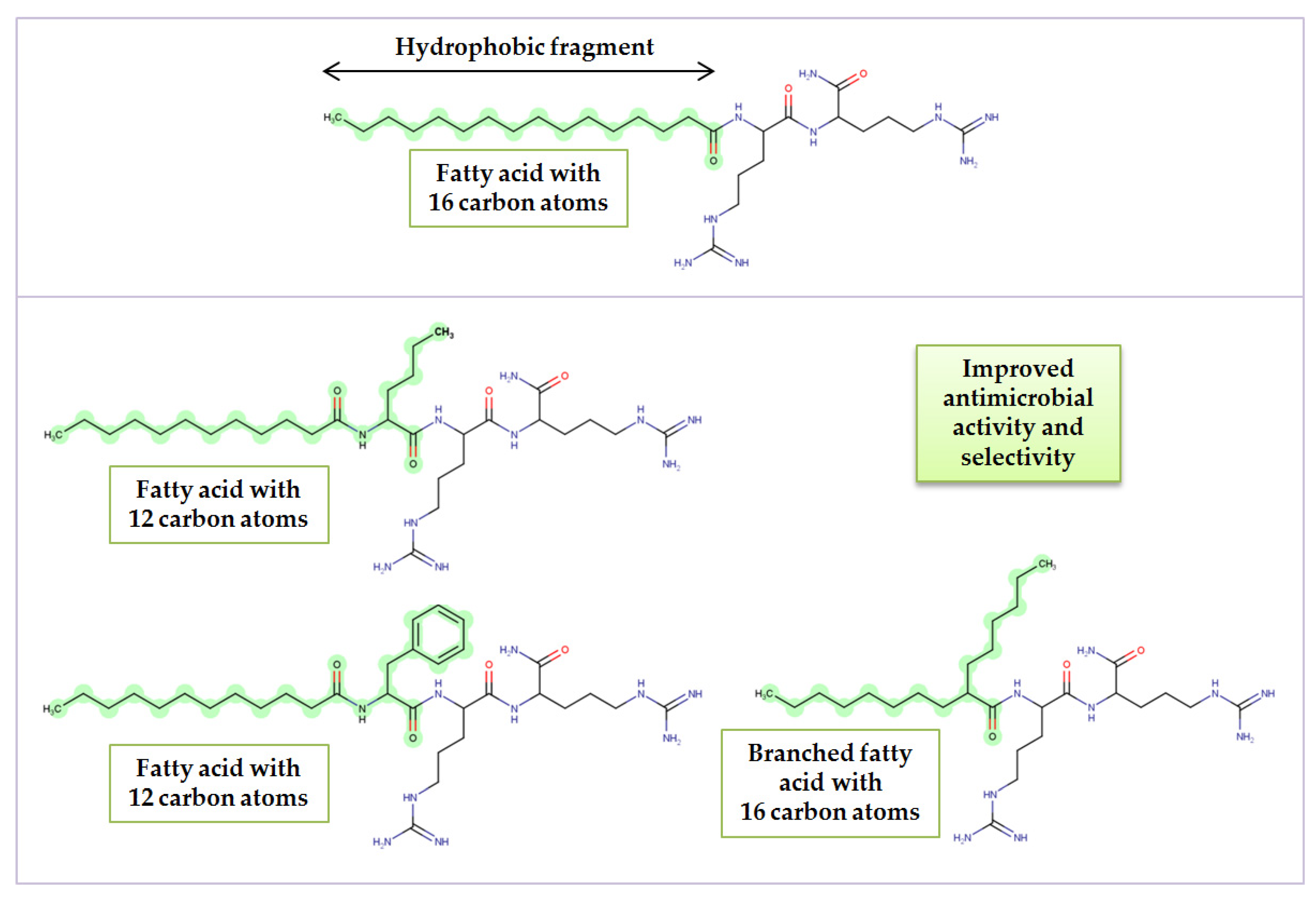
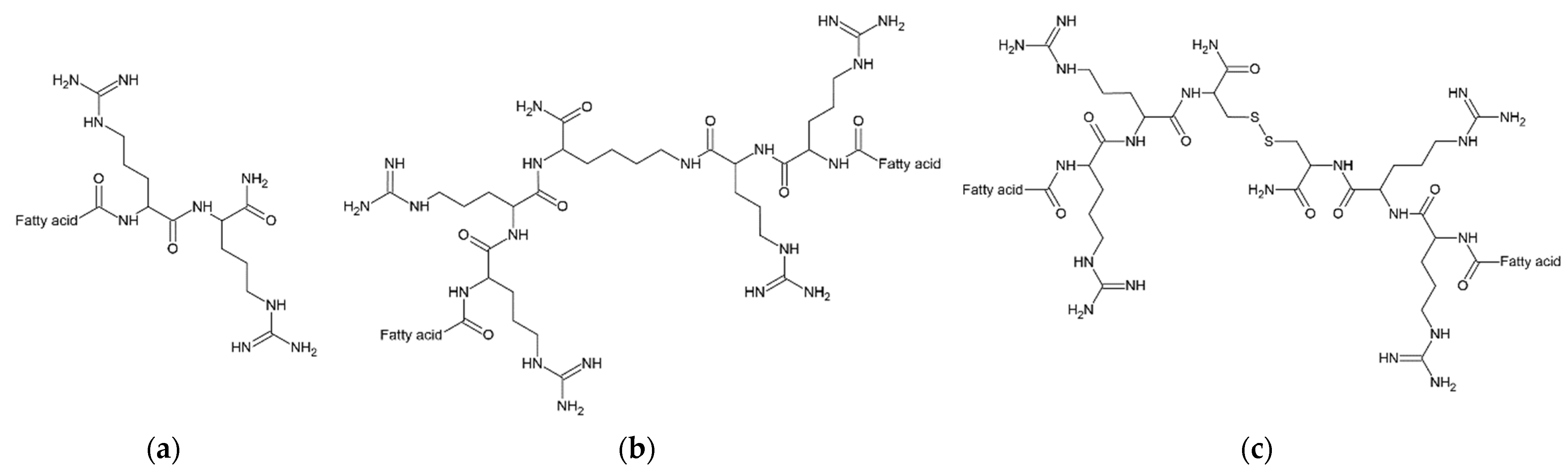
3.1. Lipopeptide Synthesis
3.2. Determination of Lipopeptide Hydrophobicity with RP-HPLC
3.3. Critical Aggregation Concentration Measurements
3.4. Antimicrobial Activity
3.4.1. Cultivation of Microorganisms
3.4.2. Activity against Planktonic Cultures
3.4.3. Activity against Biofilm
3.4.4. Antimicrobial Activity in the Presence of Serum
3.5. Hemolysis Assay
3.6. MTS Assay
3.7. Bacterial Membrane Permeabilization Assays
3.8. Serum Stability
3.8.1. Incubation in Serum
3.8.2. Calibration Curve
3.8.3. UPLC-HRMS Analytical Parameters
3.9. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AMPs | antimicrobial peptides |
| APL | area per lipid |
| ATCC | American Type Culture Collection |
| AU | absorbance unit |
| BSA | bovine serum albumin |
| CAC | critical aggregation concentration |
| CENTA | (6R,7R)-3-{[(3-Carboxy-4-nitrophenyl)sulfanyl]methyl}-8-oxo-7-[(2-thienylacetyl)amino]-5-thia-1-azabicyclo [4.2.0]oct-2-ene-2-carboxylic acid |
| CFU | colony forming units |
| CLSI | Clinical and Laboratory Standards Institute |
| CMC | critical micelle concentration |
| DCM | dichloromethane |
| DIC | N,N′-diisopropylcarbodiimide |
| DMF | N,N-dimethylformamide |
| EDT | 1,2-ethaneditiol |
| EDTA | ethylenediaminetetraacetic acid |
| ESI | electrospray-ionization |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| GSH | reduced glutathione |
| HaCaT | immortalized human keratinocytes cell line |
| HC50 | surfactant concentration causing 50% hemolysis |
| hRBCs | human red blood cells |
| HSA | human serum albumin |
| IC50 | surfactant concentration causing 50% inhibition of the growth |
| IM | inner membrane |
| LC-MS | liquid chromatography-mass spectrometry |
| MBEC | minimum biofilm eradication concentration |
| MD | molecular dynamics |
| MHB | Mueller–Hinton broth |
| MIC | minimum inhibitory concentration |
| MRSA | methicillin-resistant Staphylococcus aureus |
| MTS | 4-{5-[3-(Carboxymethoxy)phenyl]-3-(4,5-dimethyl-1,3-thiazol-2-yl)-2H-tetrazol-3-ium-2-yl}benzenesulfonate; tetrazolium inner salt |
| nd | not determined |
| NHS | normal human serum |
| OM | outer membrane |
| ONP | ortho-nitrophenol |
| ONPG | o-nitrophenyl-β-d-galactopyranoside |
| OxymaPure | ethyl (2E)-cyano(hydroxyimino)acetate |
| PBS | phosphate-buffer saline |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol |
| Q-TOF | quadrupole time-of-flight |
| RP-HPLC | reverse-phase high-performance liquid chromatography |
| SDA | Sabouraud dextrose agar |
| SI | selectivity index |
| tBu | tert-butyl |
| TFA | trifluoroacetic acid |
| TIS | triisopropylsilane |
| TNB- | 2-nitro-5-thiobenzoate anion |
| UPLC-HRMS | ultra-performance liquid chromatography-high-resolution mass spectrometry |
| USCLs | ultrashort cationic lipopeptides |
| VRE | vancomycin-resistant enterococci |
| WHO | World Health Organization |
References
- Rounds, T.; Straus, S.K. Lipidation of antimicrobial peptides as a design strategy for future alternatives to antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef] [PubMed]
- WHO Model List of Essential Medicines, 20th List; World Health Organization: Geneva, Switzerland, 2017.
- The 2019 WHO AWaRe Classification of Antibiotics for Evaluation and Monitoring of Use; World Health Organization: Geneva, Switzerland, 2019.
- Kaye, K.S.; Pogue, J.M.; Tran, T.B.; Nation, R.L.; Li, J. Agents of Last Resort: Polymyxin Resistance. Infect. Dis. Clin. N. Am. 2016, 30, 391–414. [Google Scholar] [CrossRef]
- Eisenstein, B.I.; Oleson, F.B., Jr.; Baltz, R.H. Daptomycin: From the Mountain to the Clinic, with Essential Help from Francis Tally, MD. Clin. Infect. Dis. 2010, 50, S10–S15. [Google Scholar] [CrossRef]
- Landman, D.; Georgescu, C.; Martin, D.A.; Quale, J. Polymyxins revisited. Clin. Microbiol. Rev. 2008, 21, 449–465. [Google Scholar] [CrossRef] [PubMed]
- King, S.T.; Walker, E.D.; Cannon, C.G.; Finley, R.W. Daptomycin-induced rhabdomyolysis and acute liver injury. Scand. J. Infect. Dis. 2014, 46, 537–540. [Google Scholar] [CrossRef]
- He, W.; Zhang, Y.; Chen, H.; Zhao, C.; Wang, H. Efficacy and safety of daptomycin for the treatment of infectious disease: A meta-analysis based on randomized controlled trials. J. Antimicrob. Chemother. 2014, 69, 3181–3189. [Google Scholar] [CrossRef] [PubMed]
- Gales, A.C.; Jones, R.N.; Sader, H.S. Global assessment of the antimicrobial activity of polymyxin B against 54 731 clinical isolates of Gram-negative bacilli: Report from the SENTRY antimicrobial surveillance programme (2001–2004). Clin. Microbiol. Infect. 2006, 12, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Farrell, D.J.; Flamm, R.K.; Jones, R.N. Daptomycin activity tested against 164457 bacterial isolates from hospitalised patients: Summary of 8 years of a Worldwide Surveillance Programme (2005–2012). Int. J. Antimicrob. Agents 2014, 43, 465–469. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Perlin, D.S. Echinocandin resistance: An emerging clinical problem? Curr. Opin. Infect. Dis. 2014, 27, 484–492. [Google Scholar] [CrossRef]
- WHO | Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017.
- Beyer, P.; Paulin, S. Priority pathogens and the antibiotic pipeline: An update. Bull. World Health Organ. 2020, 98, 151. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, X.; Yang, Q.; Qiao, S. Antimicrobial peptides as potential alternatives to antibiotics in food animal industry. Int. J. Mol. Sci. 2016, 17, 603. [Google Scholar] [CrossRef]
- Neubauer, D.; Jaśkiewicz, M.; Bauer, M.; Gołacki, K.; Kamysz, W. Ultrashort Cationic Lipopeptides–Effect of N-Terminal Amino Acid and Fatty Acid Type on Antimicrobial Activity and Hemolysis. Molecules 2020, 25, 257. [Google Scholar] [CrossRef]
- Du, X.; Lu, Y.; Li, L.; Wang, J.; Yang, Z. Synthesis and unusual properties of novel alkylbenzene sulfonate gemini surfactants. Colloids Surfaces A Physicochem. Eng. Asp. 2006, 290, 132–137. [Google Scholar] [CrossRef]
- Monger, F.M.; Littau, C.A. Gemini Surfactants: Synthesis and Properties. J. Am. Chem. Soc. 1991, 113, 1451–1452. [Google Scholar] [CrossRef]
- Chau, H.N.; Kawase, T.; Oida, T. Preparation of dicarboxylic acid—Type Gemini surfactant via Diels-Alder reaction & ozone oxidation. J. Oleo Sci. 2013, 62, 409–414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Damen, M.; Cristóbal-Lecina, E.; Sanmartí, G.C.; Van Dongen, S.F.M.; García Rodríguez, C.L.; Dolbnya, I.P.; Nolte, R.J.M.; Feiters, M.C. Structure-delivery relationships of lysine-based gemini surfactants and their lipoplexes. Soft Matter 2014, 10, 5702–5714. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.; Torres, J.L.; Manresa, A.; Solans, C.; Infante, M.A.R. Synthesis, aggregation, and biological properties of a new class of gemini cationic amphiphilic compounds from arginine, bis(args). Langmuir 1996, 12, 5296–5301. [Google Scholar] [CrossRef]
- Cardoso, A.M.; Morais, C.M.; Cruz, A.R.; Silva, S.G.; Do Vale, M.L.; Marques, E.F.; De Lima, M.C.P.; Jurado, A.S. New serine-derived gemini surfactants as gene delivery systems. Eur. J. Pharm. Biopharm. 2015, 89, 347–356. [Google Scholar] [CrossRef]
- Kamboj, R.; Singh, S.; Bhadani, A.; Kataria, H.; Kaur, G. Gemini imidazolium surfactants: Synthesis and their biophysiochemical study. Langmuir 2012, 28, 11969–11978. [Google Scholar] [CrossRef]
- Bhadani, A.; Kafle, A.; Koura, S.; Sakai, K.; Sakai, H.; Abe, M. Physicochemical Evaluation of Micellar Solution and Lyotropic Phases Formed by Self-Assembled Aggregates of Morpholinium Geminis. ACS Omega 2017, 2, 5324–5334. [Google Scholar] [CrossRef] [PubMed]
- Quagliotto, P.; Viscardi, G.; Barolo, C.; Barni, E.; Bellinvia, S.; Fisicaro, E.; Compari, C. Gemini pyridinium surfactants: Synthesis and conductometric study of a novel class of amphiphiles. J. Org. Chem. 2003, 68, 7651–7660. [Google Scholar] [CrossRef]
- Menger, F.M.; Keiper, J.S.; Azov, V. Gemini surfactants with acetylenic spacers. Langmuir 2000, 16, 2062–2067. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, Z.; Xu, D.; Hou, L.; Zhao, W.; Nie, X.; Zhou, L.; Zhao, J. Synthesis of three gemini betaine surfactants and their surface active properties. J. Taiwan Inst. Chem. Eng. 2017, 74, 7–13. [Google Scholar] [CrossRef]
- Yoshimura, T.; Nyuta, K.; Esumi, K. Zwitterionic heterogemini surfactants containing ammonium and carboxylate headgroups. 1. Adsorption and micellization. Langmuir 2005, 21, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Laska, U.; Wilk, K.A.; Maliszowska, I.; Syper, L. Novel glucose-derived gemini surfactants with a 1,1′-ethylenebisurea spacer: Preparation, thermotropic behavior, and biological properties. J. Surfactants Deterg. 2006, 9, 115–124. [Google Scholar] [CrossRef]
- Yoshimura, T.; Ishihara, K.; Esumi, K. Sugar-based gemini surfactants with peptide bonds-synthesis, adsorption, micellization, and biodegradability. Langmuir 2005, 21, 10409–10415. [Google Scholar] [CrossRef] [PubMed]
- Bunton, C.A.; Robinson, L.; Stam, M.F.; Schaak, J. Catalysis of Nucleophilic Substitutions by Micelles of Dicationic Detergents. J. Org. Chem. 1971, 36, 2346–2350. [Google Scholar] [CrossRef]
- Tan, H.; Xiao, H. Synthesis and antimicrobial characterization of novel l-lysine gemini surfactants pended with reactive groups. Tetrahedron Lett. 2008, 49, 1759–1761. [Google Scholar] [CrossRef]
- Pinazo, A.; Diz, M.; Solans, C.; Pés, M.A.; Erra, P.; Infante, M.R. Synthesis and properties of cationic surfactants containing a disulfide bond. J. Am. Oil Chem. Soc. 1993, 70, 37–42. [Google Scholar] [CrossRef]
- Zana, R.; Talmon, Y. Dependence of aggregate morphology on structure of dimeric surfactants. Nature 1993, 362, 228–230. [Google Scholar] [CrossRef]
- Emara, M.M.; Abdel-Salam, F.H.; Ali, R.A.; Turky, A.S.; Elghayish, M.M. Synthesis and Evaluation of Surface Activity of Gemini Borate Surfactants Based on Glucose Moiety. J. Dispers. Sci. Technol. 2016, 37, 733–742. [Google Scholar] [CrossRef]
- Singh, R.K.; Kukrety, A.; Saxena, R.C.; Thakre, G.D.; Atray, N.; Ray, S.S. Novel Triazine Schiff Base-Based Cationic Gemini Surfactants: Synthesis and Their Evaluation as Antiwear, Antifriction, and Anticorrosive Additives in Polyol. Ind. Eng. Chem. Res. 2016, 55, 2520–2526. [Google Scholar] [CrossRef]
- Damen, M.; Groenen, A.J.J.; van Dongen, S.F.M.; Nolte, R.J.M.; Scholte, B.J.; Feiters, M.C. Transfection by cationic gemini lipids and surfactants. MedChemComm 2018, 9, 1404–1425. [Google Scholar] [CrossRef] [PubMed]
- Damen, M.; Aarbiou, J.; van Dongen, S.F.M.; Buijs-Offerman, R.M.; Spijkers, P.P.; van den Heuvel, M.; Kvashnina, K.; Nolte, R.J.M.; Scholte, B.J.; Feiters, M.C. Delivery of DNA and siRNA by novel gemini-like amphiphilic peptides. J. Control. Release 2010, 145, 33–39. [Google Scholar] [CrossRef]
- Micich, T.J.; Linfield, W.M. Wetting properties of nonionics from branched fatty diamides. J. Am. Oil Chem. Soc. 1988, 65, 820–825. [Google Scholar] [CrossRef]
- Li, H.; Yang, H.; Yan, Y.; Wang, Q.; He, P. Synthesis and solution properties of cationic gemini surfactants with long unsaturated tails. Surf. Sci. 2010, 604, 1173–1178. [Google Scholar] [CrossRef]
- Sakai, K.; Kaji, M.; Takamatsu, Y.; Tsuchiya, K.; Torigoe, K.; Tsubone, K.; Yoshimura, T.; Esumi, K.; Sakai, H.; Abe, M. Fluorocarbon-hydrocarbon gemini surfactant mixtures in aqueous solution. Colloids Surf. A Physicochem. Eng. Asp. 2009, 333, 26–31. [Google Scholar] [CrossRef]
- Hussain, S.M.S.; Kamal, M.S.; Solling, T.; Murtaza, M.; Fogang, L.T. Surface and thermal properties of synthesized cationic poly(ethylene oxide) gemini surfactants: The role of the spacer. RSC Adv. 2019, 9, 30154–30163. [Google Scholar] [CrossRef]
- Kim, B.K.; Doh, K.O.; Bae, Y.U.; Seu, Y.B. Synthesis and optimization of cholesterol-based diquaternary ammonium gemini surfactant (Chol-GS) as a new gene delivery vector. J. Microbiol. Biotechnol. 2011, 21, 93–99. [Google Scholar] [CrossRef]
- Bajaj, A.; Paul, B.; Indi, S.S.; Kondaiah, P.; Bhattacharya, S. Effect of the hydrocarbon chain and polymethylene spacer lengths on gene transfection efficacies of gemini lipids based on aromatic backbone. Bioconjug. Chem. 2007, 18, 2144–2158. [Google Scholar] [CrossRef]
- Rosen, M.J.; Tracy, D.J. Gemini surfactants. J. Surfactants Deterg. 1998, 1, 547–554. [Google Scholar] [CrossRef]
- Bombelli, C.; Giansanti, L.; Luciani, P.; Mancini, G. Gemini Surfactant Based Carriers in Gene and Drug Delivery. Curr. Med. Chem. 2008, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, J.; Ma, J.; Li, M.; Li, X. Physicochemical and antimicrobial activities of cationic gemini surfactants with polyether siloxane linked group. J. Mol. Liq. 2017, 242, 8–15. [Google Scholar] [CrossRef]
- Meyer, V.R. Reversed-Phase Chromatography. In Practical High-Performance Liquid Chromatography; John Wiley & Sons, Ltd.: Chichester, UK, 2010; pp. 173–193. [Google Scholar]
- Klevens, H.B. Structure and aggregation in dilate solution of surface active agents. J. Am. Oil Chem. Soc. 1953, 30, 74–80. [Google Scholar] [CrossRef]
- Garcia, M.T.; Kaczerewska, O.; Ribosa, I.; Brycki, B.; Materna, P.; Drgas, M. Hydrophilicity and flexibility of the spacer as critical parameters on the aggregation behavior of long alkyl chain cationic gemini surfactants in aqueous solution. J. Mol. Liq. 2017, 230, 453–460. [Google Scholar] [CrossRef]
- Wettig, S.D.; Li, X.; Verrall, R.E. Thermodynamic and aggregation properties of gemini surfactants with ethoxylated spacers in aqueous solution. Langmuir 2003, 19, 3666–3670. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Wang, Y.; Yan, H.; Li, P.; Thomas, R.K. Effect of the Nature of the Spacer on the Aggregation Properties of Gemini Surfactants in an Aqueous Solution. Langmuir 2004, 20, 53–56. [Google Scholar] [CrossRef]
- Parikh, K.; Singh, S.; Desai, A.; Kumar, S. An interplay between spacer nature and alkyl chain length on aqueous micellar properties of cationic Gemini surfactants: A multi-technique approach. J. Mol. Liq. 2019, 278, 290–298. [Google Scholar] [CrossRef]
- Chaney, M.O.; Steinrauf, L.K. The crystal and molecular structure of tetragonal L -cystine. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 711–716. [Google Scholar] [CrossRef]
- Pisárčik, M.; Pupák, M.; Devínsky, F.; Almásy, L.; Tian, Q.; Bukovský, M. Urea-based gemini surfactants: Synthesis, aggregation behaviour and biological activity. Colloids Surfaces A Physicochem. Eng. Asp. 2016, 497, 385–396. [Google Scholar] [CrossRef]
- Kar, T.; Debnath, S.; Das, D.; Shome, A.; Das, P.K. Organogelation and hydrogelation of low-molecular-weight amphiphilic dipeptides: pH responsiveness in phase-selective gelation and dye removal. Langmuir 2009, 25, 8639–8648. [Google Scholar] [CrossRef] [PubMed]
- Mozrzymas, A. On the hydrophobic chains effect on critical micelle concentration of cationic gemini surfactants using molecular connectivity indices. Mon. Chem. 2020, 151, 525–531. [Google Scholar] [CrossRef]
- Hu, Z.; Zhu, H.; Wang, J.; Cao, D. Surface Activities of Three Anionic Gemini Surfactants Derived from Cyanuric Chloride: Effect of a Branched Hydrophobic Chain. J. Surfactants Deterg. 2016, 19, 487–492. [Google Scholar] [CrossRef]
- Rosen, M.J.; Kunjappu, J.T. Micelle Formation by Surfactants. In Surfactants and Interfacial Phenomena; John Wiley & Sons, Inc.: New York, NY, USA, 2012; pp. 123–201. [Google Scholar]
- Greber, K.E.; Dawgul, M.; Kamysz, W.; Sawicki, W. Cationic Net Charge and Counter Ion Type as Antimicrobial Activity Determinant Factors of Short Lipopeptides. Front. Microbiol. 2017, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Armas, F.; Pacor, S.; Ferrari, E.; Guida, F.; Pertinhez, T.A.; Romani, A.A.; Scocchi, M.; Benincasa, M. Design, antimicrobial activity and mechanism of action of Arg-rich ultra-short cationic lipopeptides. PLoS ONE 2019, 14, e0212447. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.A.; Pinazo, A.; Carilla, J.; Infante, M.R.; Alsina, M.A.; Haro, I.; Clapés, P. Interaction of Antimicrobial Arginine-Based Cationic Surfactants with Liposomes and Lipid Monolayers. Langmuir 2004, 20, 3379–3387. [Google Scholar] [CrossRef]
- Pérez, L.; Garcia, M.T.; Ribosa, I.; Vinardell, M.P.; Manresa, A.; Infante, M.R. Biological properties of arginine-based gemini cationic surfactants. Environ. Toxicol. Chem. 2002, 21, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Bartoszewska, S.; Kamysz, W.; Sikorska, E. Effect of self-assembly on antimicrobial activity of double-chain short cationic lipopeptides. Bioorg. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Brzozowski, K.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 2242–2251. [Google Scholar] [CrossRef]
- Paduszynska; Maciejewska; Neubauer; Golacki; Szymukowicz; Bauer; Kamysz Influence of Short Cationic Lipopeptides with Fatty Acids of Different Chain Lengths on Bacterial Biofilms Formed on Polystyrene and Hydrogel Surfaces. Pharmaceutics 2019, 11, 506. [CrossRef]
- Mitjans, M.; Martínez, V.; Clapés, P.; Pérez, L.; Infante, M.R.; Vinardell, M.P. Low Potential Ocular Irritation of Arginine-Based Gemini Surfactants and Their Mixtures with Nonionic and Zwitterionic Surfactants. Pharm. Res. 2003, 20, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.; Sánchez, L.; Busquets, M.A.; Infante, M.R.; Pilar Vinardell, M.; Mitjans, M. Disturbance of erythrocyte lipid bilayer by amino acid-based surfactants. Amino Acids 2007, 33, 459–462. [Google Scholar] [CrossRef]
- Manaargadoo-Catin, M.; Ali-Cherif, A.; Pougnas, J.L.; Perrin, C. Hemolysis by surfactants—A review. Adv. Colloid Interface Sci. 2016, 228, 1–16. [Google Scholar] [CrossRef]
- Tavano, L.; Infante, M.R.; Riya, M.A.; Pinazo, A.; Vinardell, M.P.; Mitjans, M.; Manresa, M.A.; Perez, L. Role of aggregate size in the hemolytic and antimicrobial activity of colloidal solutions based on single and gemini surfactants from arginine. Soft Matter 2013, 9, 306–319. [Google Scholar] [CrossRef]
- Cao, M.; Cao, C.; Zhou, P.; Wang, N.; Wang, D.; Wang, J.; Xia, D.; Xu, H. Self-assembly of amphiphilic peptides: Effects of the single-chain-to-gemini structural transition and the side chain groups. Colloids Surf. A Physicochem. Eng. Asp. 2015, 469, 263–270. [Google Scholar] [CrossRef]
- Briggs, D.B.; Jones, C.M.; Mashalidis, E.H.; Nuñez, M.; Hausrath, A.C.; Wysocki, V.H.; Tsao, T.S. Disulfide-dependent self-assembly of adiponectin octadecamers from trimers and presence of stable octadecameric adiponectin lacking disulfide bonds in vitro. Biochemistry 2009, 48, 12345–12357. [Google Scholar] [CrossRef][Green Version]
- Bej, R.; Dey, P.; Ghosh, S. Disulfide chemistry in responsive aggregation of amphiphilic systems. Soft Matter 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Maiti, B.; Kumar, K.; Moitra, P.; Kondaiah, P.; Bhattacharya, S. Reduction Responsive Nanovesicles Derived from Novel α-Tocopheryl-Lipoic Acid Conjugates for Efficacious Drug Delivery to Sensitive and Drug Resistant Cancer Cells. Bioconjug. Chem. 2018, 29, 255–266. [Google Scholar] [CrossRef]
- Oyinloye, B.E.; Adenowo, A.F.; Kappo, A.P. Reactive oxygen species, apoptosis, antimicrobial peptides and human inflammatory diseases. Pharmaceuticals 2015, 8, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Y.; Huang, J.L.; Ramirez, D.C.; Chignell, C.F. Role of reduced glutathione efflux in apoptosis of immortalized human keratinocytes induced by UVA. J. Biol. Chem. 2003, 278, 8058–8064. [Google Scholar] [CrossRef]
- Benavides, T.; Mitjans, M.; Martínez, V.; Clapés, P.; Infante, M.R.; Clothier, R.H.; Vinardell, M.P. Assessment of primary eye and skin irritants by in vitro cytotoxicity and phototoxicity models: An in vitro approach of new arginine-based surfactant-induced irritation. Toxicology 2004, 197, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.; Pinazo, A.; Pons, R.; Infante, M. Gemini surfactants from natural amino acids. Adv. Colloid Interface Sci. 2014, 205, 134–155. [Google Scholar] [CrossRef]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.P.; Black, D.S.; Walsh, W.R.; Kumar, N. A new era of antibiotics: The clinical potential of antimicrobial peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef] [PubMed]
- Effendy, I.; Maibach, H.I. Detergent and skin irritation. Clin. Dermatol. 1996, 14, 15–21. [Google Scholar] [CrossRef]
- Patel, V.M.; Schwartz, R.A.; Lambert, W.C. Topical antibiotics in pregnancy: A review of safety profiles. Dermatol. Ther. 2019, 32, e12951. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef] [PubMed]
- Bionda, N.; Fleeman, R.M.; De La Fuente-Núñez, C.; Rodriguez, M.C.; Reffuveille, F.; Shaw, L.N.; Pastar, I.; Davis, S.C.; Hancock, R.E.W.; Cudic, P. Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur. J. Med. Chem. 2016, 108, 354–363. [Google Scholar] [CrossRef]
- Pinazo, A.; Pons, R.; Marqués, A.; Farfan, M.; da Silva, A.; Perez, L. Biocompatible Catanionic Vesicles from Arginine-Based Surfactants: A New Strategy to Tune the Antimicrobial Activity and Cytotoxicity of Vesicular Systems. Pharmaceutics 2020, 12, 857. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.B.; Northeved, H.; Pramod Kumar, E.K.; Permin, A.; Gjetting, T.; Andresen, T.L.; Larsen, S.; Wegener, K.M.; Lykkesfeldt, J.; Jantzen, K.; et al. In vivo toxicity of cationic micelles and liposomes. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nie, Y.; Zhu, R.; Shi, S.; Luo, K.; He, B.; Yang, Y.; Yang, J.; Gu, Z. Preparation and gene delivery of alkaline amino acids-based cationic liposomes. Arch. Pharm. Res. 2008, 31, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Kamel, A.O.; Wettig, S.D. Interactions between DNA and Gemini surfactant: Impact on gene therapy: Part i. Nanomedicine 2016, 11, 289–306. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. Arginine-Rich Cationic Polypeptides Amplify Lipopolysaccharide-Induced Monocyte Activation. Infect. Immun. 2002, 70, 6904–6910. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maisetta, G.; Di Luca, M.; Esin, S.; Florio, W.; Brancatisano, F.L.; Bottai, D.; Campa, M.; Batoni, G. Evaluation of the inhibitory effects of human serum components on bactericidal activity of human beta defensin 3. Peptides 2008, 29, 1–6. [Google Scholar] [CrossRef]
- Lee, B.L.; Sachdeva, M.; Chambers, H.F. Effect of protein binding of daptomycin on MIC and antibacterial activity. Antimicrob. Agents Chemother. 1991, 35, 2505–2508. [Google Scholar] [CrossRef]
- Svenson, J.; Brandsdal, B.O.; Stensen, W.; Svendsen, J.S. Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J. Med. Chem. 2007, 50, 3334–3339. [Google Scholar] [CrossRef]
- Findlay, B.; Szelemej, P.; Zhanel, G.G.; Schweizer, F.; Beyermann, M. Guanidylation and Tail Effects in Cationic Antimicrobial Lipopeptoids. PLoS ONE 2012, 7, e41141. [Google Scholar] [CrossRef] [PubMed]
- Jaśkiewicz, M.; Neubauer, D.; Kamysz, W. Comparative Study on Antistaphylococcal Activity of Lipopeptides in Various Culture Media. Antibiotics 2017, 6, 15. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Richie, D.L.; Ghannoum, M.A.; Isham, N.; Thompson, K.V.; Ryder, N.S. Nonspecific effect of mycograb on amphotericin B MIC. Antimicrob. Agents Chemother. 2012, 56, 3963–3964. [Google Scholar] [CrossRef][Green Version]
- Ghobrial, O.; Derendorf, H.; Hillman, J.D. Human serum binding and its effect on the pharmacodynamics of the lantibiotic MU1140. Eur. J. Pharm. Sci. 2010, 41, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef]
- Bauer, J.; Siala, W.; Tulkens, P.M.; Van Bambeke, F. A combined pharmacodynamic quantitative and qualitative model reveals the potent activity of daptomycin and delafloxacin against Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2013, 57, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Paduszynska, M.A.; Greber, K.E.; Paduszynski, W.; Sawicki, W.; Kamysz, W. Activity of temporin a and short lipopeptides combined with gentamicin against biofilm formed by staphylococcus aureus and pseudomonas aeruginosa. Antibiotics 2020, 9, 566. [Google Scholar] [CrossRef]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef]
- Cruz, C.D.; Shah, S.; Tammela, P. Defining conditions for biofilm inhibition and eradication assays for Gram-positive clinical reference strains. BMC Microbiol. 2018, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, E.G.; Toma, L.; Provot, C.; Ascenzioni, F.; Sperduti, I.; Prignano, G.; Gallo, M.T.; Pimpinelli, F.; Bordignon, V.; Bernardi, T.; et al. Development of an in vitro Assay, Based on the BioFilm Ring Test®, for Rapid Profiling of Biofilm-Growing Bacteria. Front. Microbiol. 2016, 7, 1429. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and resistance of pseudomonas aeruginosabiofilms to antimicrobial agents-how P. aeruginosaCan escape antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef]
- Grosso-Becerra, M.V.; González-Valdez, A.; Granados-Martínez, M.J.; Morales, E.; Servín-González, L.; Méndez, J.L.; Delgado, G.; Morales-Espinosa, R.; Ponce-Soto, G.Y.; Cocotl-Yañez, M.; et al. Pseudomonas aeruginosa ATCC 9027 is a non-virulent strain suitable for mono-rhamnolipids production. Appl. Microbiol. Biotechnol. 2016, 100, 9995–10004. [Google Scholar] [CrossRef]
- Zhang, Y.; Miller, R.M. Enhanced octadecane dispersion and biodegradation by a Pseudomonas rhamnolipid surfactant (biosurfactant). Appl. Environ. Microbiol. 1992, 58, 3276–3282. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Bradbury, M.; Murphy, A.B. Draft genome sequence of Pseudomonas aeruginosa ATCC 9027 (DSM 1128), an important rhamnolipid surfactant producer and sterility testing strain. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA Chelates Cations and Induces Antibiotic Resistance in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef]
- Billings, N.; Ramirez Millan, M.; Caldara, M.; Rusconi, R.; Tarasova, Y.; Stocker, R.; Ribbeck, K. The Extracellular Matrix Component Psl Provides Fast-Acting Antibiotic Defense in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2013, 9, e1003526. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Conover, M.; Lu, H.; Parsek, M.R.; Bayles, K.; Wozniak, D.J. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009, 5, 1000354. [Google Scholar] [CrossRef] [PubMed]
- Swedan, S.; Shubair, Z.; Almaaytah, A. Synergism of cationic antimicrobial peptide WLBU2 with antibacterial agents against biofilms of multi-drug resistant Acinetobacter baumannii and Klebsiella pneumoniae. Infect. Drug Resist. 2019, 12, 2019–2030. [Google Scholar] [CrossRef]
- Prax, M.; Bertram, R. Metabolic aspects of bacterial persisters. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Michiels, J.E.; Van Den Bergh, B.; Verstraeten, N.; Fauvart, M.; Michiels, J. In vitro emergence of high persistence upon periodic aminoglycoside challenge in the ESKAPE pathogens. Antimicrob. Agents Chemother. 2016, 60, 4630–4637. [Google Scholar] [CrossRef]
- Cavalheiro, M.; Teixeira, M.C. Candida Biofilms: Threats, challenges, and promising strategies. Front. Med. 2018, 5, 28. [Google Scholar] [CrossRef]
- Mitchell, K.F.; Taff, H.T.; Cuevas, M.A.; Reinicke, E.L.; Sanchez, H.; Andes, D.R. Role of matrix β-1,3 glucan in antifungal resistance of non-albicans Candida biofilms. Antimicrob. Agents Chemother. 2013, 57, 1918–1920. [Google Scholar] [CrossRef]
- Martins, M.; Uppuluri, P.; Thomas, D.P.; Cleary, I.A.; Henriques, M.; Lopez-Ribot, J.L.; Oliveira, R. Presence of extracellular DNA in the candida albicans biofilm matrix and its contribution to biofilms. Mycopathologia 2010, 169, 323–331. [Google Scholar] [CrossRef]
- Al-Fattani, M.A.; Douglas, L.J. Biofilm matrix of Candida albicans and Candida tropicalis: Chemical composition and role in drug resistance. J. Med. Microbiol. 2006, 55, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Mathé, L.; Van Dijck, P. Recent insights into Candida albicans biofilm resistance mechanisms. Curr. Genet. 2013, 59, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Parahitiyawa, N.B.; Samaranayake, Y.H.; Samaranayake, L.P.; Ye, J.; Tsang, P.W.K.; Cheung, B.P.K.; Yau, J.Y.Y.; Yeung, S.K.W. Interspecies variation in Candida biofilm formation studied using the Calgary biofilm device. APMIS 2006, 114, 298–306. [Google Scholar] [CrossRef]
- Neubauer, D.; Jaśkiewicz, M.; Sikorska, E.; Bartoszewska, S.; Bauer, M.; Kapusta, M.; Narajczyk, M.; Kamysz, W. Effect of Disulfide Cyclization of Ultrashort Cationic Lipopeptides on Antimicrobial Activity and Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7208. [Google Scholar] [CrossRef]
- Nugent, K.M.; Couchot, K.R.; Gray, L.D. Effect of Candida morphology on amphotericin B susceptibility. Antimicrob. Agents Chemother. 1987, 31, 335–336. [Google Scholar] [CrossRef]
- Suci, P.A.; Tyler, B.J. Action of chlorhexidine digluconate against yeast and filamentous forms in an early-stage Candida albicans biofilm. Antimicrob. Agents Chemother. 2002, 46, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.A. UV Atlas of Organic Compounds; Verlag Chemie: London, UK, 1971. [Google Scholar]
- Sala, A.; Cabassi, C.S.; Santospirito, D.; Polverini, E.; Flisi, S.; Cavirani, S.; Taddei, S. Novel Naja atra cardiotoxin 1 (CTX-1) derived antimicrobial peptides with broad spectrum activity. PLoS ONE 2018, 13, e0190778. [Google Scholar] [CrossRef] [PubMed]
- Ilić, N.; Novković, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Otte, A.; Xiang, M.; Liu, D.; Pinal, R. Investigation of Film with β-Galactosidase Designed for Stabilization and Handling in Dry Configuration. Molecules 2015, 20, 17180–17193. [Google Scholar] [CrossRef]
- Liu, B.; Huang, H.; Yang, Z.; Liu, B.; Gou, S.; Zhong, C.; Han, X.; Zhang, Y.; Ni, J.; Wang, R. Design of novel antimicrobial peptide dimer analogues with enhanced antimicrobial activity in vitro and in vivo by intermolecular triazole bridge strategy. Peptides 2017, 88, 115–125. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, C.; Li, T.; Zhang, M.; Liu, Y.; Gauthier, M.A.; Zhao, Y.; Wu, C. The Interplay of Disulfide Bonds, α-Helicity, and Hydrophobic Interactions Leads to Ultrahigh Proteolytic Stability of Peptides. Biomacromolecules 2015, 16, 2347–2355. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.J.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef]
- Kowalczyk, R.; Harris, P.W.R.; Williams, G.M.; Yang, S.H.; Brimble, M.A. Peptide lipidation—A synthetic strategy to afford peptide based therapeutics. Adv. Exp. Med. Biol. 2017, 1030, 185–227. [Google Scholar]
- Zhang, L.; Bulaj, G. Converting Peptides into Drug Leads by Lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [Google Scholar] [CrossRef] [PubMed]
- Wanjari, P.P.; Sangwai, A.V.; Ashbaugh, H.S. Confinement induced conformational changes in n-alkanes sequestered within a narrow carbon nanotube. Phys. Chem. Chem. Phys. 2012, 14, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Ding, L.; Liu, J.; Zhao, B.; Deng, Q.; Yan, T. Synthesis and physiochemical properties of novel gemini surfactants with phenyl-1,4-bis(carbamoylmethyl) spacer. RSC Adv. 2015, 5, 74764–74773. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Y. Aggregation behavior of gemini surfactants and their interaction with macromolecules in aqueous solution. Phys. Chem. Chem. Phys. 2011, 13, 1939–1956. [Google Scholar] [CrossRef]
- Pisárčik, M.; Pupák, M.; Lukáč, M.; Devínsky, F.; Hubčík, L.; Bukovský, M.; Horváth, B. The synthesis, self-assembled structures, and microbicidal activity of cationic gemini surfactants with branched tridecyl chains. Molecules 2019, 24, 4380. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Grossfield, A. Thermodynamics of Micelle Formation and Membrane Fusion Modulate Antimicrobial Lipopeptide Activity. Biophys. J. 2015, 109, 750–759. [Google Scholar] [CrossRef]
- Neue, U.D.; Niederlaender, C.L.; Petersen, J.S. Liquid Chromatography Stationary Phases with Reduced Silanol Interactions. U.S. Patent US5374755A, 20 December 1994. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standards-Second Edition, in CLSI document M27-2A 2002; CLSI: Wayne, PA, USA, 2002; Volume 22, ISBN 1562384694. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests f or Bacteria That Grow Aerobically; Approved Standard, 9th ed.; CLSI: Wayne, PA, USA, 2012; Volume 32, ISBN 1562387839. [Google Scholar]
- Avrahami, D.; Shai, Y. A New Group of Antifungal and Antibacterial Lipopeptides Derived from Non-membrane Active Peptides Conjugated to Palmitic Acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.N.; Wilson, H.W.; Novick, W.J.; Barry, A.L.; Thornsberry, C. In vitro evaluation of CENTA, a new beta-lactamase-susceptible chromogenic cephalosporin reagent. J. Clin. Microbiol. 1982, 15, 954–958. [Google Scholar] [CrossRef]
- Abbassi, F.; Lequin, O.; Piesse, C.; Goasdoué, N.; Foulon, T.; Nicolas, P.; Ladram, A. Temporin-SHf, a new type of phe-rich and hydrophobic ultrashort antimicrobial peptide. J. Biol. Chem. 2010, 285, 16880–16892. [Google Scholar] [CrossRef]
- Arcidiacono, S.; Soares, J.W.; Meehan, A.M.; Marek, P.; Kirby, R. Membrane permeability and antimicrobial kinetics of cecropin P1 against Escherichia coli. J. Pept. Sci. 2009, 15, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.A.; Baroni, M.C.; Taddei, S.; Ghidini, F.; Sansoni, P.; Cavirani, S.; Cabassi, C.S. In vitro activity of novel in silico -developed antimicrobial peptides against a panel of bacterial pathogens. J. Pept. Sci. 2013, 19, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Aspmo, S.I. Serum stability of peptides. Methods Mol. Biol. 2008, 494, 177–186. [Google Scholar] [PubMed]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016; Available online: https://ambermd.org/doc12/Amber16.pdf (accessed on 15 March 2021).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Periole, X.; Marrink, S.-J. The Martini Coarse-Grained Force Field. Biomol. Simul. 2013, 533–565. [Google Scholar]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Winger, M.; Trzesniak, D.; Baron, R.; Van Gunsteren, W.F. On using a too large integration time step in molecular dynamics simulations of coarse-grained molecular models. Phys. Chem. Chem. Phys. 2009, 11, 1–8. [Google Scholar] [CrossRef][Green Version]
- Gapsys, V.; de Groot, B.L.; Briones, R. Computational analysis of local membrane properties. J. Comput.-Aided Mol. Des. 2013, 27, 845–858. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Lipopeptide Fragment | Spacer | Adjusted Retention Time (min) | CAC mM (μg/mL) | Surface Tension at CAC (mN/m) |
|---|---|---|---|---|---|
| A1a | C8-RR-NH2 | - | 2.93 | nd | nd |
| A2a | C10-RR-NH2 | - | 7.74 | nd | nd |
| A3 | C12-RR-NH2 | - | 12.15 | 7.48 (3827.3) | 38 |
| A4 | C14-RR-NH2 | - | 16.31 | 1.67 (899.7) | 35 |
| B1a | C8-RR- | l-Lysine amide | 8.11 | nd | nd |
| B2 | C10-RR- | l-Lysine amide | 13.73 | 3.69 (3980.0) | 36 |
| B3 | C12-RR- | l-Lysine amide | 19.09 | 0.87 (987.3) | 41 |
| B4 | C14-RR- | l-Lysine amide | 25.33 | 0.31 (365.7) | 40 |
| B5 | C8-FRR- | l-Lysine amide | 15.19 | 2.47 (3247.9) | 39 |
| B6 | C8-NleRR- | l-Lysine amide | 13.96 | 3.59 (4482.4) | 37 |
| B7b | C8(4)-RR- | l-Lysine amide | 14.68 | nd | nd |
| C1a | C8-RR- | l-Cystine diamide | 8.26 (0.15) c | nd | nd |
| C2 | C10-RR- | l-Cystine diamide | 13.94 (0.21) c | 3.30 (3832.7) | 43 |
| C3 | C12-RR- | l-Cystine diamide | 19.24 (0.15) c | 0.25 (305.4) | 43 |
| C4 | C14-RR- | l-Cystine diamide | 25.30 (−0.03) c | 0.06 (71.1) | 47 |
| C5 | C8-FRR- | l-Cystine diamide | 15.42 (0.23) c | 2.58 (3632.7) d | 37 |
| C6 | C8-NleRR- | l-Cystine diamide | 14.29 (0.33) c | 4.28 (5737.7) | 35 |
| C7 | C8(4)-RR- | l-Cystine diamide | 14.83 (0.15) c | 6.21 (7628.1) | 35 |
| Compound | HC50 | IC50 | Enterococcus faecium ATCC 700221 | Staphylococcus aureus ATCC 33591 | Staphylococcus aureus ATCC 25923 | Klebsiella pneumoniae ATCC 700603 | Acinetobacter baumannii ATCC BAA 1605 | Pseudomonas aeruginosa ATCC 9027 | Klebsiella aerogenes ATCC 13048 | C. albicans ATCC 10231 | C. glabrata ATCC 15126 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | >256 * | >500 | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) |
| A2 | >256 * | >500 | 256 (−/−) | 256 (−/−) | 256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) | >256 (−/−) |
| A3 | >256 * | 153.50 ± 8.13 | 32 (>8/4.80) | 32 (>8/4.80) | 32 (>8/4.80) | >256 (−/−) | 256 (−/−) | 128 (>2/1.20) | >256 (−/−) | 64 (>4/2.40) | 128 (>2/1.20) |
| A4 | 68.43 ± 1.41 * | 46.48 ± 3.65 | 8 (6.02/5.81) | 8 (6.02/5.81) | 8 (6.02/5.81) | 256 (−/−) | 64 (0.75/0.73) | 32 (1.50/1.45) | 256 (−/−) | 8 (6.02/5.81) | 16 (3.01/2.91) |
| B1 | >256 | 378.40 ± 93.71 | 64 (>4/5.91) | 8 (>32/47.30) | 4 (>64/94.60) | >256 (−/−) | 256 (−/−) | 32 (>8/11.83) | >256 (−/−) | 64 (>4/5.91) | 64 (>4/5.91) |
| B2 | >256 | 150.03 ± 3.15 | 2 (>128/75.02) | 1 (>256/150.03) | 1 (>256/150.03) | 128 (>2/1.17) | 8 (>32/18.75) | 8 (>32/18.75) | 128 (>2/1.17) | 8 (>32/18.75) | 4 (>64/37.51) |
| B3 | 56.29 ± 6.50 | 26.42 ± 4.24 | 2 (28.15/13.21) | 2 (28.15/13.21) | 4 (14.07/6.61) | 64 (0.88/0.41) | 8 (7.04/3.30) | 4 (14.07/6.61) | 32 (1.76/0.83) | 2 (28.15/13.21) | 2 (28.15/13.21) |
| B4 | 44.12 ± 4.70 | 32.39 ± 4.49 | 8 (5.52/4.05) | 64 (0.69/0.51) | 64 (0.69/0.51) | 32 (1.38/1.01) | 64 (0.69/0.51) | 32 (1.38/1.01) | 32 (1.38/1.01) | 4 (11.03/8.10) | 2 (22.06/8.10) |
| B5 | 181.38 ± 8.99 | 169.13 ± 32.80 | 4 (45.35/42.28) | 4 (45.35/42.28) | 2 (90.69/84.57) | 128 (1.42/1.32) | 32 (5.69/5.29) | 8 (22.67/21.14) | 256 (−/−) | 16 (11.34/10.57) | 16 (11.34/10.57) |
| B6 | >256 | 164.17 ± 14.65 | 2 (>128/82.09) | 4 (>64/41.04) | 2 (>128/82.09) | 64 (>4/2.57) | 32 (>8/5.13) | 4 (>64/41.04) | 256 (−/−) | 16 (>16/10.26) | 16 (>16/10.26) |
| B7 | >256 | 78.78 ± 6.93 | 1 (>256/78.78) | 0.5 (>512/157.56) | 1 (>256/78.78) | 32 (>8/2.46) | 32 (>8/2.46) | 4 (>64/19.70) | 16 (>16/4.92) | 8 (>32/9.85) | 16 (>16/4.92) |
| C1 | >256 | >500 | 32 (>8/>15.63) | 16 (>16/>31.25) | 8 (>32/>62.50) | >256 (−/−) | 256 (−/−) | 64 (>4/>7.81) | >256 (−/−) | 128 (>2/>3.91) | 64 (>4/>7.81) |
| C2 | >256 | 231.17 ± 24.37 | 2 (>128/115.59) | 1 (>256/231.17) | 2 (>128/115.59) | >256 (−/−) | 16 (>16/14.45) | 8 (>32/28.90) | 128 (>2/1.81) | 4 (>64/57.79) | 8 (>32/28.90) |
| C3 | 36.24 ± 2.35 | 119.47 ± 6.03 | 2 (18.12/59.74) | 4 (9.06/29.87) | 4 (9.06/29.87) | >256 (−/−) | 64 (0.57/1.87) | 8 (4.53/14.93) | 32 (1.13/3.73) | 1 (36.24/119.47) | 2 (18.12/59.74) |
| C4 | 20.89 ± 1.06 | 54.52 ± 15.40 | 8 (2.61/6.82) | 32 (0.65/1.70) | 64 (0.33/0.85) | >256 (−/−) | 32 (0.65/1.70) | 64 (0.33/0.85) | 128 (0.16/0.43) | 2 (10.45/27.26) | 1 (20.89/54.52) |
| C5 | 117.06 ± 4.60 | 146.63 ± 14.95 | 2 (58.53/73.32) | 2 (58.53/73.32) | 2 (58.53/73.32) | 32 (3.66/4.58) | 32 (3.66/4.58) | 8 (14.63/18.33) | 128 (0.91/1.15) | 8 (14.63/18.33) | 8 (14.63/18.33) |
| C6 | >256 | 248.83 ± 18.81 | 2 (>128/124.42) | 2 (>128/124.42) | 2 (>128/124.42) | 64 (>4/3.89) | 32 (>8/7.78) | 8 (>32/31.10) | 256 (−/−) | 16 (>16/15.55) | 16 (>16/15.55) |
| C7 | >256 | 163.98 ± 13.66 | 2 (>128/81.99) | 2 (>128/81.99) | 2 (>128/81.99) | 32 (>8/5.12) | 32 (>8/5.12) | 4 (>64/41.00) | 16 (>16/10.25) | 16 (>16/10.25) | 16 (>16/10.25) |
| Code | cLogP (Dimer) | cLogP (Corresponding Monomer) |
|---|---|---|
| C1 | −0.05 ± 0.92 | −0.43 ± 0.75 |
| C2 | 2.07 ± 0.92 | 0.64 ± 0.75 |
| C3 | 4.20 ± 0.92 | 1.70 ± 0.75 |
| C4 | 6.32 ± 0.92 | 2.76 ± 0.75 |
| C5 | 3.09 ± 0.97 | 1.15 ± 0.85 |
| C6 | 2.60 ± 0.96 | 0.90 ± 0.82 |
| C7 | 3.83 ± 0.93 | 1.52 ± 0.75 |
| MIC (µg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | S. aureus ATCC 33591 | P. aeruginosa ATCC 9027 | C. glabrata ATCC 15126 | ||||||
| Serum Concentration % (v/v) | 0% | 1% | 10% | 0% | 1% | 10% | 0% | 1% | 10% |
| A1 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >256 |
| A2 | 256 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >256 |
| A3 | 32 | 64 | 128 | 128 | 256 | >256 | 128 | 256 | >256 |
| A4 | 8 | 16 | 64 | 32 | 128 | 256 | 16 | 64 | 256 |
| B1 | 8 | 32 | >256 | 32 | >256 | >256 | 64 | >256 | >256 |
| B2 | 1 | 32 | 64 | 8 | 64 | 256 | 4 | 64 | >256 |
| B3 | 2 | 8 | 16 | 4 | 32 | 64 | 2 | 16 | >256 |
| B4 | 64 | 16 | 64 | 32 | 128 | >256 | 2 | 32 | >256 |
| B5 | 4 | 64 | >256 | 8 | 128 | >256 | 16 | 64 | 256 |
| B6 | 4 | 128 | >256 | 4 | 256 | >256 | 16 | 128 | >256 |
| B7 | 0.5 | 4 | 8 | 4 | 64 | 128 | 16 | 32 | 64 |
| C1 | 16 | >256 | >256 | 64 | >256 | >256 | 64 | >256 | >256 |
| C2 | 1 | 64 | >256 | 8 | 128 | >256 | 8 | 128 | >256 |
| C3 | 4 | 32 | 32 | 8 | 64 | 256 | 2 | 32 | >256 |
| C4 | 32 | 16 | 64 | 64 | 128 | 256 | 1 | 32 | >256 |
| C5 | 2 | 64 | 256 | 8 | 128 | >256 | 8 | 128 | >256 |
| C6 | 2 | 128 | >256 | 8 | 256 | >256 | 16 | 128 | >256 |
| C7 | 2 | 32 | 64 | 4 | 128 | 256 | 16 | 64 | 256 |
| Compound | Enterococcus faecium ATCC 700221 | Staphylococcus aureus ATCC 33591 | Staphylococcus aureus ATCC 25923 | Klebsiella pneumoniae ATCC 700603 | Acinetobacter baumannii ATCC BAA 1605 | Pseudomonas aeruginosa ATCC 9027 | Klebsiella aerogenes ATCC 13048 | Candida albicans ATCC 10231 | Candida glabrata ATCC 15126 |
|---|---|---|---|---|---|---|---|---|---|
| A1 | >512 | >512 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| A2 | 512 | 512 | 512 | >512 | >512 | >512 | >512 | 512 | >512 |
| A3 | 32 | 64 | 64 | 512 | 512 | 512 | 512 | 128 | 512 |
| A4 | 8 | 8 | 16 | 256 | 256 | 512 | 256 | 64 | 256 |
| B1 | 128 | 32 | 32 | >512 | 512 | 512 | >512 | 256 | >512 |
| B2 | 4 | 4 | 8 | 256 | 128 | 512 | 256 | 32 | 512 |
| B3 | 16 | 16 | 64 | 128 | 256 | 512 | 128 | 32 | 64 |
| B4 | 64 | 128 | 256 | 512 | 512 | >512 | 256 | 64 | 64 |
| B5 | 8 | 4 | 16 | 256 | 256 | 512 | 256 | 32 | 256 |
| B6 | 8 | 4 | 16 | 256 | 256 | 256 | 256 | 256 | 512 |
| B7 | 8 | 4 | 8 | 128 | 64 | 256 | 64 | 64 | 256 |
| C1 | 64 | 32 | 64 | >512 | 512 | 512 | >512 | 256 | >512 |
| C2 | 4 | 8 | 32 | 512 | 256 | 512 | 256 | 32 | 512 |
| C3 | 16 | 32 | 128 | 256 | 512 | 512 | 256 | 16 | 128 |
| C4 | 64 | 128 | 512 | 512 | 512 | >512 | 512 | 16 | 64 |
| C5 | 8 | 8 | 64 | 256 | 512 | 512 | 256 | 128 | 256 |
| C6 | 8 | 8 | 32 | 256 | 512 | 512 | 256 | 256 | 512 |
| C7 | 4 | 8 | 32 | 256 | 256 | 512 | 128 | 64 | 128 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neubauer, D.; Jaśkiewicz, M.; Bauer, M.; Olejniczak-Kęder, A.; Sikorska, E.; Sikora, K.; Kamysz, W. Biological and Physico-Chemical Characteristics of Arginine-Rich Peptide Gemini Surfactants with Lysine and Cystine Spacers. Int. J. Mol. Sci. 2021, 22, 3299. https://doi.org/10.3390/ijms22073299
Neubauer D, Jaśkiewicz M, Bauer M, Olejniczak-Kęder A, Sikorska E, Sikora K, Kamysz W. Biological and Physico-Chemical Characteristics of Arginine-Rich Peptide Gemini Surfactants with Lysine and Cystine Spacers. International Journal of Molecular Sciences. 2021; 22(7):3299. https://doi.org/10.3390/ijms22073299
Chicago/Turabian StyleNeubauer, Damian, Maciej Jaśkiewicz, Marta Bauer, Agata Olejniczak-Kęder, Emilia Sikorska, Karol Sikora, and Wojciech Kamysz. 2021. "Biological and Physico-Chemical Characteristics of Arginine-Rich Peptide Gemini Surfactants with Lysine and Cystine Spacers" International Journal of Molecular Sciences 22, no. 7: 3299. https://doi.org/10.3390/ijms22073299
APA StyleNeubauer, D., Jaśkiewicz, M., Bauer, M., Olejniczak-Kęder, A., Sikorska, E., Sikora, K., & Kamysz, W. (2021). Biological and Physico-Chemical Characteristics of Arginine-Rich Peptide Gemini Surfactants with Lysine and Cystine Spacers. International Journal of Molecular Sciences, 22(7), 3299. https://doi.org/10.3390/ijms22073299