
Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Calmodulin—Ubiquitous Ca2+ Sensor in Neurons
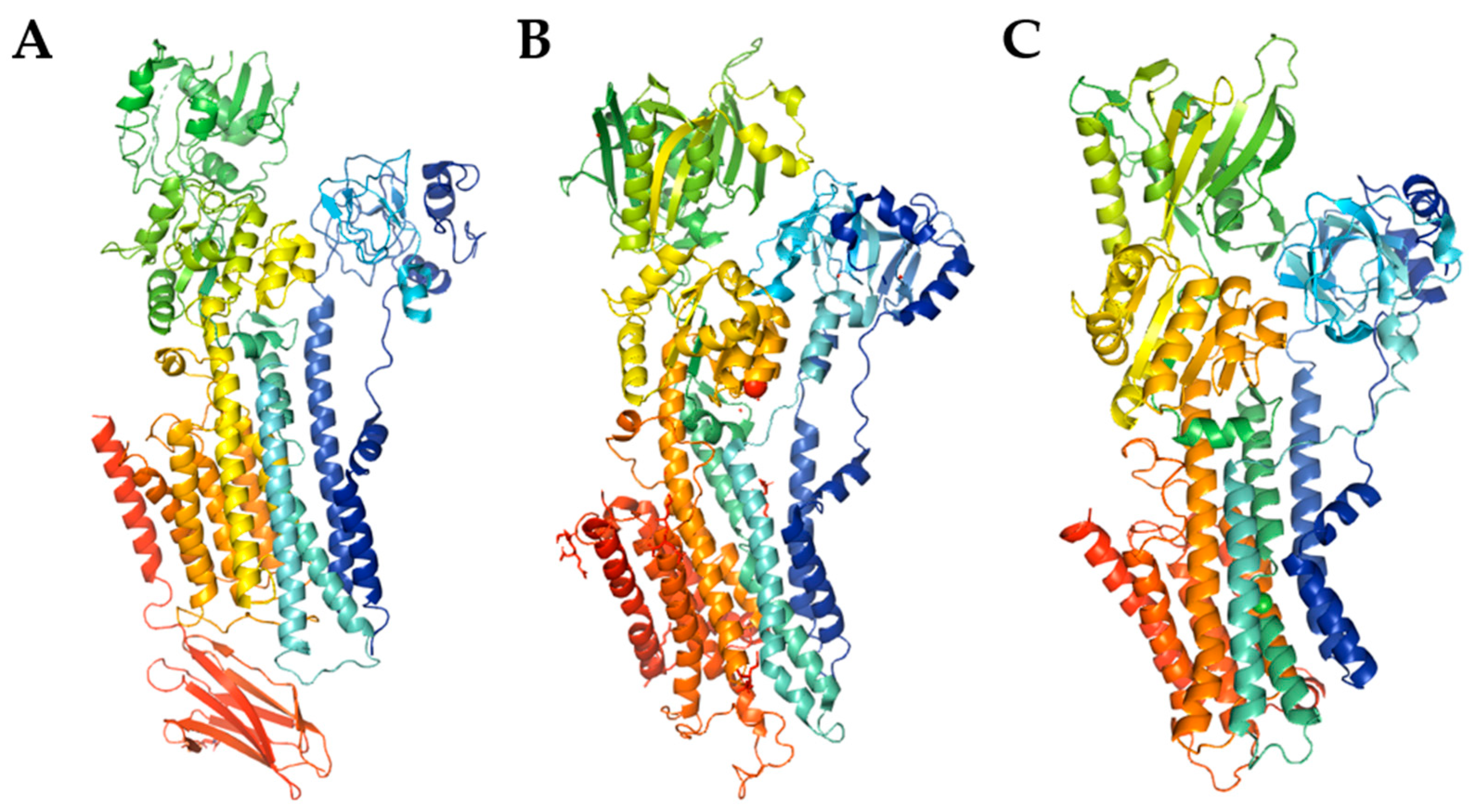
3. Plasma Membrane Ca2+-ATPase (PMCA)—The Only Calcium Pump Directly Regulated by Calmodulin
3.1. PMCA in Neuropathology
3.1.1. PMCA in Aging
3.1.2. PMCA in Alzheimer’s Disease
3.1.3. PMCA in Parkinson’s Disease
3.1.4. PMCA in Schizophrenia and Bipolar Disorder
3.1.5. PMCA in Cerebellar Disorders

{kind=link}
| Species | Mutation | Phenotype | Reference | |
|---|---|---|---|---|
| PMCA2 | Mouse | G283S (Dfw) | Vestibular/motor imbalance | [107] |
| Mouse | I655N (Elfin) | Ataxia | [113] | |
| Mouse | S877F (Obv) | Ataxia | [114] | |
| Mouse | E629K (Tmy) | Ataxia | [115] | |
| Mouse | E412K (Wri) | Abnormal movements | [116] | |
| Human | V1143F | Ataxia | [111] | |
| PMCA3 | Human | G1107D | Ataxia | [93] |
| Human | R482H | Ataxia | [97] | |
| Rat | R35C | Ataxia | [117] | |
| Human | G773R | Ataxia | [99] |
3.2. PMCA-Interacting Proteins in Mental Diseases
4. The Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA)
4.1. SERCA Pumps in Neuropathology
4.2. Calmodulin-Controlled Regulation of SERCA Pumps
5. Secretory Pathway Ca2+-ATPase (SPCA)—The Golgi-Resident Ca2+/Mn2+ Pump
5.1. SPCA Pumps in Neuropathology
5.2. Calmodulin-Controlled Regulation of Calcium in Golgi Apparatus
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Brini, M.; Carafoli, E. Calcium Pumps in Health and Disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Bonifas, J.M.; Beech, J.; Bench, G.; Shigihara, T.; Ogawa, H.; Ikeda, S.; Mauro, T.M.; Epstein, E.H., Jr. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat. Genet. 2000, 24, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Sudbrak, R.; Brown, J.M.; Dobson-Stone, C.; Carter, S.A.; Ramser, J.; White, J.; Healy, E.; Dissanayake, M.; Larrègue, M.; Perrussel, M.; et al. Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca2+ pump. Hum. Mol. Genet. 2000, 9, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Strehler, E.E.; Zacharias, D.A. Role of Alternative Splicing in Generating Isoform Diversity Among Plasma Membrane Calcium Pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Chi, X.; Ren, K.; Huang, G.; Zhou, G.; Yan, N.; Lei, J.; Zhou, Q. Structure of the human plasma membrane Ca. Nat. Commun. 2018, 9, 3623. [Google Scholar] [CrossRef]
- Toyoshima, C.; Iwasawa, S.; Ogawa, H.; Hirata, A.; Tsueda, J.; Inesi, G. Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nat. Cell Biol. 2013, 495, 260–264. [Google Scholar] [CrossRef]
- Friedberg, F.; Rhoads, A.R.; Friedberg, A.R. Evolutionary Aspects of Calmodulin. IUBMB Life 2001, 51, 215–221. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Ikura, M. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef]
- Vetter, S.W.; Leclerc, E. Novel aspects of calmodulin target recognition and activation. JBIC J. Biol. Inorg. Chem. 2003, 270, 404–414. [Google Scholar] [CrossRef]
- Jiang, X.; Lautermilch, N.J.; Watari, H.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc. Natl. Acad. Sci. USA 2007, 105, 341–346. [Google Scholar] [CrossRef]
- Kortvely, E.; Gulya, K. Calmodulin, and various ways to regulate its activity. Life Sci. 2004, 74, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A. The multifunctional role of phospho-calmodulin in pathophysiological processes. Biochem. J. 2018, 475, 4011–4023. [Google Scholar] [CrossRef]
- Cobb, J.A.; Roberts, D.M. Structural Requirements for N-Trimethylation of Lysine 115 of Calmodulin. J. Biol. Chem. 2000, 275, 18969–18975. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.R. Calmodulin methylation: Another layer of regulation in calcium signaling. Plant Cell 2013, 25, 4284. [Google Scholar] [CrossRef][Green Version]
- Thulin, E.; Andersson, A.; Drakenberg, T.; Forsén, S.; Vogel, H.J. Metal ion and drug binding to proteolytic fragments of calmodulin: Proteolytic cadmium-113 and proton nuclear magnetic resonance studies. Biochem. J. 1984, 23, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Grabarek, Z. Structural Basis for Diversity of the EF-hand Calcium-binding Proteins. J. Mol. Biol. 2006, 359, 509–525. [Google Scholar] [CrossRef]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix–loop–helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef]
- Kawasaki, H.; Soma, N.; Kretsinger, R.H. Molecular Dynamics Study of the Changes in Conformation of Calmodulin with Calcium Binding and/or Target Recognition. Sci. Rep. 2019, 9, 10688. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.H.; Brohus, M.; Nyegaard, M.; Overgaard, M.T. Human Calmodulin Mutations. Front. Mol. Neurosci. 2018, 11, 396. [Google Scholar] [CrossRef]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef] [PubMed]
- Mruk, K.; Farley, B.M.; Ritacco, A.W.; Kobertz, W.R. Calmodulation meta-analysis: Predicting calmodulin binding via canonical motif clustering. J. Gen. Physiol. 2014, 144, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bähler, M.; Rhoads, A. Calmodulin signaling via the IQ motif. FEBS Lett. 2001, 513, 107–113. [Google Scholar] [CrossRef]
- Biber, A.; Schmid, G.; Hempel, K. Calmodulin content in specific brain areas. Exp. Brain Res. 1984, 56, 323–326. [Google Scholar] [CrossRef]
- Calì, T.; Brini, M.; Carafoli, E. Regulation of Cell Calcium and Role of Plasma Membrane Calcium ATPases. Int. Rev. Cell Mol. Biol. 2017, 332, 259–296. [Google Scholar] [CrossRef]
- Schatzmann, H.J. ATP-dependent Ca+-Extrusion from human red cells. Cell. Mol. Life Sci. 1966, 22, 364–365. [Google Scholar] [CrossRef]
- Boczek, T.; Radzik, T.; Ferenc, B.; Zylinska, L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. Int. J. Mol. Sci. 2019, 20, 6338. [Google Scholar] [CrossRef]
- Guerini, D.; García-Martin, E.; Gerber, A.; Volbracht, C.; Leist, M.; Merino, C.G.; Carafoli, E. The Expression of Plasma Membrane Ca2+ Pump Isoforms in Cerebellar Granule Neurons Is Modulated by Ca2+. J. Biol. Chem. 1999, 274, 1667–1676. [Google Scholar] [CrossRef]
- Padányi, R.; Pászty, K.; Hegedűs, L.; Varga, K.; Papp, B.; Penniston, J.T.; Enyedi, Á. Multifaceted plasma membrane Ca2+ pumps: From structure to intracellular Ca2+ handling and cancer. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1863, 1351–1363. [Google Scholar] [CrossRef]
- Monteith, G.R.; Wanigasekara, Y.; Roufogalis, B.D. The plasma membrane calcium pump, its role and regulation: New complexities and possibilities. J. Pharmacol. Toxicol. Methods 1998, 40, 183–190. [Google Scholar] [CrossRef]
- Falchetto, R.; Vorherr, T.; Brunner, J.; Carafoli, E. The plasma membrane Ca2+ pump contains a site that interacts with its calmodulin-binding domain. J. Biol. Chem. 1991, 266, 2930–2936. [Google Scholar] [CrossRef]
- Burette, A.; Rockwood, J.M.; Strehler, E.E.; Weinberg, R.J. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J. Comp. Neurol. 2003, 467, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Eakin, T.J.; Antonelli, M.C.; Malchiodi, E.L.; Baskin, D.G.; Stahl, W.L. Localization of the plasma membrane Ca2+-ATPase isoform PMCA3 in rat cerebellum, choroid plexus and hippocampus. Mol. Brain Res. 1995, 29, 71–80. [Google Scholar] [CrossRef]
- Zacharias, D.; Kappen, C. Developmental expression of the four plasma membrane calcium ATPase (Pmca) genes in the mouse. Biochim. Biophys. Acta BBA Gen. Subj. 1999, 1428, 397–405. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Brandt, P.C.; Sisken, J.E.; Neve, R.L.; Vanaman, T.C. Blockade of plasma membrane calcium pumping ATPase isoform I impairs nerve growth factor-induced neurite extension in pheochromocytoma cells. Proc. Natl. Acad. Sci. USA 1996, 93, 13843–13848. [Google Scholar] [CrossRef] [PubMed]
- Szemraj, J.; Kawecka, I.; Bartkowiak, J.; Zylińska, L. The effect of antisense oligonucleotide treatment of plasma membrane Ca(+2)-ATPase in PC12 cells. Cell. Mol. Biol. Lett. 2004, 9, 451–464. [Google Scholar]
- Boczek, T.; Lisek, M.; Kowalski, A.; Pikula, S.; Niewiarowska, J.; Wiktorska, M.; Zylinska, L. Downregulation of PMCA2 or PMCA3 reorganizes Ca2+ handling systems in differentiating PC12 cells. Cell Calcium 2012, 52, 433–444. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Cross talk among PMCA, calcineurin and NFAT transcription factors in control of calmodulin gene expression in differentiating PC12 cells. Biochim. Biophys. Acta BBA Bioenerg. 2017, 1860, 502–515. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Kowalski, A.; Wiktorska, M.; Zylinska, L. Silencing of Plasma Membrane Ca2+-ATPase Isoforms 2 and 3 Impairs Energy Metabolism in Differentiating PC12 Cells. BioMed Res. Int. 2014, 2014, 735106. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Lisek, M.; Ferenc, B.; Kowalski, A.; Stepinski, D.; Wiktorska, M.; Zylinska, L. Plasma Membrane Ca2+-ATPase Isoforms Composition Regulates Cellular pH Homeostasis in Differentiating PC12 Cells in a Manner Dependent on Cytosolic Ca2+ Elevations. PLoS ONE 2014, 9, e102352. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Usachev, Y.M.; Thayer, S.A.; Strehler, E.E.; Windebank, A.J. Plasma membrane calcium ATPase plays a role in reducing Ca2+-mediated cytotoxicity in PC12 cells. J. Neurosci. Res. 2001, 64, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Usachev, Y.M.; Demarco, S.J.; Campbell, C.; Strehler, E.E.; Thayer, S.A. Bradykinin and ATP Accelerate Ca2+ Efflux from Rat Sensory Neurons via Protein Kinase C and the Plasma Membrane Ca2+ Pump Isoform 4. Neuron 2002, 33, 113–122. [Google Scholar] [CrossRef]
- Michaelis, M.; Johe, K.; Kitos, T. Age-dependent alterations in synaptic membrane systems for Ca2+ regulation. Mech. Ageing Dev. 1984, 25, 215–225. [Google Scholar] [CrossRef]
- Michaelis, M.; Bigelow, D.; Schöneich, C.; Williams, T.; Ramonda, L.; Yin, D.; Hühmer, A.; Yao, Y.; Gao, J.; Squier, T. Decreased plasma membrane calcium transport activity in aging brain. Life Sci. 1996, 59, 405–412. [Google Scholar] [CrossRef]
- Michaelis, M.L. Ca2+Handling Systems and Neuronal Aging. Ann. N. Y. Acad. Sci. 1989, 568, 89–94. [Google Scholar] [CrossRef]
- Zaidi, A.; Gao, J.; Squier, T.C.; Michaelis, M.L. Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol. Aging 1998, 19, 487–495. [Google Scholar] [CrossRef]
- McCarthy, M.R.; Thompson, A.R.; Nitu, F.; Moen, R.J.; Olenek, M.J.; Klein, J.C.; Thomas, D.D. Impact of methionine oxidation on calmodulin structural dynamics. Biochem. Biophys. Res. Commun. 2015, 456, 567–572. [Google Scholar] [CrossRef]
- Zaidi, A.; Barŕon, L.; Sharov, V.S.; Schöneich, C.; Michaelis, E.K.; Michaelis, M.L. Oxidative Inactivation of Purified Plasma Membrane Ca2+-ATPase by Hydrogen Peroxide and Protection by Calmodulin. Biochemistry 2003, 42, 12001–12010. [Google Scholar] [CrossRef]
- Zaidi, A.; Fernandes, D.; Bean, J.L.; Michaelis, M.L. Effects of paraquat-induced oxidative stress on the neuronal plasma membrane Ca2+-ATPase. Free. Radic. Biol. Med. 2009, 47, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Michaelis, M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free. Radic. Biol. Med. 1999, 27, 810–821. [Google Scholar] [CrossRef]
- Kip, S.N.; Strehler, E.E. Rapid Downregulation of NCX and PMCA in Hippocampal Neurons Following H2O2 Oxidative Stress. Ann. N. Y. Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fernandes, D.; Mehta, N.; Bean, J.L.; Michaelis, M.L.; Zaidi, A. Partitioning of the plasma membrane Ca2+-ATPase into lipid rafts in primary neurons: Effects of cholesterol depletion. J. Neurochem. 2007, 102, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Bechtel, M.D.; Galeva, N.A.; Williams, T.D.; Michaelis, E.K.; Michaelis, M.L. Decreases in plasma membrane Ca2+-ATPase in brain synaptic membrane rafts from aged rats. J. Neurochem. 2012, 123, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Corbacho, I.; Vázquez-Hernández, M.; Ávila, J.; Sepúlveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Marcos, D.; Sepúlveda, M.R.; Pérez, M.; Ávila, J.; Mata, A.M. Altered Ca2+ dependence of synaptosomal plasma membrane Ca2+ -ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid β-peptide to calmodulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Wong, L.; Bergeron, C.; Baimbridge, K.G. Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 171–179. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; Van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Lu, X.; Bernard, A.; Khrestchatisky, M.; Baudry, M. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem. 2008, 79, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Padilla, R.; Maccioni, R.; Avila, J. Calmodulin binds to a tubulin binding site of the microtubule-associated protein tau. Mol. Cell. Biochem. 1990, 97, 35–41. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wolff, J. Calmodulin binds to both microtubule-associated protein 2 and tau proteins. J. Biol. Chem. 1984, 259, 1226–1230. [Google Scholar] [CrossRef]
- Baudier, J.; Mochly-Rosen, D.; Newton, A.; Lee, S.H.; Koshland, D.E.; Cole, R.D. Comparison of S100b protein with calmodulin: Interactions with melittin and microtubule-associated. tau. proteins and inhibition of phosphorylation of. tau. proteins by protein kinase C. Biochem. J. 1987, 26, 2886–2893. [Google Scholar] [CrossRef]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef]
- Zaidi, A. Plasma membrane Ca2+-ATPases: Targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 2010, 1, 271–280. [Google Scholar] [CrossRef]
- Brendel, A.; Renziehausen, J.; Behl, C.; Hajieva, P. Downregulation of PMCA2 increases the vulnerability of midbrain neurons to mitochondrial complex I inhibition. NeuroToxicology 2014, 40, 43–51. [Google Scholar] [CrossRef]
- Mocko, J.B.; Kern, A.; Moosmann, B.; Behl, C.; Hajieva, P. Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 120–129. [Google Scholar] [CrossRef]
- Fernandes, D.; Zaidi, A.; Bean, J.; Hui, D.; Michaelis, M.L. RNAi- induced silencing of the plasma membrane Ca2+- ATPase 2 in neuronal cells: Effects on Ca2+ homeostasis and cell viability. J. Neurochem. 2007, 102, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Moeller, I.; Erdjument-Bromage, H.; Tempst, P.; Lauring, B. Parkinson’s Disease-associated α-Synuclein Is a Calmodulin Substrate. J. Biol. Chem. 2003, 278, 17379–17387. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, Y.; Wang, P.; Gu, L.; Wang, L.; Yang, H.; Wei, Q.; Li, Z.; Luo, J. The interaction between calcineurin and α-synuclein is regulated by calcium and calmodulin. Biochem. Biophys. Res. Commun. 2018, 496, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; Soste, M.; Cappelleti, V.; Fanning, S.; Van Rossum, D.B.; Whitesell, L.; Huang, Y.; Chung, C.Y.; Baru, V.; Zaichick, S.; et al. FKBP12 contributes to α-synuclein toxicity by regulating the calcineurin-dependent phosphoproteome. Proc. Natl. Acad. Sci. USA 2017, 114, E11313–E11322. [Google Scholar] [CrossRef]
- Holton, M.; Yang, D.; Wang, W.; Mohamed, T.M.; Neyses, L.; Armesilla, A.L. The interaction between endogenous calcineurin and the plasma membrane calcium-dependent ATPase is isoform specific in breast cancer cells. FEBS Lett. 2007, 581, 4115–4119. [Google Scholar] [CrossRef] [PubMed]
- Kosiorek, M.; Podszywalow-Bartnicka, P.; Zylinska, L.; Zablocki, K.; Pikula, S. Interaction of plasma membrane Ca2+-ATPase isoform 4 with calcineurin A: Implications for catecholamine secretion by PC12 cells. Biochem. Biophys. Res. Commun. 2011, 411, 235–240. [Google Scholar] [CrossRef]
- Boczek, T.; Ferenc, B.; Lisek, M.; Zylinska, L. Regulation of GAP43/calmodulin complex formation via calcineurin-dependent mechanism in differentiated PC12 cells with altered PMCA isoforms composition. Mol. Cell. Biochem. 2015, 407, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, 5. [Google Scholar] [CrossRef]
- Lidow, M.S. Calcium signaling dysfunction in schizophrenia: A unifying approach. Brain Res. Rev. 2003, 43, 70–84. [Google Scholar] [CrossRef]
- Kluge, H.; Kühne, G. Preliminary findings on calmodulin-stimulated Ca2+-ATPase of erythrocyte ghosts in psychotic patients. Eur. Arch. Psychiatry Clin. Neurosci. 1985, 235, 57–59. [Google Scholar] [CrossRef]
- Martins-De-Souza, D.; Gattaz, W.F.; Schmitt, A.; Rewerts, C.; Marangoni, S.; Novello, J.C.; Maccarrone, G.; Turck, C.W.; Dias-Neto, E. Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J. Neural. Transm. 2008, 116, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Föcking, M.; Lopez, L.M.; English, J.A.; Dicker, P.; Wolff, A.; Brindley, E.; Wynne, K.; Cagney, G.; Cotter, D.R. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol. Psychiatry 2014, 20, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.L.; Kassir, S.; Goodnick, P.J.; Fieve, R.R.; Chrisomalis, L.; Feliciano, M.; Szypula, D. Calmodulin-activated calcium ATPase in bipolar illness. Neuropsychobiology 1988, 20, 169–173. [Google Scholar] [CrossRef]
- Meltzer, H.L.; Kassir, S. Abnormal calmodulin-activated CaATPase in manic-depressive subjects. J. Psychiatr. Res. 1982, 17, 29–35. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Lisek, M.; Boczek, T.; Ferenc, B.; Zylinska, L. Regional brain dysregulation of Ca2+-handling systems in ketamine-induced rat model of experimental psychosis. Cell Tissue Res. 2015, 363, 609–620. [Google Scholar] [CrossRef]
- Lisek, M.; Ferenc, B.; Studzian, M.; Pulaski, L.; Guo, F.; Zylinska, L.; Boczek, T. Glutamate Deregulation in Ketamine-Induced Psychosis—A Potential Role of PSD95, NMDA Receptor and PMCA Interaction. Front. Cell. Neurosci. 2017, 11, 181. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Plasma membrane Ca2+-ATPase is a novel target for ketamine action. Biochem. Biophys. Res. Commun. 2015, 465, 312–317. [Google Scholar] [CrossRef]
- Adaikkan, C.; Taha, E.; Barrera, I.; David, O.; Rosenblum, K. Calcium/Calmodulin-Dependent Protein Kinase II and Eukaryotic Elongation Factor 2 Kinase Pathways Mediate the Antidepressant Action of Ketamine. Biol. Psychiatry 2018, 84, 65–75. [Google Scholar] [CrossRef]
- Xiao, Y.; Luo, H.; Yang, W.Z.; Zeng, Y.; Shen, Y.; Ni, X.; Shi, Z.; Zhong, J.; Liang, Z.; Fu, X.; et al. A Brain Signaling Framework for Stress-Induced Depression and Ketamine Treatment Elucidated by Phosphoproteomics. Front. Cell. Neurosci. 2020, 14, 48. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Mata, A.M. Localization of intracellular and plasma membrane Ca2+-ATPases in the cerebellum. Cerebellum 2005, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.E.; Sillitoe, R.V. Interactions between Purkinje Cells and Granule Cells Coordinate the Development of Functional Cerebellar Circuits. Neuroscience 2020. [Google Scholar] [CrossRef] [PubMed]
- Zanni, G.; Calì, T.; Kalscheuer, V.M.; Ottolini, D.; Barresi, S.; Lebrun, N.; Montecchi-Palazzi, L.; Hu, H.; Chelly, J.; Bertini, E.; et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 14514–14519. [Google Scholar] [CrossRef] [PubMed]
- Zanni, G.; Bertini, E.S. X-linked disorders with cerebellar dysgenesis. Orphanet J. Rare Dis. 2011, 6, 24. [Google Scholar] [CrossRef]
- Hsueh, Y.-P. Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann. Neurol. 2009, 66, 438–443. [Google Scholar] [CrossRef]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef]
- Calì, T.; Lopreiato, R.; Shimony, J.; Vineyard, M.; Frizzarin, M.; Zanni, G.; Zanotti, G.; Brini, M.; Shinawi, M.; Carafoli, E. A Novel Mutation in Isoform 3 of the Plasma Membrane Ca2+ Pump Impairs Cellular Ca2+ Homeostasis in a Patient with Cerebellar Ataxia and Laminin Subunit 1α Mutations. J. Biol. Chem. 2015, 290, 16132–16141. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Mosca, S.J.; Tétreault, M.; Dempsey, J.C.; Ishak, G.E.; Hartley, T.; Phelps, I.G.; Lamont, R.E.; O’Day, D.R.; Basel, D.; et al. Mutations in LAMA1 Cause Cerebellar Dysplasia and Cysts with and without Retinal Dystrophy. Am. J. Hum. Genet. 2014, 95, 227–234. [Google Scholar] [CrossRef]
- Vicario, M.; Calì, T.; Cieri, D.; Vallese, F.; Bortolotto, R.; Lopreiato, R.; Zonta, F.; Nardella, M.; Micalizzi, A.; Lefeber, D.J.; et al. A novel PMCA3 mutation in an ataxic patient with hypomorphic phosphomannomutase 2 (PMM2) heterozygote mutations: Biochemical characterization of the pump defect. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 3303–3312. [Google Scholar] [CrossRef]
- Westphal, V.; Peterson, S.; Patterson, M.; Tournay, A.; Blumenthal, A.; Treacy, E.P.; Freeze, H.H. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet. Med. 2001, 3, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Briones, P.; Vilaseca, M.A.; Schollen, E.; Ferrer, I.; Maties, M.; Busquets, C.; Artuch, R.; Gort, L.; Marco, M.; Van Schaftingen, E.; et al. Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J. Inherit. Metab. Dis. 2003, 25, 635–646. [Google Scholar] [CrossRef]
- Andreotti, G.; de Vaca, I.C.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef]
- Bortolozzi, M.; Mammano, F. PMCA2 pump mutations and hereditary deafness. Neurosci. Lett. 2018, 663, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; De Mario, A.; Primerano, S.; Brini, M.; Carafoli, E. Hair cells, plasma membrane Ca2+ ATPase and deafness. Int. J. Biochem. Cell Biol. 2012, 44, 679–683. [Google Scholar] [CrossRef]
- Carafoli, E. The plasma membrane calcium pump in the hearing process: Physiology and pathology. Sci. China Life Sci. 2011, 54, 686–690. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kozel, P.J.; Friedman, R.A.; Erway, L.C.; Yamoah, E.N.; Liu, L.H.; Riddle, T.; Duffy, J.J.; Doetschman, T.; Miller, M.L.; Cardell, E.L.; et al. Balance and Hearing Deficits in Mice with a Null Mutation in the Gene Encoding Plasma Membrane Ca2+-ATPase Isoform 2. J. Biol. Chem. 1998, 273, 18693–18696. [Google Scholar] [CrossRef] [PubMed]
- Street, V.A.; McKee-Johnson, J.W.; Fonseca, R.C.; Tempel, B.L.; Noben-Trauth, K. Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat. Genet. 1998, 19, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Matsumura, Y.; Inoue, K.; Ichikawa, R.; Takayama, C. Abnormal synaptic architecture in the cerebellar cortex of a new dystonic mutant mouse, Wriggle Mouse Sagami. Neurosci. Res. 1993, 16, 39–48. [Google Scholar] [CrossRef]
- Ueno, T.; Kameyama, K.; Hirata, M.; Ogawa, M.; Hatsuse, H.; Takagaki, Y.; Ohmura, M.; Osawa, N.; Kudo, Y. A mouse with a point mutation in plasma membrane Ca2+-ATPase isoform 2 gene showed the reduced Ca2+ influx in cerebellar neurons. Neurosci. Res. 2002, 42, 287–297. [Google Scholar] [CrossRef]
- Empson, R.M.; Turner, P.R.; Nagaraja, R.Y.; Beesley, P.W.; Knopfel, T. Reduced expression of the Ca2+transporter protein PMCA2 slows Ca2+dynamics in mouse cerebellar Purkinje neurones and alters the precision of motor coordination. J. Physiol. 2010, 588, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Zanni, G.; Vallese, F.; Santorelli, F.; Grinzato, A.; Cieri, D.; Berto, P.; Frizzarin, M.; Lopreiato, R.; Zonta, F.; et al. A V1143F mutation in the neuronal-enriched isoform 2 of the PMCA pump is linked with ataxia. Neurobiol. Dis. 2018, 115, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E.; Calì, T. The plasma membrane calcium pumps: Focus on the role in (neuro)pathology. Biochem. Biophys. Res. Commun. 2017, 483, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Z.; Xiong, X.; Gu, X.; Gao, X.; Gao, X. Identification of a novel point mutation of mouse Atp2b2 induced by N-ethyl-N-nitrosourea mutagenesis. Exp. Anim. 2011, 60, 71–78. [Google Scholar] [CrossRef]
- Spiden, S.L.; Bortolozzi, M.; Di Leva, F.; De Angelis, M.H.; Fuchs, H.; Lim, D.; Ortolano, S.; Ingham, N.J.; Brini, M.; Carafoli, E.; et al. The Novel Mouse Mutation Oblivion Inactivates the PMCA2 Pump and Causes Progressive Hearing Loss. PLoS Genet. 2008, 4, e1000238. [Google Scholar] [CrossRef]
- Bortolozzi, M.; Brini, M.; Parkinson, N.; Crispino, G.; Scimemi, P.; De Siati, R.D.; Di Leva, F.; Parker, A.; Ortolano, S.; Arslan, E.; et al. The Novel PMCA2 Pump Mutation Tommy Impairs Cytosolic Calcium Clearance in Hair Cells and Links to Deafness in Mice. J. Biol. Chem. 2010, 285, 37693–37703. [Google Scholar] [CrossRef]
- Takahashi, K.; Kitamura, K. A Point Mutation in a Plasma Membrane Ca2+-ATPase Gene Causes Deafness in Wriggle Mouse Sagami. Biochem. Biophys. Res. Commun. 1999, 261, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, K.P.; Paul, S.; Cali’, T.; Lopreiato, R.; Karan, S.; Frizzarin, M.; Ames, D.; Zanni, G.; Brini, M.; Dansithong, W.; et al. Spontaneous shaker rat mutant—A new model for X-linked tremor/ataxia. Dis. Model. Mech. 2016, 9, 553–562. [Google Scholar] [CrossRef]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The Plasma Membrane Calcium Pump: New Ways to Look at an Old Enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [PubMed]
- Lisek, M.; Boczek, T.; Zylinska, L. Calcium as a Trojan horse in mental diseases—The role of PMCA and PMCA-interacting proteins in bipolar disorder and schizophrenia. Neurosci. Lett. 2018, 663, 48–54. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nat. Cell Biol. 2014, 506, 179–184. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.-J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.J.; Mielnik, C.A.; Koene, R.; Newburn, E.; Ramsey, A.J.; Lipska, B.K.; McCullumsmith, R.E. Postsynaptic Density-95 Isoform Abnormalities in Schizophrenia. Schizophr. Bull. 2017, 43, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Garside, M.L.; Turner, P.R.; Austen, B.; Strehler, E.E.; Beesley, P.W.; Empson, R.M. Molecular interactions of the plasma membrane calcium ATPase 2 at pre- and post-synaptic sites in rat cerebellum. Neuroscience 2009, 162, 383–395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamamoto, H.; Hagino, Y.; Kasai, S.; Ikeda, K. Specific Roles of NMDA Receptor Subunits in Mental Disorders. Curr. Mol. Med. 2015, 15, 193–205. [Google Scholar] [CrossRef]
- Strehler, E.E. Plasma Membrane Calcium ATPases as Novel Candidates for Therapeutic Agent Development. J. Pharm. Pharm. Sci. 2013, 16, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-G.; Zhu, X.-H.; Nemes, A.D.; Zhu, D.-Y. Neuronal nitric oxide synthase and affective disorders. IBRO Rep. 2018, 5, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Nitric Oxide in Health and Disease of the Nervous System. Antioxid. Redox Signal. 2009, 11, 541–553. [Google Scholar] [CrossRef]
- Becker, M.; Mastropasqua, F.; Reising, J.P.; Maier, S.; Ho, M.-L.; Rabkina, I.; Li, D.; Neufeld, J.; Ballenberger, L.; Myers, L.; et al. Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl. Psychiatry 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Voit, T.; Kramer, H.; Thomas, C.; Wechsler, W.; Reichmann, H.; Lenard, H.G. Mypopathy in Williams-Beuren syndrome. Eur. J. Nucl. Med. Mol. Imaging 1991, 150, 521–526. [Google Scholar] [CrossRef]
- Tarpey, P.; Parnau, J.; Blow, M.; Woffendin, H.; Bignell, G.; Cox, C.; Cox, J.; Davies, H.; Edkins, S.; Holden, S.; et al. Mutations in the DLG3 Gene Cause Nonsyndromic X-Linked Mental Retardation. Am. J. Hum. Genet. 2004, 75, 318–324. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Meador-Woodruff, J.H. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr. Res. 2005, 78, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Marcello, E.; Di Luca, M. Postsynaptic density–membrane associated guanylate kinase proteins (PSD–MAGUKs) and their role in CNS disorders. Neuroscience 2009, 158, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Clifton, N.E.; Trent, S.; Thomas, K.L.; Hall, J. Regulation and Function of Activity-Dependent Homer in Synaptic Plasticity. Mol. Neuropsychiatry 2019, 5, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, T.; Leiter, L.M.; Gerber, D.J.; Gainetdinov, R.R.; Sotnikova, T.D.; Zeng, H.; Caron, M.G.; Tonegawa, S. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. USA 2003, 100, 8987–8992. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Kimaro, W.H.; Etet, P.F.S. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef]
- Mathieu, F.; Miot, S.; Etain, B.; El Khoury, M.-A.; Chevalier, F.; Bellivier, F.; Leboyer, M.; Giros, B.; Tzavara, E.T. Association between the PPP3CC gene, coding for the calcineurin gamma catalytic subunit, and bipolar disorder. Behav. Brain Funct. 2008, 4, 2. [Google Scholar] [CrossRef]
- Donohoe, G.; Walters, J.; Morris, D.W.; Quinn, E.M.; Judge, R.; Norton, N.; Giegling, I.; Hartmann, A.M.; Möller, H.-J.; Muglia, P.; et al. Influence of NOS1 on Verbal Intelligence and Working Memory in Both Patients with Schizophrenia and Healthy Control Subjects. Arch. Gen. Psychiatry 2009, 66, 1045–1054. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Kurinnaia, O.S.; Yurov, Y.B. An Interstitial 20q11.21 Microdeletion Causing Mild Intellectual Disability and Facial Dysmorphisms. Case Rep. Genet. 2013, 2013, 353028. [Google Scholar] [CrossRef]
- Foote, M.; Zhou, Y. 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Boil. 2012, 3, 152–164. [Google Scholar]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Brandl, C.J.; DeLeon, S.; Martin, D.R.; MacLennan, D.H. Adult forms of the Ca2+ATPase of sarcoplasmic reticulum. Expression in developing skeletal muscle. J. Biol. Chem. 1987, 262, 3768–3774. [Google Scholar] [CrossRef]
- Verkhratsky, A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Hidalgo-Sánchez, M.; Mata, A.M. Localization of endoplasmic reticulum and plasma membrane Ca2+-ATPases in subcellular fractions and sections of pig cerebellum. Eur. J. Neurosci. 2004, 19, 542–551. [Google Scholar] [CrossRef]
- Morita, M.; Kudo, Y. Growth factors upregulate astrocyte [Ca2+]i oscillation by increasing SERCA2b expression. Glia 2010, 58, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.C.; Witkovsky, P.; Avshalumov, M.V.; Rice, M.E. Mobilization of Calcium from Intracellular Stores Facilitates Somatodendritic Dopamine Release. J. Neurosci. 2009, 29, 6568–6579. [Google Scholar] [CrossRef] [PubMed]
- Arredouani, A.; Guiot, Y.; Jonas, J.-C.; Liu, L.H.; Nenquin, M.; Pertusa, J.A.; Rahier, J.; Rolland, J.-F.; Shull, G.E.; Stevens, M.; et al. SERCA3 Ablation Does Not Impair Insulin Secretion but Suggests Distinct Roles of Different Sarcoendoplasmic Reticulum Ca2+ Pumps for Ca2+ Homeostasis in Pancreatic -cells. Diabetes 2002, 51, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Wuytack, F.; Raeymaekers, L.; De Smedt, H.; Eggermont, J.A.; Missiaen, L.; Bosch, L.V.D.; De Jaegere, S.; Verboomen, H.; Plessers, L.; Casteels, R. Ca2+-Transport ATPases and Their Regulation in Muscle and Brain. Ann. N. Y. Acad. Sci. 1992, 671, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wuytack, F.; Dode, L.; Baba-Aissa, F.; Raeymaekers, L. The SERCA3-type of organellar Ca2+pumps. Biosci. Rep. 1995, 15, 299–306. [Google Scholar] [CrossRef]
- Campbell, A.M.; Wuytack, F.; Fambrough, D.M. Differential distribution of the alternative forms of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase, SERCA2b and SERCA2a, in the avian brain. Brain Res. 1993, 605, 67–76. [Google Scholar] [CrossRef]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; De Greef, C.; Missiaen, L.; Casteels, R. Distribution of the organellar Ca2+ transport ATPase SERCA2 isoforms in the cat brain. Brain Res. 1996, 743, 141–153. [Google Scholar] [CrossRef]
- Salvador, J.M.; Berengena, M.; Sepúlveda, M.R.; Mata, A.M. Distribution of the intracellular Ca(2+)-ATPase isoform 2b in pig brain subcellular fractions and cross-reaction with a monoclonal antibody raised against the enzyme isoform. J. Biochem. 2001, 129, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Britzolaki, A.; Saurine, J.; Flaherty, E.; Thelen, C.; Pitychoutis, P.M. The SERCA2: A Gatekeeper of Neuronal Calcium Homeostasis in the Brain. Cell. Mol. Neurobiol. 2018, 38, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. Adv. Exp. Med. Biol. 2020, 1131, 131–161. [Google Scholar] [CrossRef]
- Chami, M.; Oulès, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Bréchot, P. Role of SERCA1 Truncated Isoform in the Proapoptotic Calcium Transfer from ER to Mitochondria during ER Stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Bussiere, R.; Oulès, B.; Mary, A.; Vaillant-Beuchot, L.; Martin, C.; El Manaa, W.; Vallée, D.; Duplan, E.; Paterlini-Bréchot, P.; Da Costa, C.A.; et al. Upregulation of the Sarco-Endoplasmic Reticulum Calcium ATPase 1 Truncated Isoform Plays a Pathogenic Role in Alzheimer’s Disease. Cells 2019, 8, 1539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bruce, A.T.; Tu, C.; Ma, K.; Zeng, L.; Zheng, P.; Liu, Y.; Liu, Y. Protein aggregation of SERCA2 mutants associated with Darier disease elicits ER stress and apoptosis in keratinocytes. J. Cell Sci. 2011, 124, 3568–3580. [Google Scholar] [CrossRef]
- Wang, S.-L.; Yang, S.-F.; Chen, C.-C.; Tsai, P.-T.; Chai, C.-Y. Darier’s disease associated with bipolar affective disorder: A case report. Kaohsiung J. Med. Sci. 2002, 18, 622–626. [Google Scholar]
- Nakamura, T.; Kazuno, A.; Nakajima, K.; Kusumi, I.; Tsuboi, T.; Kato, T. Loss of function mutations in ATP2A2 and psychoses: A case report and literature survey. Psychiatry Clin. Neurosci. 2016, 70, 342–350. [Google Scholar] [CrossRef]
- Gordon-Smith, K.; Green, E.; Grozeva, D.; Tavadia, S.; Craddock, N.; Jones, L. Genotype-phenotype correlations in Darier disease: A focus on the neuropsychiatric phenotype. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, e32679. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Sugiura, K.; Nakamura, Y.; Fujio, Y.; Konohana, I.; Akiyama, M. Darier’s Disease Complicated by Schizophrenia Caused by a Novel ATP2A2 Mutation. Acta Derm. Venereol. 2016, 96, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Simmerman, H.K.B.; Jones, L.R. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. [Google Scholar] [CrossRef]
- Shaikh, S.A.; Sahoo, S.K.; Periasamy, M. Phospholamban and sarcolipin: Are they functionally redundant or distinct regulators of the Sarco(Endo)Plasmic Reticulum Calcium ATPase? J. Mol. Cell. Cardiol. 2016, 91, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of Cardiac Contractility by the Phopholamban/SERCA2a Regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ Pumps of the Endoplasmic Reticulum and Golgi Apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef] [PubMed]
- Stammers, A.N.; Susser, S.E.; Hamm, N.C.; Hlynsky, M.W.; Kimber, D.E.; Kehler, D.S.; Duhamel, T.A. The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 2015, 93, 843–854. [Google Scholar] [CrossRef]
- Münch, G.; Bölck, B.; Karczewski, P.; Schwinger, R.H. Evidence for Calcineurin-mediated Regulation of SERCA 2a Activity in Human Myocardium. J. Mol. Cell. Cardiol. 2002, 34, 321–334. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood–Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 570045. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive Glia Inflammatory Signaling Pathways and Epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. α-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.-H.; Kim, H.; Park, S.M.; Joe, E.-H.; Jou, I. Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol. Commun. 2019, 7, 68. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Pu, J. Leucine-Rich Repeat Kinase 2 in Parkinson’s Disease: Updated from Pathogenesis to Potential Therapeutic Target. Eur. Neurol. 2018, 79, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 pathobiology in Parkinson’s disease. J. Neurochem. 2014, 131, 554–565. [Google Scholar] [CrossRef]
- Mandal, D.; Rulli, S.J.; Rao, R. Packing Interactions between Transmembrane Helices Alter Ion Selectivity of the Yeast Golgi Ca2+/Mn2+-ATPase PMR1. J. Biol. Chem. 2003, 278, 35292–35298. [Google Scholar] [CrossRef]
- Xiang, M.; Mohamalawari, D.; Rao, R. A Novel Isoform of the Secretory Pathway Ca2+,Mn2+-ATPase, hSPCA2, Has Unusual Properties and Is Expressed in the Brain. J. Biol. Chem. 2005, 280, 11608–11614. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Aschner, M. Manganese toxicity in the central nervous system: The glutamine/glutamate-γ-aminobutyric acid cycle. J. Intern. Med. 2013, 273, 466–477. [Google Scholar] [CrossRef]
- Van Baelen, K.; Dode, L.; Vanoevelen, J.; Callewaert, G.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Raeymaekers, L.; Wuytack, F. The Ca2+/Mn2+ pumps in the Golgi apparatus. Biochim. Biophys. Acta BBA Bioenerg. 2004, 1742, 103–112. [Google Scholar] [CrossRef]
- Wootton, L.L.; Argent, C.C.; Wheatley, M.; Michelangeli, F. The expression, activity and localisation of the secretory pathway Ca2+-ATPase (SPCA1) in different mammalian tissues. Biochim. Biophys. Acta BBA Biomembr. 2004, 1664, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.R.; Vanoevelen, J.; Raeymaekers, L.; Mata, A.M.; Wuytack, F. Silencing the SPCA1 (Secretory Pathway Ca2+-ATPase Isoform 1) Impairs Ca2+ Homeostasis in the Golgi and Disturbs Neural Polarity. J. Neurosci. 2009, 29, 12174–12182. [Google Scholar] [CrossRef] [PubMed]
- Murín, R.; Verleysdonk, S.; Raeymaekers, L.; Kaplan, P.; Lehotský, J. Distribution of Secretory Pathway Ca2+ ATPase (SPCA1) in Neuronal and Glial Cell Cultures. Cell. Mol. Neurobiol. 2006, 26, 1353–1363. [Google Scholar] [CrossRef]
- Vanoevelen, J.; Dode, L.; Van Baelen, K.; Fairclough, R.J.; Missiaen, L.; Raeymaekers, L.; Wuytack, F. The Secretory Pathway Ca2+/Mn2+-ATPase 2 Is a Golgi-localized Pump with High Affinity for Ca2+ Ions. J. Biol. Chem. 2005, 280, 22800–22808. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.R.; Berrocal, M.; Marcos, D.; Wuytack, F.; Mata, A.M. Functional and immunocytochemical evidence for the expression and localization of the secretory pathway Ca2+-ATPase isoform 1 (SPCA1) in cerebellum relative to other Ca2+pumps. J. Neurochem. 2007, 103, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Micaroni, M.; Giacchetti, G.; Plebani, R.; Xiao, G.G.; Federici, L. ATP2C1 gene mutations in Hailey–Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016, 7, e2259. [Google Scholar] [CrossRef]
- Chen, J.; Smaardijk, S.; Mattelaer, C.-A.; Pamula, F.; Vandecaetsbeek, I.; Vanoevelen, J.; Wuytack, F.; Lescrinier, E.; Eggermont, J.; Vangheluwe, P. An N-terminal Ca2+-binding motif regulates the secretory pathway Ca2+/Mn2+-transport ATPase SPCA1. J. Biol. Chem. 2019, 294, 7878–7891. [Google Scholar] [CrossRef]
- Laude, A.J.; Simpson, A.W.M. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Missiaen, L.; Dode, L.; Vanoevelen, J.; Raeymaekers, L.; Wuytack, F. Calcium in the Golgi apparatus. Cell Calcium 2007, 41, 405–416. [Google Scholar] [CrossRef]
- Brunskill, E.W.; Potter, A.S.; Distasio, A.; Dexheimer, P.; Plassard, A.; Aronow, B.J.; Potter, S.S. A gene expression atlas of early craniofacial development. Dev. Biol. 2014, 391, 133–146. [Google Scholar] [CrossRef]
- Ramos-Castañeda, J.; Park, Y.-N.; Liu, M.; Hauser, K.; Rudolph, H.; Shull, G.E.; Jonkman, M.F.; Mori, K.; Ikeda, S.; Ogawa, H.; et al. Deficiency of ATP2C1, a Golgi Ion Pump, Induces Secretory Pathway Defects in Endoplasmic Reticulum (ER)-associated Degradation and Sensitivity to ER Stress. J. Biol. Chem. 2005, 280, 9467–9473. [Google Scholar] [CrossRef]
- Brown, J.M.; García-García, M.J. Secretory pathway calcium ATPase 1 (SPCA1) controls mouse neural tube closure by regulating cytoskeletal dynamics. Development 2018, 145, dev170019. [Google Scholar] [CrossRef]
- Okunade, G.W.; Miller, M.L.; Azhar, M.; Andringa, A.; Sanford, L.P.; Doetschman, T.; Prasad, V.; Shull, G.E. Loss of the Atp2c1 Secretory Pathway Ca2+-ATPase (SPCA1) in Mice Causes Golgi Stress, Apoptosis, and Midgestational Death in Homozygous Embryos and Squamous Cell Tumors in Adult Heterozygotes. J. Biol. Chem. 2007, 282, 26517–26527. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hu, Z.; Zeng, L.; Lu, W.; Tang, X.; Zhang, J.; Li, T. Golgi apparatus and neurodegenerative diseases. Int. J. Dev. Neurosci. 2008, 26, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Lehotský, J.; Racay, P.; Pavlíková, M.; Tatarková, Z.; Urban, P.; Chomová, M.; Kovalská, M.; Kaplán, P. Cross-talk of intracellular calcium stores in the response to neuronal ischemia and ischemic tolerance. Gen. Physiol. Biophys. 2009, 28, 104–114. [Google Scholar]
- Hu, Z.-P.; Li, L.-H.; Tian, X.-R. The key target of neuroprotection after the onset of ischemic stroke: Secretory pathway Ca2+-ATPase 1. Neural Regen. Res. 2015, 10, 1271–1278. [Google Scholar] [CrossRef]
- Pavlíková, M.; Tatarková, Z.; Sivoňová, M.; Kaplan, P.; Križanová, O.; Lehotský, J. Alterations Induced by Ischemic Preconditioning on Secretory Pathways Ca2+-ATPase (SPCA) Gene Expression and Oxidative Damage After Global Cerebral Ischemia/Reperfusion in Rats. Cell. Mol. Neurobiol. 2009, 29, 909–916. [Google Scholar] [CrossRef]
- Lu, T.; Hu, Z.; Zeng, L.; Jiang, Z. Changes in secretory pathway Ca2+-ATPase 2 following focal cerebral ischemia/reperfusion injury. Neural Regen. Res. 2013, 8, 76–82. [Google Scholar]
- Olanow, C.W. Manganese-Induced Parkinsonism and Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2004, 1012, 209–223. [Google Scholar] [CrossRef]
- Aschner, M.; Erikson, K.M.; Hernández, E.H.; Tjalkens, R. Manganese and its Role in Parkinson’s Disease: From Transport to Neuropathology. Neuro Mol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, M.R.; Wuytack, F.; Mata, A.M. High levels of Mn2+inhibit secretory pathway Ca2+/Mn2+-ATPase (SPCA) activity and cause Golgi fragmentation in neurons and glia. J. Neurochem. 2012, 123, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Aschner, M.; Guilarte, T.R.; Dydak, U.; Criswell, S.R.; Zheng, W. Pathophysiology of manganese-associated neurotoxicity. NeuroToxicology 2012, 33, 881–886. [Google Scholar] [CrossRef]
- Gubert, P.; Boas, G.R.V.; Paes, M.M.; Santamaría, A.; Lee, E.; Tinkov, A.A.; Bowman, A.B.; Aschner, M. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert Rev. Neurother. 2020, 20, 1109–1121. [Google Scholar] [CrossRef]
- Tong, Y.; Yang, H.; Tian, X.; Wang, H.; Zhou, T.; Zhang, S.; Yu, J.; Zhang, T.; Fan, D.; Guo, X.; et al. High Manganese, A Risk for Alzheimer’s Disease: High Manganese Induces Amyloid-β Related Cognitive Impairment. J. Alzheimers Dis. 2014, 42, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; Morley, J.W.; Buskila, Y. Neuromodulation of Glial Function During Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 278. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Perea, G.; Sur, M.; Araque, A. Neuron-glia networks: Integral gear of brain function. Front. Cell. Neurosci. 2014, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte–neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Caudal, L.C.; Gobbo, D.; Scheller, A.; Kirchhoff, F. The Paradox of Astroglial Ca2+ Signals at the Interface of Excitation and Inhibition. Front. Cell. Neurosci. 2020, 14, 609947. [Google Scholar] [CrossRef]
- Porasuphatana, S.; Tsai, P.; Rosen, G.M. The generation of free radicals by nitric oxide synthase. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2003, 134, 281–289. [Google Scholar] [CrossRef]
- Moreno, J.A.; Sullivan, K.A.; Carbone, D.L.; Hanneman, W.H.; Tjalkens, R.B. Manganese potentiates nuclear factor-κB-dependent expression of nitric oxide synthase 2 in astrocytes by activating soluble guanylate cyclase and extracellular responsive kinase signaling pathways. J. Neurosci. Res. 2008, 86, 2028–2038. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.Y.; Caballero, A.; Dec, A.; Cass, D.K.; Simak, N.; Sunu, E.; Park, M.J.; Blume, S.R.; Sammut, S.; Park, D.J.; et al. Inhibition of Striatal Soluble Guanylyl Cyclase-cGMP Signaling Reverses Basal Ganglia Dysfunction and Akinesia in Experimental Parkinsonism. PLoS ONE 2011, 6, e27187. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. [Google Scholar] [CrossRef]
- Deng, S.; Liu, H.; Qiu, K.; You, H.; Lei, Q.; Lu, W. Role of the Golgi Apparatus in the Blood-Brain Barrier: Golgi Protection May Be a Targeted Therapy for Neurological Diseases. Mol. Neurobiol. 2017, 55, 4788–4801. [Google Scholar] [CrossRef]
- Parys, J.B.; Vervliet, T. New Insights in the IP3 Receptor and Its Regulation. Calcium Signal. 2020, 1131, 243–270. [Google Scholar]
- Kesherwani, V.; Agrawal, S.K. Regulation of Inositol 1,4,5-triphosphate receptor, type 1 (IP3R1) in hypoxic/reperfusion injury of white matter. Neurol. Res. 2012, 34, 504–511. [Google Scholar] [CrossRef]
- Seo, M.-D.; Enomoto, M.; Ishiyama, N.; Stathopulos, P.B.; Ikura, M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1853, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef] [PubMed]

| PMCA1 | PMCA2 | PMCA3 | PMCA4 | |
|---|---|---|---|---|
| Tissue Distribution | Ubiquitous | Restricted (brain) | Restricted (brain) | Ubiquitous |
| Developmental Expression/Switch | Isoform switch fetal/adult | Isoform switch fetal/adult | Isoform switch fetal/adult | Isoform switch fetal/adult |
| Affinity CaM (Kd nM) | 40–50 | 2–4 | 8 | 3–40 |
| A Protein Interacting with PMCA | Associated Disease | PMCA Domain Involved in the Interaction | A Protein Domain Involved in the Interaction | Functional Importance of the Interaction |
|---|---|---|---|---|
| NOS | AD, BP, MDD, ALS, anxiety, stroke, HD [126,127] | PDZ-domain binding sequence | PDZ domain | Decline in NOS activity, down-regulation of NO production |
| CASK | Microcephaly with pontine and cerebellar hypoplasia, X-linked intellectual disability, ASD [128] | PDZ-domain binding sequence | PDZ domain | Down-regulation the T- dependent transcriptional activity |
| CLP36 | Williams-Beuren syndrome [129] | PDZ-domain binding sequence | PDZ domain | Pump translocation during platelet activation |
| MAGUK | AD, PD, stroke, X-linked mental retardation, BD, MDD, SZ [130,131,132] | PDZ-domain binding sequence | PDZ domain | Biding enables the localization of PMCAs in specific membrane domains and local control of Ca2+ concentration |
| Ania3/Homer | SZ, ASD, MDD, suicide attempt, cocaine dependence, opiate abuse [133] | PDZ-domain binding sequence | PDZ domain | Stabilization of PMCA in domains near the sites of calcium influx into the cell |
| Calcineurin | AD, HD, SZ, PD, ALS, BD epilepsy [134,135,136] | catalytic domain | Amino acids 58–143 | Inhibition of the phosphatase activity of calcineurin, decrease in the activity of the transcription factor NFAT |
| Syntrophin α1 | SZ, mild intellectual disability [137,138] | catalytic domain | Amino acids 399–447 | The formation of a triple complex with PMCA and NOS-1 inhibits the production of NO |
| ε 14-3-3 | AD, BP, PD, SZ [139] | the N-terminal region | Amino acids 2–92 | Inhibition of PMCA activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczek, T.; Sobolczyk, M.; Mackiewicz, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases. Int. J. Mol. Sci. 2021, 22, 2785. https://doi.org/10.3390/ijms22062785
Boczek T, Sobolczyk M, Mackiewicz J, Lisek M, Ferenc B, Guo F, Zylinska L. Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases. International Journal of Molecular Sciences. 2021; 22(6):2785. https://doi.org/10.3390/ijms22062785
Chicago/Turabian StyleBoczek, Tomasz, Marta Sobolczyk, Joanna Mackiewicz, Malwina Lisek, Bozena Ferenc, Feng Guo, and Ludmila Zylinska. 2021. "Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases" International Journal of Molecular Sciences 22, no. 6: 2785. https://doi.org/10.3390/ijms22062785
APA StyleBoczek, T., Sobolczyk, M., Mackiewicz, J., Lisek, M., Ferenc, B., Guo, F., & Zylinska, L. (2021). Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases. International Journal of Molecular Sciences, 22(6), 2785. https://doi.org/10.3390/ijms22062785