
Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
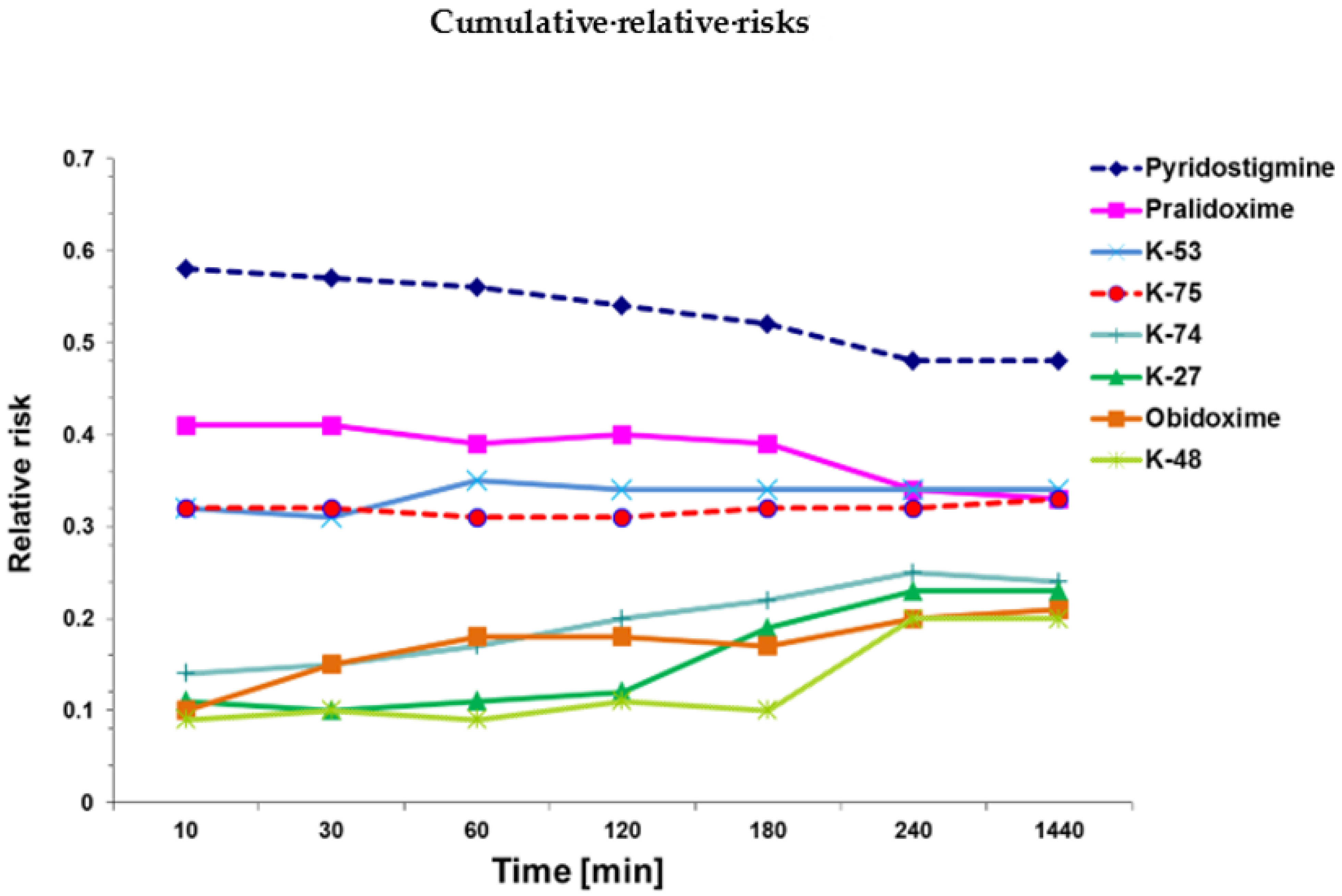
2.1. Mortality Rates
2.2. Cox Survival Analysis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Animals
4.2.1. Choice of Dosage for Pretreatment
- Reference group: only azinphos-methyl exposure.
- Pyridostigmine: 1 µmol/rat = 0.26 mg/rat (= 1.0 mg/kg average body weight).
- Pralidoxime: 30 µmol/rat = 5.2 mg/rat (= 20 mg/kg average body weight).
- Obidoxime: 25 µmol/rat = 9.0 mg/rat (=35 mg/kg average body weight).
- K-27: 60 µmol/rat = 26.8 mg/rat (=103 mg/kg average body weight).
- K-48: 25 µmol/rat = 11.5 mg/rat (=44 mg/kg average body weight).
- K-53: 3 µmol/rat = 1.37 mg/rat (=5.3 mg/kg average body weight).
- K-74: 3 µmol/rat = 1.38 mg/rat (=5.3 mg/kg average body weight).
- K-75: 3 µmol/rat = 1.37 mg/rat (=5.3 mg/kg average body weight).
4.2.2. Pretreatment and Azinphos-Methyl Exposure

{kind=link}
| Molecular Weight | Injected Dose (µmol/rat) | Injected Dose (mg/rat) | Injected Dose (mg/kg Average Body Weight) | IC50 Human (µM] | IC50 Rat (µM) | LD50/LD01 (µmol/rat) | |
|---|---|---|---|---|---|---|---|
| Azinphos-methyl | 317.3 | 5, 10, 15 | 1.59, 3.18, 4.77 | 6.14, 12.28, 18.42 | 189 * | NA | 3.2/0.4 * |
| Pyridostigmine | 172.60 | 30 | 0.26 | 1.0 | 0.33 | NA | 7.2/3.7 |
| Pralidoxime | 172.60 | 30 | 5.2 | 20 | 592 | 412 | 180/117 |
| Obidoxime | 359.21 | 25 | 9.0 | 35 | 702 | 193 | 132/107 |
| K-27 | 446.16 | 60 | 26.8 | 103 | 414 | 1054 | 350/250 |
| K-45 | 460.16 | 25 | 11.5 | 44 | 461 | 643 | 140/110 |
| K-53 | 458.15 | 3 | 1.37 | 5.3 | 115 | 83 | 21/13 |
| K-74 | 460.16 | 3 | 1.38 | 5.3 | 103 | 66 | 28/13 |
| K-75 | 458.15 | 3 | 1.37 | 5.3 | 63 | 101 | 51/13 |
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Casida, J.E.; Quistad, G.B. Golden Age of Insecticide Research: Past, Present, or Future? Annu. Rev. Èntomol. 1998, 43, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Young, R.A.; Watson, A. Organophosphate nerve agents. In Handbook of Toxicology of Chemical Warfare Agents, 3rd ed.; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2020; Chapter 8; pp. 97–126. [Google Scholar] [CrossRef]
- Dworkin, J.; Prescott, M.; Jamal, R.; Hardawan, S.A.; Abdullah, A.; Galea, S. The Long-Term Psychosocial Impact of a Surprise Chemical Weapons Attack on Civilians in Halabja, Iraqi Kurdistan. J. Nerv. Ment. Dis. 2008, 196, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Macilwain, C. Study proves Iraq used nerve gas. Nature 1993, 363, 3. [Google Scholar] [CrossRef] [PubMed]
- Riley, B. The toxicology and treatment of injuries from chemical warfare agents. Curr. Anaesth. Crit. Care 2003, 14, 149–154. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Balali-Mood, K. Neurotoxic disorders of organophosphorus compounds and their managements. Arch. Iran. Med. 2008, 11, 65–89. [Google Scholar]
- Delfino, R.T.; Ribeiro, T.S.; Figueroa-Villar, J.D. Organophosphorus compounds as chemical warfare agents: A review. J. Braz. Chem. Soc. 2009, 20, 407–428. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Morita, H.; Nakajima, T. Sarin experiences in Japan: Acute toxicity and long-term effects. J. Neurol. Sci. 2006, 249, 76–85. [Google Scholar] [CrossRef]
- Kostadinov, R.; Kanev, K.; Dimov, D. Chemical Terrorism, History and Threat Assessment. Med. Manag. Chem. Biol. Casualties 2010, 8, 77–84. [Google Scholar]
- Brooks, J.; Erickson, T.B.; Kayden, S.; Ruiz, R.; Wilkinson, S.; Burklejr, F.M., Jr. Responding to chemical weapons violations in Syria: Legal, health, and humanitarian recommendations. Confl. Health 2018, 12, 12. [Google Scholar] [CrossRef]
- Chowdhary, S.; Bhattacharyya, R.; Banerjee, D. Acute organophosphorus poisoning. Clin. Chim. Acta 2014, 431, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Petroianu, G.; Toomes, L.M.; Petroianu, A.; Bergler, W.; Rüfer, R. Control of blood pressure, heart rate and haematocrit during high-dose intravenous paraoxon exposure in mini pigs. J. Appl. Toxicol. 1998, 18, 293–298. [Google Scholar] [CrossRef]
- Eddleston, M.; Buckley, N.A.; Eyer, P.; Dawson, A.H. Management of acute organophosphorus pesticide poisoning. Lancet 2008, 371, 597–607. [Google Scholar] [CrossRef]
- Masson, P.; Nachon, F. Cholinesterase reactivators and bioscavengers for pre- and post-exposure treatments of organophosphorus poisoning. J. Neurochem. 2017, 142 (Suppl. 2), 26–40. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Petroianu, G.A. Reversible cholinesterase inhibitors as pretreatment for exposure to organophosphates. A review. J. Appl. Toxicol. 2019, 39, 101–116. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Lorke, D.E. Reversible cholinesterase inhibitors as pre-treatment for exposure to organophosphates: Assessment using azinphos-methyl. J. Appl. Toxicol. 2015, 35, 493–499. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Optimal Pre-treatment for Acute Exposure to the Organophosphate Dicrotophos. Curr. Pharm. Des. 2017, 23, 3432–3439. [Google Scholar] [CrossRef]
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Shafiullah, M.; Kuca, K.; Petroianu, G.A. Pretreatment for acute exposure to diisopropylfluorophosphate: In vivo efficacy of various acetylcholinesterase inhibitors. J. Appl. Toxicol. 2011, 31, 515–523. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Combined Pre- and Posttreatment of Paraoxon Exposure. Molecules 2020, 25, 1521. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Nurulain, S.M.; Shafiullah, M.; Hasan, M.Y.; Kuča, K.; Lorke, D.E. Usefulness of administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: Assessment using paraoxon. J. Appl. Toxicol. 2013, 33, 894–900. [Google Scholar] [CrossRef]
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Shafiullah, M.; Kuča, K.; Petroianu, G.A. Acetylcholinesterase inhibitors as pretreatment before acute exposure to organophosphates: Assessment using methyl-paraoxon. CNS Neurol. Disord. Drug Targets 2012, 11, 1052–1060. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Prophylactic administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: Assessment using terbufos sulfone. J. Appl. Toxicol. 2014, 34, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration FDA. Approves Pyridostigmine Bromide as Pretreatment against Nerve Gas. Available online: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm130342.htm (accessed on 15 February 2021).
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Sheen, R.; Kuča, K.; Petroianu, G.A. Entry of two new asymmetric bispyridinium oximes (K-27 and K-48) into the rat brain: Comparison with obidoxime. J. Appl. Toxicol. 2007, 27, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Kalasz, H.; Petroianu, G.A.; Tekes, K. Entry of Oximes into the Brain: A Review. Curr. Med. Chem. 2008, 15, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Petroianu, G.A. The Experimental Oxime K027—A Promising Protector from Organophosphate Pesticide Poisoning. A Review Comparing K027, K048, Pralidoxime, and Obidoxime. Front. Neurosci. 2019, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Cabal, J. In vitro reactivation of tabun-inhibited acetylcholinesterase using new oximes—K027, K005, K033 and K048. Central Eur. J. Public Health 2004, 12, S59–S61. [Google Scholar]
- Kuca, K.; Cabal, J.; Kassa, J. A Comparison of the Potency of Newly Developed Oximes (K005, K027, K033, K048) and Currently Used Oximes (Pralidoxime, Obidoxime, HI-6) to Reactivate Sarin-Inhibited Rat Brain Acetylcholinesterase by In Vitro Methods. J. Toxicol. Environ. Health Part A 2005, 68, 677–686. [Google Scholar] [CrossRef]
- Kuca, K.; Kassa, J. In vitro reactivation of acetylcholinesterase using the oxime K027. Vet. Hum. Toxicol. 2004, 46, 15–18. [Google Scholar]
- Kuca, K.; Musilek, K.; Jun, D.; Pohanka, M.; Ghosh, K.K.; Hrabinova, M. Oxime K027: Novel low-toxic candidate for the universal reactivator of nerve agent- and pesticide-inhibited acetylcholinesterase. J. Enzym. Inhib. Med. Chem. 2010, 25, 509–512. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Musilek, K.; Petroianu, G.A. Eight new bispyridinium oximes in comparison with the conventional oximes pralidoxime and obidoxime:in vivoefficacy to protect from diisopropylfluorophosphate toxicity. J. Appl. Toxicol. 2008, 28, 920–928. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Lorke, D.E.; Kalász, H. Comparison of the Ability of Pyridinium Aldoximes to Reactivate Human Red Blood Cell Acetylcholinesterases Inhibited by ethyl- and methyl-paraoxon. Curr. Org. Chem. 2012, 16, 1359–1369. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Lorke, D.E. Pyridinium oxime reactivators of cholinesterase inhibited by diisopropyl-fluorophosphate (DFP): Predictive value of in-vitro testing for in-vivo efficacy. Mini Rev. Med. Chem. 2008, 8, 1328–1342. [Google Scholar] [CrossRef]
- Kayouka, M.; Houzé, P.; Lejay, M.; Baud, F.J.; Kuca, K. Safety and Efficacy of New Oximes to Reverse Low Dose Diethyl-Paraoxon-Induced Ventilatory Effects in Rats. Molecules 2020, 25, 3056. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Petroianu, G.A. Treatment of Organophosphate Poisoning with Experimental Oximes: A Review. Curr. Org. Chem. 2019, 23, 628–639. [Google Scholar] [CrossRef]
- Kassa, J.; Kuca, K.; Cabal, J.; Paar, M. A Comparison of the Efficacy of New Asymmetric Bispyridinium Oximes (K027, K048) with Currently Available Oximes against Tabun by In Vivo Methods. J. Toxicol. Environ. Health Part A 2006, 69, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Bielavsky, J.; Cabal, J.; Bielavska, M. Synthesis of a potential reactivator of acetylcholinesterase—1-(4-hydroxyiminomethylpyridinium)-3-(carbamoylpyridinium)propane dibromide. Tetrahedron Lett. 2003, 44, 3123–3125. [Google Scholar] [CrossRef]
- Kuca, K.; Musilova, L.; Paleček, J.; Církva, V.; Paar, M.; Musilek, K.; Hrabinová, M.; Pohanka, M.; Karasová, J.Ž.; Jun, D. Novel Bisquaternary Oximes—Reactivation of Acetylcholinesterase and Butyrylcholinesterase Inhibited by Paraoxon. Molecules 2009, 14, 4915–4921. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Kuca, K.; Dohnal, V.; Jun, D.; Marek, J.; Koleckar, V. Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator. Molecules 2007, 12, 1755–1761. [Google Scholar] [CrossRef]
- Lorke, D.E.; Hasan, M.Y.; Arafat, K.; Kuča, K.; Musilek, K.; Schmitt, A.; Petroianu, G.A. In vitro oxime protection of human red blood cell acetylcholinesterase inhibited by diisopropyl-fluorophosphate. J. Appl. Toxicol. 2008, 28, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Oximes as pretreatment before acute exposure to paraoxon. J. Appl. Toxicol. 2019, 39, 1506–1515. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life tables. J. R. Stat. Soc. B 1972, 34, 189–220. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Five Experimental Bispyridinium Oximes in Comparison with the Conventional Oximes Pralidoxime and Obidoxime: In Vivo Efficacy to Protect from Azinphos-methyl-induced Toxicity. J. Environ. Immunol. Toxicol. 2013, 1, 44. [Google Scholar] [CrossRef]
- Lewis, C.M. Azinphos-Methyl (Guthion) Risk Characterization Document (Revision No. 1). Available online: http://www.cdpr.ca.gov/docs/risk/rcd/azmrcdre.pdf (accessed on 4 March 2021).
- Belenguer, V.; Martinez-Capel, F.; Masiá, A.; Picó, Y. Patterns of presence and concentration of pesticides in fish and waters of the Júcar River (Eastern Spain). J. Hazard. Mater. 2014, 265, 271–279. [Google Scholar] [CrossRef]
- Schulz, R. Field Studies on Exposure, Effects, and Risk Mitigation of Aquatic Nonpoint-Source Insecticide Pollution: A Review. J. Environ. Qual. 2004, 33, 419–448. [Google Scholar] [CrossRef]
- Stoner, K.A.; Eitzer, B.D. Using a Hazard Quotient to Evaluate Pesticide Residues Detected in Pollen Trapped from Honey Bees (Apis mellifera) in Connecticut. PLoS ONE 2013, 8, e77550. [Google Scholar] [CrossRef]
- Buratti, F.M.; Volpe, M.T.; Fabrizi, L.; Meneguz, A.; Vittozzi, L.; Testai, E. Kinetic parameters of OPT pesticide desulfuration by c-DNA expressed human CYPs. Environ. Toxicol. Pharmacol. 2002, 11, 181–190. [Google Scholar] [CrossRef]
- Dubois, K.P.; Thursh, D.R.; Murphy, S.D. Studies on the toxicity and pharmacologic actions of the dimethoxy ester of benzotriazine dithiophosphoric acid (DBD, guthion). J. Pharmacol. Exp. Ther. 1957, 119, 208–218. [Google Scholar] [PubMed]
- Pasquet, J.; Mazuret, A.; Fournel, J.; Koenig, F.H. Acute oral and percutaneous toxicity of phosalone in the rat, in comparison with azinphosmethyl and parathion. Toxicol. Appl. Pharmacol. 1976, 37, 85–92. [Google Scholar] [CrossRef]
- Vrdoljak, A.L.; Čalić, M.; Radić, B.; Berend, S.; Jun, D.; Kuča, K.; Kovarik, Z. Pretreatment with pyridinium oximes improves antidotal therapy against tabun poisoning. Toxicology 2006, 228, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Koelle, G.B. Protection of cholinesterase against irreversible inactivation by di-isopropyl fluorophosphate in vitro. J. Pharmacol. Exp. Ther. 1946, 88, 232–237. [Google Scholar] [PubMed]
- Koster, R. Synergisms and antagonisms between physostigmine and di-isopropyl fluorophosphate in cats. J. Pharmacol. Exp. Ther. 1946, 88, 39–46. [Google Scholar]
- Lenina, O.A.; Zueva, I.V.; Zobov, V.V.; Semenov, V.E.; Masson, P.; Petrov, K.A. Slow-binding reversible inhibitor of acetylcholinesterase with long-lasting action for prophylaxis of organophosphate poisoning. Sci. Rep. 2020, 10, 16611. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, D.; Friedman, A.; Seidman, S.; Soreq, H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 1998, 393, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Erb, C.; Gazit, R.; Pavlovsky, L.; Kaufer, D.; Friedman, A.; Glick, D.; Ben-Arie, N.; Soreq, H. Alternative Splicing and Neuritic mRNA Translocation Under Long-Term Neuronal Hypersensitivity. Science 2002, 295, 508–512. [Google Scholar] [CrossRef]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Prado, A.; Petroianu, G.A.; Lorke, D.E.; Chambers, J.W. A trivalent approach for determining in vitro toxicology: Examination of oxime K027. J. Appl. Toxicol. 2015, 35, 219–227. [Google Scholar] [CrossRef]
- Jun, D.; Kuca, K.; Stodülka, P.; Koleckar, V.; Doležal, B.; Simon, P.; Veverka, M. HPLC Analysis of HI-6 Dichloride and Dimethanesulfonate—Antidotes against Nerve Agents and Organophosphorus Pesticides. Anal. Lett. 2007, 40, 2783–2787. [Google Scholar] [CrossRef]
- Jun, D.; Stodulka, P.; Kuca, K.; Koleckar, V.; Dolezal, B.; Simon, P.; Veverka, M. TLC analysis of intermediates arising during the preparation of oxime HI-6 dimethanesulfonate. J. Chromatogr. Sci. 2008, 46, 316–319. [Google Scholar] [CrossRef]
- Lorke, D.E.; Petroianu, G.A. Minireview: Does in-vitro testing of oximes help predict their in-vivo action after paraoxon exposure? J. Appl. Toxicol. 2009, 29, 459–469. [Google Scholar] [CrossRef] [PubMed]

| Substance | Structure |
|---|---|
| Azinphos-methyl |  |
| Pyridostigmine |  |
| Pralidoxime |  |
| Obidoxime |  |
| K-27 |  |
| K-48 |  |
| K-53 |  |
| K-74 |  |
| K-75 |  |
| Groups (G) | 10 min | 30 min | 1 h | 2 h | 3 h | 4 h | 24 h | 48 h |
|---|---|---|---|---|---|---|---|---|
| G1: Azinphos-methyl only | 71/92/92 | 71/92/96 | 71/92/100 | 71/92/100 | 71/92/100 | 71/92/100 | 75/92/100 | 75/92/100 |
| G2: Pyridostigmine pretreatment | 29/50/71 | 42/54/75 | 42/54/79 | 42/58/83 | 42/58/88 | 42/58/88 | 46/58/88 | 46/58/88 |
| G3: Pralidoxime pretreatment | 21/33/54 | 21/50/58 | 21/50/67 | 25/50/71 | 25/63/71 | 25/63/71 | 25/63/71 | 25/63/71 |
| G4: Obidoxime pretreatment | 4/8/21 | 4/8/21 | 4/8/42 | 4/29/42 | 4/29/54 | 4/29/54 | 17/46/63 | 17/46/67 |
| G5: K-27 pretreatment | 21/0/4 | 21/4/13 | 21/4/13 | 21/4/25 | 29/4/29 | 29/42/29 | 50/46/46 | 50/50/46 |
| G6: K-48 pretreatment | 0/8/8 | 8/13/13 | 8/13/21 | 8/13/21 | 8/13/38 | 8/13/38 | 25/50/58 | 25/54/58 |
| G7: K-53 pretreatment | 13/33/29 | 29/33/42 | 29/33/50 | 29/46/63 | 29/50/63 | 29/50/63 | 38/54/71 | 38/54/71 |
| G8: K-74 pretreatment | 0/8/17 | 17/8/25 | 17/8/38 | 17/13/46 | 17/33/50 | 17/50/50 | 33/58/54 | 33/58/54 |
| G9: K-75 pretreatment | 4/17/33 | 33/21/50 | 38/21/58 | 38/25/58 | 38/38/58 | 42/42/58 | 50/50/63 | 50/58/67 |
| Groups | Relative Risk (RR) | 95% CI | p-Value |
|---|---|---|---|
| Azinphos-methyl only | 1 | reference | reference |
| Pyridostigmine + azinphos | 0.52 ± 0.10 | 0.36–0.68 | ≤0. 01 |
| Pralidoxime + azinphos | 0.37 ± 0.03 | 0.33–0.41 | ≤0. 01 a |
| Obidoxime+ azinphos | 0.21 ± 0.09 | 0.06–0.36 | ≤0. 01 a, b |
| K-27 + azinphos | 0.23 ± 0.02 | 0.20–0.25 | ≤0.01 a, b, c |
| K-48 + azinphos | 0.20 ± 0.03 | 15–0.24 | ≤0.01 a, b, c |
| K-53 + azinphos | 0.37 ± 0.14 | 0.14–0.59 | ≤0.01 |
| K-74 + azinphos | 0.26 ± 0.10 | 0.09–0.42 | ≤0.01 a |
| K-75 + azinphos | 0.35 ± 0.03 | 0.30–0.39 | ≤0.01 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. Int. J. Mol. Sci. 2021, 22, 3072. https://doi.org/10.3390/ijms22063072
Lorke DE, Nurulain SM, Hasan MY, Kuča K, Petroianu GA. Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. International Journal of Molecular Sciences. 2021; 22(6):3072. https://doi.org/10.3390/ijms22063072
Chicago/Turabian StyleLorke, Dietrich E., Syed M. Nurulain, Mohamed Y. Hasan, Kamil Kuča, and Georg A. Petroianu. 2021. "Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl" International Journal of Molecular Sciences 22, no. 6: 3072. https://doi.org/10.3390/ijms22063072
APA StyleLorke, D. E., Nurulain, S. M., Hasan, M. Y., Kuča, K., & Petroianu, G. A. (2021). Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. International Journal of Molecular Sciences, 22(6), 3072. https://doi.org/10.3390/ijms22063072