
Endothelial Glycocalyx as a Regulator of Fibrotic Processes

 ,
,  ,
, 
 and
and 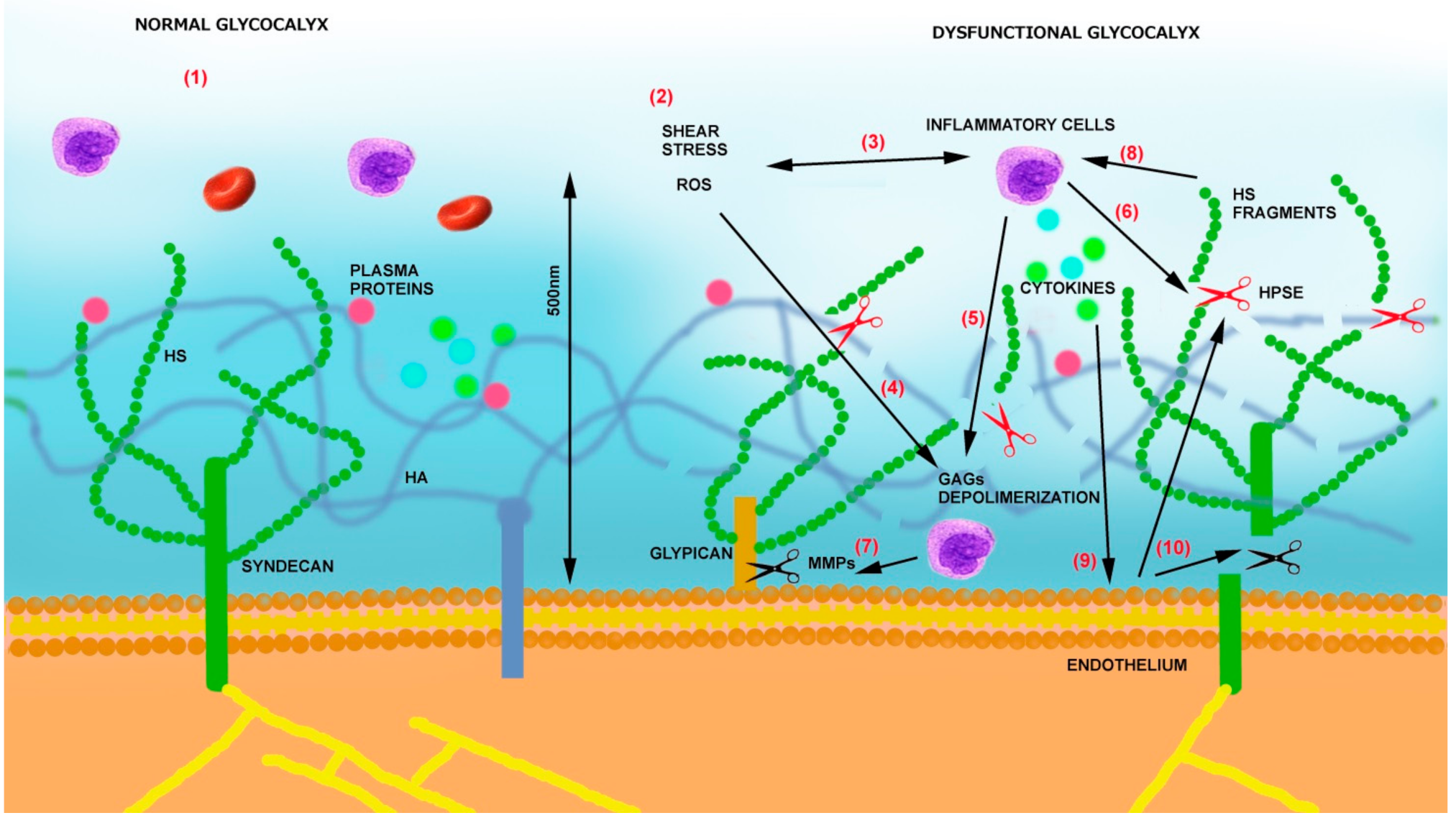{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endothelial Glycocalyx Structure and Functions
3. Mechanisms of Endothelial Glycocalyx Damage
- (1)
- (2)
- By releasing proinflammatory cytokines and chemokines bound to GAGs, it sustains inflammation, oxidative stress, and additional glycocalyx damage [61].
- (3)
- Heparanase is also able to sustain inflammation by activating Toll-like receptors (TLRs) on macrophages via heparan sulfate fragments, leading to the activation of nuclear factor-κB (NF-κB), which results in the expression of additional inflammatory cytokines (TNF-α, IL-1β, and IL-8) [62]. The same cytokines can then sustain heparanase expression on endothelial cells [54] as well as the production of MMPs [63] and ROS.
- (4)
- Heparanase also contributes to glycocalyx damage, thus increasing their procoagulant state by increasing tissue factor (TF) and modulating tissue factor pathway inhibitor (TFPI) [64].
- (5)
4. Glycocalyx Dysfunction Conditions
5. Role of Glycocalyx Dysfunction in Fibrosis
6. Therapeutic Strategies to Preserve Glycocalyx
- Resuscitation fluids (fresh frozen plasma, plasma albumin, and hydroxyethyl starch) may influence glycocalyx shedding [118].
- It has been proved that anesthetic sevoflurane attenuates glycocalyx degradation in guinea pig hearts in a myocardial I/R injury model [78].
- Glucocorticoid: hydrocortisone reduced coronary resistance, vascular permeability, tissue edema, the release of lactate, uric acid, purines, and histamine, which were accompanied by severe degradation induced by TNF-α [119]. Dexamethasone suppressed the expression of MMPs and rescued the expression of ZO-1 and syndecan-1 in sepsis [120].
- Elevated levels of oxidative stress are present in the serum of CKD patients [121]; moreover, antioxidant elements such as ascorbic acid are reduced, limiting NO bioactivity [122]. Some strategies aimed at reducing oxidative stress have been tested. In a rat model of angiotensin-II-induced hypertension, the administration of green tea extract restored endothelium vasodilatation through ROS scavenging [123]. Additionally, the use of the antioxidant N-acetylcysteine reduces oxidative stress in a hyperglycemic state and, by doing so, reduces endothelial activation [124].
- Heparin and heparinoids may act toward several mechanisms. Firstly, heparins, by binding to endothelial cells, participate in the reconstitution of the glycocalyx and recover its negative charge [125]. It has also been reported that heparins increase heparan sulfate production and sustain its sulfation pattern [126]. Secondly, heparins are able to control multiple inflammatory effects. Heparins are able to protect cells from ROS, and they bind complement, growth factors and cytokines (i.e., interferon-γ and FGF-2), and P- and L-selectin (inhibiting leukocyte adhesion) [127]. Third, heparins protect endothelial cells from high-glucose damage by preventing the interaction of advanced glycosylation end products with their receptors [128], reducing membrane disruption and cell death [127]. Lastly, heparins are heparanase inhibitors, and thus they can modulate all the effects of this enzyme in direct glycocalyx degradation but also in inflammation and fibrosis [127]. Heparins are also able to bind and inhibit NF-κB [128] and thus regulate inflammatory cytokines but probably also the same heparanase and syndecan-4 expression involved in the development of fibrosis [88,116]. In this situation, a promising agent is sulodexide, a mixture of 80% fast-moving heparin and 20% of dermatan sulfate. Sulodexide has antithrombotic, profibrinolytic, anti-inflammatory, antioxidant, and anti-ischemic properties. In addition, its proposed mode of action is the inhibition of heparanase and also the modulation of MMP-9 production [127]. Animal models revealed multifaceted effects of sulodexide on endothelial functions [127,129], and, in clinical evaluation, sulodexide was able to partially restore endothelial glycocalyx and vascular permeability in patients with type 2 diabetes [130,131].
- Another element that could help to maintain glycocalyx integrity in diabetes is atrasentan and metformin. Atrasentan, antagonizing endothelin-1, reduces the glomerular expression of heparanase and its activator cathepsin-L [73]. The mechanism of action of metformin has not yet been clarified, but two weeks of metformin in drinking water is associated with an improvement in glycocalyx barrier properties in db/db mice [132].
- Since MMPs are central elements in glycocalyx degradation, some attempts at inhibition have been made, but more cell and animal experiments are necessary for a clinical translation. In vitro, sphingosine-1-phosphate (S1P) inhibits MMP-9 and -13 activity by activating the S1P1 receptor, which restores the endothelial glycocalyx through the activation of the PI3K pathway. S1P by inhibiting MMPs prevents the shedding of CS, HS, and the syndecan-1 ectodomain [133,134]. The use of pan-MMPs inhibitors, however, is not viable [135]. Some studies have shown that specific MMP-2 and -9 inhibition prevent the shedding of SDC-4 and HS in response to TNF-α preserving glycocalyx integrity [63,136].
- Another strategy to protect and reconstitute damaged glycocalyx is to supply endothelial cells with glycocalyx components. It has been proved, in an in vivo model, that glycocalyx damaged by hyaluronidase treatment can be partially recovered by acute infusion of hyaluronan and chondroitin sulfate [137]. It has also been proposed that the use of glycocalyx-mimetic biomaterials such as corline heparin conjugate, a structure resembling a proteoglycan, is able to protect the vasculature in thrombotic disorders and organ transplantation [138]. Additionally, elements designed to improve the compatibility between blood and polymeric biomaterials, such as the glycocalyx-mimetic dextran-modified poly(vinyl amine) surfactant, could represent useful tools to ameliorate glycocalyx structure [139].
- Possible strategies in future could implement NO production through the use of small molecules such as the protein kinase C inhibitor midostaurin, the pentacyclic triterpenoids ursolic acid and betulinic acid, the eNOS-enhancing compounds AVE9488 and AVE3085, and the polyphenolic phytoalexin transresveratrol [140].
- Giving the central role of SIRT-1 on endothelial glycocalyx preservation strategies aimed at restoration of its expression and activity are currently being tested [141]. The first generation of SIRT1 activators were plant polyphenols, such as butein, piceatannol, isoliquiritigenin, and mostly resveratrol [142,143]. Advances in sirtuin biochemistry, assays, and crystal structures allowed the development of more specific SIRT-1 modulators. Three small-molecule SIRT1 activators (SRT2104, SRT2379, and SRT3025) have been tested in clinical trials. All the compounds were well tolerated. In three studies, in elderly volunteers, healthy cigarette smokers, and type 2 diabetics, the compound SRT2104 had a beneficial effect on lipids, decreasing serum cholesterol, LDL levels, and triglycerides [139]. SRT2104 also reduced the LPS-induced release of inflammatory mediators and activation of coagulation [144]. Other studies have been carried out to test the anti-inflammatory effects of SRT2104 [141]. Starting from this evidence, the evaluation of these compounds’ effects on glycocalyx preservation and the regulation of fibrosis would be desirable to be made.
- It has been described that the patchy degradation of ESG is a result of the exocytosis of lysosome-related organelles. The control of excessive exocytosis could be achieved by sustaining NO production such as with NG-hydroxy-l-arginine, a nitric oxide intermediate [145].
- A new and promising strategy to obtain the restoration of glycocalyx is the recently described nanoliposomal carriers of preassembled glycocalyx [146]. These structures are able to bind to cells with degraded glycocalyx and restore NO production in endothelial cells, and they are able to induce a flow-induced vasodilatory response in perfused mesenteric arteries with a degraded glycocalyx [146].
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Vink, H.; Duling, B.R. Capillary endothelial surface layer selectively reduces plasma solute distribution volume. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H285–H289. [Google Scholar] [CrossRef]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Molitoris, B.A. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 2015, 35, 96–107. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Van Teeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial glycocalyx: Sweet shield of blood vessels. Trends Cardiovasc. Med. 2007, 17, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Curry, F.R. Microvascular solute and water transport. Microcirculation 2005, 12, 17–31. [Google Scholar] [CrossRef]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Lennon, F.E.; Singleton, P.A. Hyaluronan regulation of vascular integrity. Am. J. Cardiovasc. Dis. 2011, 1, 200–213. [Google Scholar] [PubMed]
- Tkachenko, E.; Rhodes, J.M.; Simons, M. Syndecans: New kids on the signaling block. Circ. Res. 2005, 96, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, T.; Whipple, C.A.; Lopez, M.E.; Gunn, J.; Young, A.; Lander, A.D.; Korc, M. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J. Clin. Investig. 2008, 118, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Ginzburg, Y.; Vlodavsky, I. Specific involvement of glypican in thrombin adhesive properties. J. Cell Biochem. 1996, 61, 278–291. [Google Scholar] [CrossRef]
- Koedam, J.A.; Cramer, E.M.; Briend, E.; Furie, B.; Furie, B.C.; Wagner, D.D. P-selectin, a granule membrane protein of platelets and endothelial cells, follows the regulated secretory pathway in AtT-20 cells. J. Cell Biol. 1992, 116, 617–625. [Google Scholar] [CrossRef]
- Jung, U.; Ley, K. Regulation of E-selectin, P-selectin, and intercellular adhesion molecule 1 expression in mouse cremaster muscle vasculature. Microcirculation 1997, 4, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J. 2006, 273, 4377–4389. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—The update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Bombeli, T.; Schwartz, B.R.; Harlan, J.M. Adhesion of activated platelets to endothelial cells: Evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J. Exp. Med. 1998, 187, 329–339. [Google Scholar] [CrossRef]
- Rüegg, C.; Mariotti, A. Vascular integrins: Pleiotropic adhesion and signaling molecules in vascular homeostasis and angiogenesis. Cell Mol. Life Sci. 2003, 60, 1135–1157. [Google Scholar] [CrossRef]
- Müller, A.M.; Hermanns, M.I.; Cronen, C.; Kirkpatrick, C.J. Comparative study of adhesion molecule expression in cultured human macro- and microvascular endothelial cells. Exp. Mol. Pathol. 2002, 73, 171–180. [Google Scholar] [CrossRef]
- Yang, X.; Meegan, J.E.; Jannaway, M.; Coleman, D.C.; Yuan, S.Y. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc. Res. 2018, 114, 1752–1763. [Google Scholar] [CrossRef]
- Lipowsky, H.H. Microvascular rheology and hemodynamics. Microcirculation 2005, 12, 5–15. [Google Scholar] [CrossRef]
- Curry, F.E. Layer upon layer: The functional consequences of disrupting the glycocalyx-endothelial barrier in vivo and in vitro. Cardiovasc. Res. 2017, 113, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflugers Arch. 2000, 440, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Lee, E.S.; Jeong, Y. In vivo Imaging of the Cerebral Endothelial Glycocalyx in Mice. J. Vasc. Res. 2017, 54, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Zhang, J.; Xin, S.Y.; Liu, C.; Wang, C.Y.; Zhao, D.; Zhang, Z.R. Mechanosensitive properties in the endothelium and their roles in the regulation of endothelial function. J. Cardiovasc. Pharmacol. 2013, 61, 461–470. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Simon, S.I.; Curry, F.R. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef]
- Rubanyi, G.M.; Romero, J.C.; Vanhoutte, P.M. Flow-induced release of endothelium-derived relaxing factor. Am. J. Physiol. 1986, 250 Pt 2, H1145–H1149. [Google Scholar] [CrossRef]
- Chappell, D.; Westphal, M.; Jacob, M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr. Opin. Anaesthesiol. 2009, 22, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H. Vascular endothelium: A vulnerable transit zone for merciless sodium. Nephrol. Dial. Transplant. 2014, 29, 240–246. [Google Scholar] [CrossRef]
- Dull, R.O.; Mecham, I.; McJames, S. Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1452–L1458. [Google Scholar] [CrossRef]
- Dull, R.O.; Cluff, M.; Kingston, J.; Hill, D.; Chen, H.; Hoehne, S.; Malleske, D.T.; Kaur, R. Lung heparan sulfates modulate K(fc) during increased vascular pressure: Evidence for glycocalyx-mediated mechanotransduction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L816–L828. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, K.B.; Arkill, K.P.; Neal, C.R.; Harper, S.J.; Foster, R.R.; Satchell, S.C.; Bates, D.O.; Salmon, A.H.J. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. Atherosclerosis 2017, 595, 5015–5035. [Google Scholar]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef]
- Dreyfuss, J.L.; Regatieri, C.V.; Jarrouge, T.R.; Cavalheiro, R.P.; Sampaio, L.O.; Nader, H.B. Heparan sulfate proteoglycans: Structure, protein interactions and cell signaling. An. Acad. Bras. Cienc. 2009, 81, 409–429. [Google Scholar] [CrossRef]
- Prior, S.M.; Cohen, M.J.; Conroy, A.S.; Nelson, M.F.; Kornblith, L.Z.; Howard, B.M.; Butenas, S. Correlation between factor (F)XIa, FIXa and tissue factor and trauma severity. J. Trauma Acute Care Surg. 2017, 82, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.B.; Lo, S.K.; Bizios, R. Thrombin-induced alterations in endothelial permeability. Ann. N. Y. Acad. Sci. 1986, 485, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Klarenbach, S.W.; Chipiuk, A.; Nelson, R.C.; Hollenberg, M.D.; Murray, A.G. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: The role of Rho-GTPases. Circ. Res. 2003, 92, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Van Golen, R.F.; Reiniers, M.J.; Vrisekoop, N.; Zuurbier, C.J.; Olthof, P.B.; van Rheenen, J.; van Gulik, T.M.; Parsons, B.J.; Heger, M. The mechanisms and physiological relevance of glycocalyx degradation in hepatic ischemia/reperfusion injury. Antioxid. Redox Signal. 2014, 21, 1098–1118. [Google Scholar] [CrossRef]
- Moseley, R.; Waddington, R.J.; Embery, G. Degradation of glycosaminoglycans by reactive oxygen species derived from stimulated polymorphonuclear leukocytes. Biochim. Biophys. Acta 1997, 1362, 221–231. [Google Scholar] [CrossRef]
- Soltés, L.; Mendichi, R.; Kogan, G.; Schiller, J.; Stankovska, M.; Arnhold, J. Degradative action of reactive oxygen species on hyaluronan. Biomacromolecules 2006, 7, 659–668. [Google Scholar] [CrossRef]
- Safrankova, B.; Gajdova, S.; Kubala, L. The potency of hyaluronan of different molecular weights in the stimulation of blood phagocytes. Mediat. Inflamm. 2010, 2010, 380948. [Google Scholar] [CrossRef]
- Castro, M.M.; Rizzi, E.; Figueiredo-Lopes, L.; Fernandes, K.; Bendhack, L.M.; Pitol, D.L.; Gerlach, R.F.; Tanus-Santos, J.E. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis 2008, 198, 320–331. [Google Scholar] [CrossRef]
- Hobeika, M.J.; Thompson, R.W.; Muhs, B.E.; Brooks, P.C.; Gagne, P.J. Matrix metalloproteinases in peripheral vascular disease. J. Vasc. Surg. 2007, 45, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Sieve, I.; Münster-Kühnel, A.K.; Hilfiker-Kleiner, D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Bogner-Flatz, V.; Braunstein, M.; Ocker, L.E.; Kusmenkov, T.; Tschoep, J.; Ney, L.; Böcker, W.; Annecke, T. On-the-Scene Hyaluronan and Syndecan-1 Serum Concentrations and Outcome after Cardiac Arrest and Resuscitation. Mediat. Inflamm. 2019, 2019, 8071619. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Gambaro, G.; Franchi, M.; Onisto, M. Role of heparanase in tumor progression: Molecular aspects and therapeutic options. Semin. Cancer Biol. 2020, 62, 86–98. [Google Scholar] [CrossRef]
- Masola, V.; Bellin, G.; Gambaro, G.; Onisto, M. Heparanase: A Multitasking Protein Involved in Extracellular Matrix (ECM) Remodeling and Intracellular Events. Cells 2018, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; van den Hoven, M.; Rops, A.; Wijnhoven, T.; van den Heuvel, L.; Lensen, J.; van Kuppevelt, T.; van Goor, H.; van der Vlag, J.; Navis, G.; et al. Induction of glomerular heparanase expression in rats with adriamycin nephropathy is regulated by reactive oxygen species and the renin-angiotensin system. J. Am. Soc. Nephrol. 2006, 17, 2513–2520. [Google Scholar] [CrossRef]
- Chen, G.; Wang, D.; Vikramadithyan, R.; Yagyu, H.; Saxena, U.; Pillarisetti, S.; Goldberg, I.J. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry 2004, 43, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Mandal, A.K.; Hiebert, L.M. Endothelial cell injury by high glucose and heparanase is prevented by insulin, heparin and basic fibroblast growth factor. Cardiovasc. Diabetol. 2005, 4, 12. [Google Scholar] [CrossRef]
- An, X.F.; Zhou, L.; Jiang, P.J.; Yan, M.; Huang, Y.J.; Zhang, S.N.; Niu, Y.F.; Ten, S.C.; Yu, J.Y. Advanced glycation end-products induce heparanase expression in endothelial cells by the receptor for advanced glycation end products and through activation of the FOXO4 transcription factor. Mol. Cell Biochem. 2011, 354, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ishai-Michaeli, R.; Eldor, A.; Vlodavsky, I. Heparanase activity expressed by platelets, neutrophils, and lymphoma cells releases active fibroblast growth factor from extracellular matrix. Cell Regul. 1990, 1, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Sulfated glycans in inflammation. Eur. J. Med. Chem. 2015, 92, 353–369. [Google Scholar] [CrossRef]
- Garsen, M.; Rops, A.L.; Rabelink, T.J.; Berden, J.H.; van der Vlag, J. The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol. Dial. Transplant. 2014, 29, 49–55. [Google Scholar] [CrossRef]
- Lever, R.; Rose, M.J.; McKenzie, E.A.; Page, C.P. Heparanase induces inflammatory cell recruitment in vivo by promoting adhesion to vascular endothelium. Am. J. Physiol. Cell. Physiol. 2014, 306, C1184–C1190. [Google Scholar] [CrossRef]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef] [PubMed]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler Thromb. Vasc. Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Ramnath, R.; Foster, R.R.; Qiu, Y.; Cope, G.; Butler, M.J.; Salmon, A.H.; Mathieson, P.W.; Coward, R.J.; Welsh, G.I.; Satchell, S.C. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: A contributor to endothelial cell glycocalyx dysfunction. FASEB J. 2014, 28, 4686–4699. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B. Heparanase procoagulant activity in cancer progression. Thromb. Res. 2016, 140 (Suppl. 1), S44–S48. [Google Scholar] [CrossRef]
- Traister, A.; Shi, W.; Filmus, J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem. J. 2008, 410, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M. Vascular Endothelial Glycocalyx Damage in COVID-19. Int. J. Mol. Sci. 2020, 21, 9712. [Google Scholar] [CrossRef] [PubMed]
- Solbu, M.D.; Kolset, S.O.; Jenssen, T.G.; Wilsgaard, T.; Løchen, M.L.; Mathiesen, E.B.; Melsom, T.; Eriksen, B.O.; Reine, T.M. Gender differences in the association of syndecan-4 with myocardial infarction: The population-based Tromsø Study. Atherosclerosis 2018, 278, 166–173. [Google Scholar] [CrossRef]
- Kataoka, H.; Ushiyama, A.; Akimoto, Y.; Matsubara, S.; Kawakami, H.; Iijima, T. Structural behavior of the endothelial glycocalyx is associated with pathophysiologic status in septic mice: An integrated approach to analyzing the behavior and function of the glycocalyx using both electron and fluorescence intravital microscopy. Anesth. Analg. 2017, 125, 874–883. [Google Scholar] [CrossRef]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chang, A.; Hack, B.K.; Eadon, M.T.; Alper, S.L. Cunningham PN. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014, 85, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Haeger, S.M.; Suflita, M.A.; Zhang, F.; Dailey, K.L.; Colbert, J.F.; Ford, J.A.; Picon, M.A.; Stearman, R.S.; Lin, L.; et al. Fibroblast Growth Factor Signaling Mediates Pulmonary Endothelial Glycocalyx Reconstitution. Am. J. Respir. Cell Mol. Biol. 2017, 56, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Freudenberger, T.; Melchior-Becker, A.; Röck, K.; Ter Braak, M.; Jastrow, H.; Kinzig, M.; Lucke, S.; Suvorava, T.; Kojda, G.; et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: Novel insights into the role of hyaluronan synthesis. Circulation 2010, 122, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Gröne, H.J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef]
- Mc Donald, K.K.; Cooper, S.; Danielzak, L.; Leask, R.L. Glycocalyx degradation induces a proinflammatory phenotype and increased leukocyte adhesion in cultured endothelial cells under flow. PLoS ONE 2016, 11, e0167576. [Google Scholar]
- Torres Filho, I.P.; Torres, L.N.; Salgado, C.; Dubick, M.A. Plasma syndecan-1 and heparan sulfate correlate with icrovascular glycocalyx degradation in hemorrhaged rats after different resuscitation fluids. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1468–H1478. [Google Scholar] [CrossRef] [PubMed]
- Yini, S.; Heng, Z.; Xin, A.; Xiaochun, M. Effect of unfractionated heparin on endothelial glycocalyx in a septic shock model. Acta Anaesthesiol. Scand. 2015, 59, 160–169. [Google Scholar] [CrossRef]
- Abassi, Z.; Hamoud, S.; Hassan, A.; Khamaysi, I.; Nativ, O.; Heyman, S.N.; Muhammad, R.S.; Ilan, N.; Singh, P.; Hammond, E.; et al. Involvement of heparanase in the pathogenesis of acute kidney injury: Nephroprotective effect of PG545. Oncotarget 2017, 8, 34191–34204. [Google Scholar] [CrossRef]
- Annecke, T.; Chappell, D.; Chen, C.; Jacob, M.; Welsch, U.; Sommerhoff, C.P.; Rehm, M.; Conzen, P.F.; Becker, B.F. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br. J. Anaesth. 2010, 104, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Bruegger, D.; Christ, F.; Conzen, P.; Thiel, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Reichart, B.; et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation 2007, 116, 1896–1906. [Google Scholar] [CrossRef]
- King, M.L.R.; Salmon, A.E.; Saleem, M.A.; Welsh, G.I.; Michel, C.C.; Satchell, S.C.; Salmon, A.H.J.; Foster, R.R. A novel assay provides sensitive measurement of physiologically relevant changes in albumin permeability in isolated human and rodent glomeruli. Kidney Int. 2018, 93, e1086–e1097. [Google Scholar]
- Diebel, L.N.; Liberati, D.M.; Martin, J.V. Acute hyperglycemia increases sepsis related glycocalyx degradation and endothelial cellular injury: A microfluidic study. Am. J. Surg. 2019, 217, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Schenning, K.J.; Anderson, S.; Alkayed, N.J.; Hutchens, M.P. Hyperglycemia abolishes the protective effect of ischemic preconditioning in glomerular endothelial cells in vitro. Physiol. Rep. 2015, 3, e12346. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Okada, H.; Tomita, H.; Sumi, K.; Kakino, Y.; Yasuda, R.; Kitagawa, Y.; Fukuta, T.; Miyake, T.; Yoshida, S.; et al. Possible involvement of Syndecan-1 in the state of COVID-19 related to endothelial injury. Thromb. J. 2021, 19, 5. [Google Scholar] [CrossRef]
- Masola, V.; Gambaro, G.; Onisto, M. Impact of Heparanse on Organ Fibrosis. Adv. Exp. Med. Biol. 2020, 1221, 669–684. [Google Scholar] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Masola, V.; Granata, S.; Pontrelli, P.; Sallustio, F.; Gesualdo, L.; Gambaro, G.; Grandaliano, G.; Lupo, A. Dialysis-related transcriptomic profiling: The pivotal role of heparanase. Exp. Biol. Med. 2014, 239, 52–64. [Google Scholar] [CrossRef]
- Masola, V.; Bellin, G.; Vischini, G.; Dall’Olmo, L.; Granata, S.; Gambaro, G.; Lupo, A.; Onisto, M.; Zaza, G. Inhibition of heparanase protects against chronic kidney dysfunction following ischemia/reperfusion injury. Oncotarget 2018, 9, 36185–36201. [Google Scholar] [CrossRef][Green Version]
- Masola, V.; Zaza, G.; Gambaro, G.; Onisto, M.; Bellin, G.; Vischini, G.; Khamaysi, I.; Hassan, A.; Hamoud, S.; Nativ, O.; et al. Heparanase: A Potential New Factor Involved in the Renal Epithelial Mesenchymal Transition (EMT) Induced by Ischemia/Reperfusion (I/R) Injury. PLoS ONE 2016, 11, e0160074. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Gambaro, G.; Tibaldi, E.; Onisto, M.; Abaterusso, C.; Lupo, A. Regulation of heparanase by albumin and advanced glycation end products in proximal tubular cells. Biochim. Biophys. Acta 2011, 1813, 1475–1482. [Google Scholar] [CrossRef]
- Gil, N.; Goldberg, R.; Neuman, T.; Garsen, M.; Zcharia, E.; Rubinstein, A.M.; van Kuppevelt, T.; Meirovitz, A.; Pisano, C.; Li, J.P.; et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes 2012, 61, 208–216. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Secchi, M.F.; Gambaro, G.; Lupo, A.; Onisto, M. Heparanase is a key player in renal fibrosis by regulating TGF-β expression and activity. Biochim. Biophys. Acta 2014, 1843, 2122–2128. [Google Scholar] [CrossRef]
- Masola, V.; Onisto, M.; Zaza, G.; Lupo, A.; Gambaro, G. A new mechanism of action of sulodexide in diabetic nephropathy: Inhibits heparanase-1 and prevents FGF-2-induced renal epithelial-mesenchymal transition. J. Transl. Med. 2012, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Bellin, G.; Dall’Olmo, L.; Granata, S.; Vischini, G.; Secchi, M.F.; Lupo, A.; Gambaro, G.; Onisto, M. Heparanase regulates the M1 polarization of renal macrophages and their crosstalk with renal epithelial tubular cells after ischemia/reperfusion injury. FASEB J. 2018, 32, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Seccia, T.M.; Barton, M.; Danser, A.H.J.; de Leeuw, P.W.; Dhaun, N.; Rizzoni, D.; Rossignol, P.; Ruilope, L.M.; van den Meiracker, A.H.; et al. Endothelial factors in the pathogenesis and treatment of chronic kidney disease Part I: General mechanisms: A joint consensus statement from the European Society of Hypertension Working Group on Endothelin and Endothelial Factors and The Japanese Society of Hypertension. J. Hypertens. 2018, 36, 451–461. [Google Scholar] [PubMed]
- Rossi, G.P.; Sacchetto, A.; Cesari, M.; Pessina, A.C. Interactions between endothelin-1 and the renin-angiotensin-aldosterone system. Cardiovasc. Res. 1999, 43, 300–307. [Google Scholar] [CrossRef]
- Clozel, M.; Salloukh, H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann. Med. 2005, 37, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Neuhofer, W.; Pittrow, D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur. J. Clin. Investig. 2006, 36 (Suppl. 3), 78–88. [Google Scholar] [CrossRef] [PubMed]
- Garsen, M.; Lenoir, O.; Rops, A.L.; Dijkman, H.B.; Willemsen, B.; van Kuppevelt, T.H.; Rabelink, T.J.; Berden, J.H.; Tharaux, P.L.; van der Vlag, J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 2016, 27, 3545–3551. [Google Scholar] [CrossRef]
- Van den Hoven, M.J.; Waanders, F.; Rops, A.L.; Kramer, A.B.; van Goor, H.; Berden, J.H.; Navis, G.; van der Vlag, J. Regulation of glomerular heparanase expression by aldosterone, angiotensin II and reactive oxygen species. Nephrol. Dial. Transplant. 2009, 24, 2637–2645. [Google Scholar] [CrossRef]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE 2011, 6, e17312. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kim, M.S.; Puthanveetil, P.; Kewalramani, G.; Deppe, S.; Ghosh, S.; Abrahani, A.; Rodrigues, B. Endothelial heparanase secretion after acute hypoinsulinemia is regulated by glucose and fatty acid. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1108–H1116. [Google Scholar] [CrossRef]
- Han, J.; Hiebert, L.M. Alteration of endothelial proteoglycan and heparanase gene expression by high glucose, insulin and heparin. Vasc. Pharmacol. 2013, 59, 112–118. [Google Scholar] [CrossRef]
- Goldberg, R.; Sonnenblick, A.; Hermano, E.; Hamburger, T.; Meirovitz, A.; Peretz, T.; Elkin, M. Heparanase augments insulin receptor signaling in breast carcinoma. Oncotarget 2017, 8, 19403–19412. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Purushothaman, A.; Babitz, S.K.; Sanderson, R.D. Heparanase enhances the insulin receptor signaling pathway to activate extracellular signal-regulated kinase in multiple myeloma. J. Biol. Chem. 2012, 287, 41288–41296. [Google Scholar] [CrossRef]
- King, G.L.; Park, K.; Li, Q. Selective Insulin Resistance and the Development of Cardiovascular Diseases in Diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes 2016, 65, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Bazyluk, A.; Malyszko, J.; Hryszko, T.; Zbroch, E. State of the art—sirtuin 1 in kidney pathology—clinical relevance. Adv. Med. Sci. 2019, 64, 356–364. [Google Scholar] [CrossRef]
- Lipphardt, M.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Fibrogenic Secretome of Sirtuin 1-Deficient Endothelial Cells: Wnt, Notch and Glycocalyx Rheostat. Front. Physiol. 2018, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R.; Xavier, S.; Chen, J.; Lin, C.H.; Ratliff, B.; Rabadi, M.; Maizel, J.; Tanokuchi, R.; Zhang, F.; Cao, J.; et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: Relevance to fibrosis of vascular senescence. J. Am. Soc. Nephrol. 2014, 25, 276–291. [Google Scholar] [CrossRef]
- Kim, H.; Baek, C.H.; Lee, R.B.; Chang, J.W.; Yang, W.S.; Lee, S.K. Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin. Int. J. Mol. Sci. 2017, 18, 305. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Pan, W.; Zhang, J.; Xu, X.; Zhang, X.; He, X.; Fan, M. Hydrogen Rich Water Attenuates Renal Injury and Fibrosis by Regulation Transforming Growth Factor-β Induced Sirt1. Biol. Pharm. Bull. 2017, 40, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Wen, D.; Zhang, M.; Xie, Q.; Ma, L.; Guan, Y.; Ren, Y.; Chen, J.; Hao, C.M. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell Biochem. 2014, 115, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 2015, 26, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.E.; Herum, K.M.; Rana, Z.A.; Skrbic, B.; Askevold, E.T.; Dahl, C.P.; Vistnes, M.; Hasic, A.; Kvaløy, H.; Sjaastad, I.; et al. Innate immune signaling induces expression and shedding of the heparan sulfate proteoglycan syndecan-4 in cardiac fibroblasts and myocytes, affecting inflammation in the pressure-overloaded heart. FEBS J. 2013, 280, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Torres, L.N.; Sondeen, J.L.; Ji, L.; Dubick, M.A.; Torres Filho, I. Evaluation of resuscitation fluids on endothelial glycocalyx, venular blood flow, and coagulation function after hemorrhagic shock in rats. J. Trauma Acute Care Surg. 2013, 75, 759–766. [Google Scholar] [CrossRef]
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89. [Google Scholar] [CrossRef]
- Cui, N.; Wang, H.; Long, Y.; Su, L.; Liu, D. Dexamethasone Suppressed LPS-Induced Matrix Metalloproteinase and Its Effect on Endothelial Glycocalyx Shedding. Mediat. Inflamm. 2015, 2015, 912726. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Canaud, B.; Eckardt, K.U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003, 18, 1272–1280. [Google Scholar] [CrossRef]
- Takahashi, N.; Morimoto, S.; Okigaki, M.; Seo, M.; Someya, K.; Morita, T.; Matsubara, H.; Sugiura, T.; Iwasaka, T. Decreased plasma level of vitamin C in chronic kidney disease: Comparison between diabetic and non-diabetic patients. Nephrol. Dial. Transplant. 2011, 26, 1252–1257. [Google Scholar] [CrossRef]
- Antonello, M.; Montemurro, D.; Bolognesi, M.; Di Pascoli, M.; Piva, A.; Grego, F.; Sticchi, D.; Giuliani, L.; Garbisa, S.; Rossi, G.P. Prevention of hypertension, cardiovascular damage and endothelial dysfunction with green tea extracts. Am. J. Hypertens. 2007, 20, 1321–1328. [Google Scholar] [CrossRef]
- Masha, A.; Brocato, L.; Dinatale, S.; Mascia, C.; Biasi, F.; Martina, V. N-acetylcysteine is able to reduce the oxidation status and the endothelial activation after a high-glucose content meal in patients with Type 2 diabetes mellitus. J. Endocrinol. Investig. 2009, 32, 352–356. [Google Scholar] [CrossRef]
- Hiebert, L.M.; Jaques, L.B. The observation of heparin on endothelium after injection. Thromb. Res. 1976, 8, 195–204. [Google Scholar] [CrossRef]
- Nader, H.B.; Toma, L.; Pinhal, M.A.; Buonassisi, V.; Colburn, P.; Dietrich, C.P. Effect of heparin and dextran sulfate on the synthesis and structure of heparan sulfate from cultured endothelial cells. Semin. Thromb. Hemost. 1991, 17 (Suppl. 1), 47–56. [Google Scholar] [PubMed]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Glycosaminoglycans, proteoglycans and sulodexide and the endothelium: Biological roles and pharmacological effects. Int. Angiol. 2014, 33, 243–254. [Google Scholar] [PubMed]
- Young, E. The anti-inflammatory effects of heparin and related compounds. Thromb. Res. 2008, 122, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Coccheri, S. Biological and clinical effects of sulodexide in arterial disorders and diseases. Int. Angiol. 2014, 33, 263–274. [Google Scholar]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.; Vink, H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef]
- Giantsos-Adams, K.; Lopez-Quintero, V.; Kopeckova, P.; Kopecek, J.; Tarbell, J.M.; Dull, R. Study of the therapeutic benefit of cationic copolymer administration to vascular endothelium under mechanical stress. Biomaterials 2011, 32, 288–294. [Google Scholar] [CrossRef]
- Eskens, B.J.; Zuurbier, C.J.; van Haare, J.; Vink, H.; van Teeffelen, J.W. Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc. Diabetol. 2013, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Adamson, R.H.; Curry, F.R.; Tarbell, J.M. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H363–H372. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, X.H.; Tarbell, J.; Fu, B. Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Exp. Cell Res. 2015, 339, 90–95. [Google Scholar] [CrossRef]
- Jacob, M.; Saller, T.; Chappell, D.; Rehm, M.; Welsch, U.; Becker, B.F. Physiological levels of A-, B- and C-type natriuretic peptide shed the endothelial glycocalyx and enhance vascular permeability. Basic Res. Cardiol. 2013, 108, 347. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Ramnath, R.; Kadoya, H.; Desposito, D.; Riquier-Brison, A.; Ferguson, J.K.; Onions, K.L.; Ogier, A.S.; ElHegni, H.; Coward, R.J.; et al. Aldosterone induces albuminuria via matrix metalloproteinase-dependent damage of the endothelial glycocalyx. Kidney Int. 2019, 95, 94–107. [Google Scholar] [CrossRef]
- Henry, C.B.; Duling, B.R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am. J. Physiol. 1999, 277, H508–H514. [Google Scholar] [CrossRef]
- Sedigh, A.; Larsson, R.; Brännström, J.; Magnusson, P.; Larsson, E.; Tufveson, G.; Lorant, T. Modifying the vessel walls in porcine kidneys during machine perfusion. J. Surg. Res. 2014, 191, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.S.; Wang, S.; Link, E.; Anderson, E.H.; Hofmann, C.; Lewandowski, J.; Kottke-Marchant, K.; Marchant, R.E. Glycocalyx-mimetic dextran-modified poly(vinyl amine) surfactant coating reduces platelet adhesion on medical-grade polycarbonate surface. Biomaterials 2006, 27, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, A.J.; Scicluna, B.P.; Moerland, P.D.; Lin, J.; Jacobson, E.W.; Vlasuk, G.P.; van der Poll, T. The Selective Sirtuin 1 Activator SRT2104 Reduces Endotoxin-Induced Cytokine Release and Coagulation Activation in Humans. Crit. Care Med. 2015, 43, e199–e202. [Google Scholar] [CrossRef]
- Zullo, J.A.; Fan, J.; Azar, T.T.; Yen, W.; Zeng, M.; Chen, J.; Ratliff, B.B.; Song, J.; Tarbell, J.M.; Goligorsky, M.S.; et al. Exocytosis of Endothelial Lysosome-Related Organelles Hair-Triggers a Patchy Loss of Glycocalyx at the Onset of Sepsis. Am. J. Pathol. 2016, 186, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, D.; Song, J.W.; Zullo, J.; Lipphardt, M.; Coneh-Gould, L.; Goligorsky, M.S. Endothelial cell dysfunction and glycocalyx—A vicious circle. Matrix Biol. 2018, 71–72, 421–431. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masola, V.; Zaza, G.; Arduini, A.; Onisto, M.; Gambaro, G. Endothelial Glycocalyx as a Regulator of Fibrotic Processes. Int. J. Mol. Sci. 2021, 22, 2996. https://doi.org/10.3390/ijms22062996
Masola V, Zaza G, Arduini A, Onisto M, Gambaro G. Endothelial Glycocalyx as a Regulator of Fibrotic Processes. International Journal of Molecular Sciences. 2021; 22(6):2996. https://doi.org/10.3390/ijms22062996
Chicago/Turabian StyleMasola, Valentina, Gianluigi Zaza, Arduino Arduini, Maurizio Onisto, and Giovanni Gambaro. 2021. "Endothelial Glycocalyx as a Regulator of Fibrotic Processes" International Journal of Molecular Sciences 22, no. 6: 2996. https://doi.org/10.3390/ijms22062996
APA StyleMasola, V., Zaza, G., Arduini, A., Onisto, M., & Gambaro, G. (2021). Endothelial Glycocalyx as a Regulator of Fibrotic Processes. International Journal of Molecular Sciences, 22(6), 2996. https://doi.org/10.3390/ijms22062996