
Calmodulin and Its Binding Proteins in Parkinson’s Disease



Abstract
1. Introduction
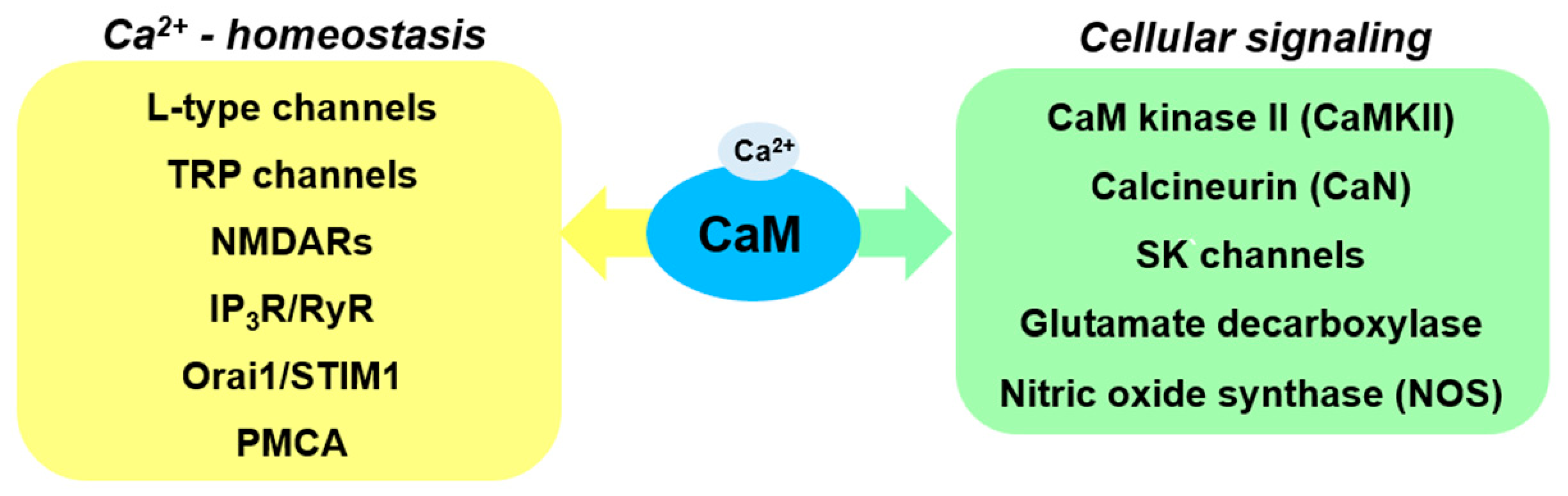
2. Ca2+ Signaling and CaM
3. CaM-Regulated Ca2+ Homeostasis
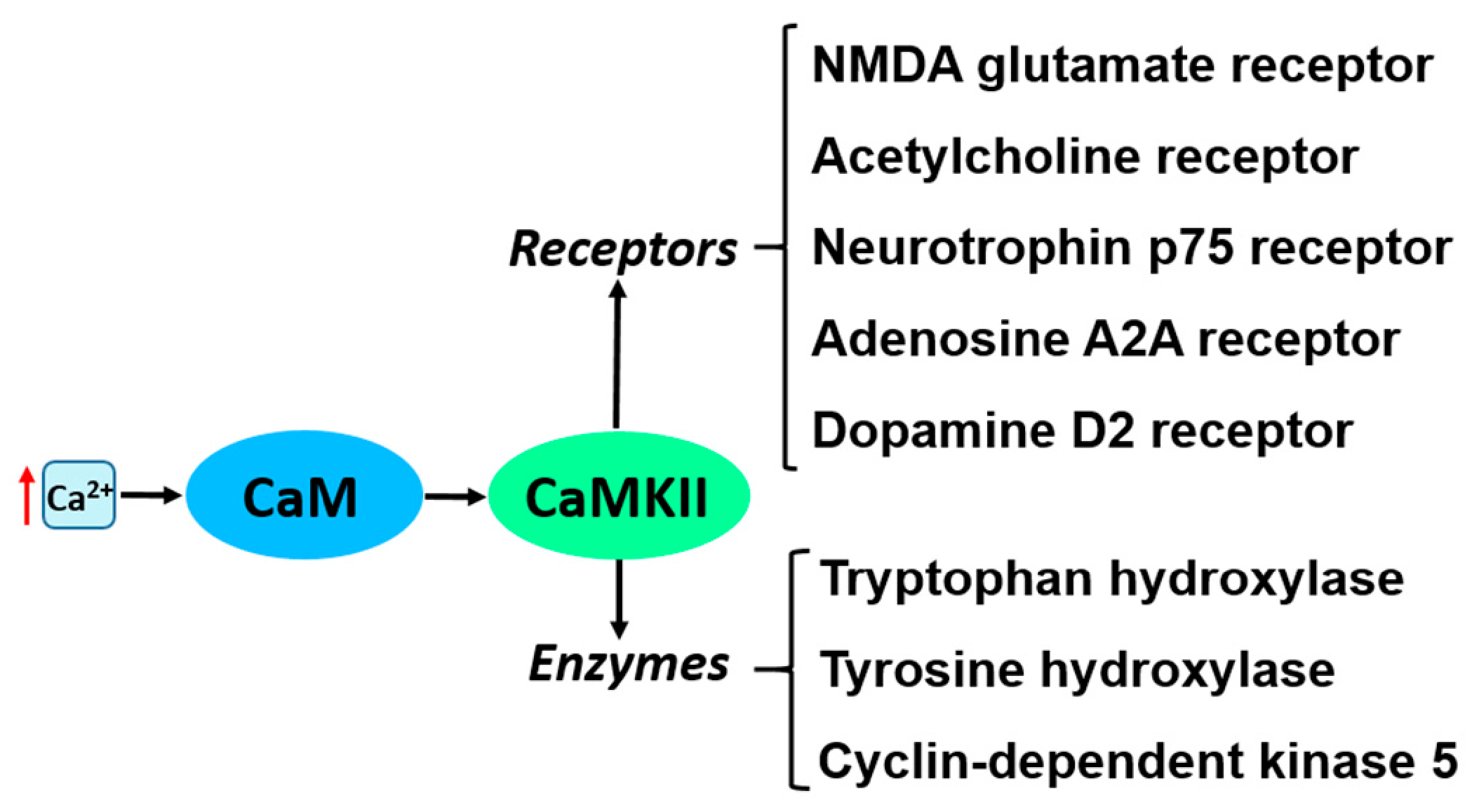
4. CaM-Dependent Protein Kinase II and Its Substrates
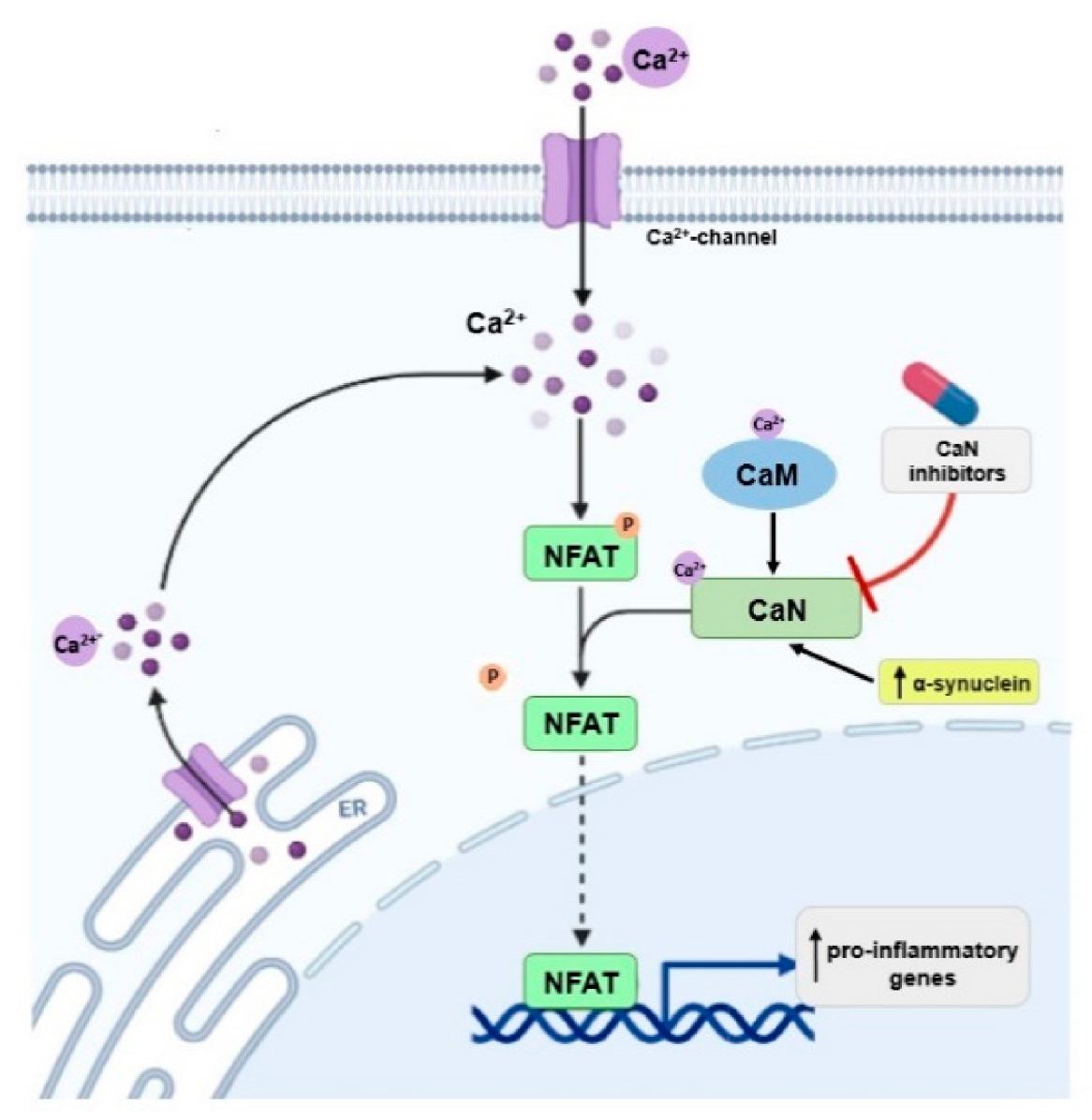
5. Involvement of Other CaM Binding Proteins in PD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: α synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 1–13. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a Partially Folded Intermediate in α-Synuclein Fibril Formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Molecular chaperones in Parkinson’s disease--present and future. J. Parkinsons Dis. 2011, 1, 299–320. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nat. Cell Biol. 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2002, 299, 256–259. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. missing article title. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Bender, P.; Snavely, L.; Rebhun, L.I.; Steward, O. Distribution and subcellular localization of calmodulin in adult and developing brain tissue. Neuroscience 1983, 10, 449–461. [Google Scholar] [CrossRef]
- Aravinda, P.; Bulbulea, S.R.; Hemalatha, N.; Babu, R.L.; Devarajua, K.S. Elevation of gene expression of calcineurin, calmodulin and calsyntenin in oxidative stress induced PC12 cells. Genes Dis. 2021, 8, 87–93. [Google Scholar] [CrossRef]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Intracellular Calcium Homeostasis and Signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J. Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 2007, 6, 933–938. [Google Scholar] [CrossRef]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci. 2009, 32, 249–256. [Google Scholar] [CrossRef]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+signaling in Parkinson’s disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.-F. Structural Basis for Calmodulin as a Dynamic Calcium Sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W. Ca2+-Binding Proteins of the EF-Hand Superfamily: Diagnostic and Prognostic Biomarkers and Novel Therapeutic Targets. Methods Mol. Biol. 2019, 1929, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and regulator of adapter/scaffold proteins. Biochem. Biophys. Acta 2018, 1865, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.-G.; Xie, H.; Pitt, G.S. Ca2+/CaM Controls Ca2+-Dependent Inactivation of NMDA Receptors by Dimerizing the NR1 C Termini. J. Neurosci. 2008, 28, 1865–1870. [Google Scholar] [CrossRef]
- Lau, S.-Y.; Procko, E.; Gaudet, R. Distinct properties of Ca2+–calmodulin binding to N- and C-terminal regulatory regions of the TRPV1 channel. J. Gen. Physiol. 2012, 140, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.-C.; Shen, Y. Calmodulin dissociates the STIM1-Orai1 complex and STIM1 oligomers. Nat. Commun. 2017, 8, 1042. [Google Scholar] [CrossRef]
- Mullins, F.M.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc. Natl. Acad. Sci. USA 2009, 106, 15495–15500. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Lode, A.J. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+. Cell Calcium 2002, 32, 321–334. [Google Scholar] [CrossRef]
- Wang, Q.-M.; Xu, Y.-Y.; Liu, S.; Ma, Z.-G. Isradipine attenuates MPTP-induced dopamine neuron degeneration by inhibiting up-regulation of L-type calcium channels and iron accumulation in the substantia nigra of mice. Oncotarget 2017, 8, 47284–47295. [Google Scholar] [CrossRef]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1.2 Ca2+ channel exacerbates the symptoms in a Parkinson’s disease model. Sci. Rep. 2019, 9, 9138. [Google Scholar] [CrossRef] [PubMed]
- Mellone, M.; Stanic, J.; Hernandez, L.F.; Iglesias, E.; Zianni, E.; Longhi, A.; Prigent, A.; Picconi, B.; Calabresi, P.; Hirsch, E.C.; et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: From experimental models to patients. Front. Cell. Neurosci. 2015, 9, 245. [Google Scholar] [CrossRef]
- Ułas, J.; Weihmuller, F.B.; Brunner, L.C.; Joyce, J.N.; Marshall, J.F.; Cotman, C.W. Selective increase of NMDA-sensitive glutamate binding in the striatum of Parkinson’s disease, Alzheimer’s disease, and mixed Parkinson’s disease/Alzheimer’s disease patients: An autoradiographic study. J. Neurosci. 1994, 14, 6317–6324. [Google Scholar] [CrossRef]
- Selvaraj, S.; Watt, J.A.; Singh, B.B. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium 2009, 46, 209–218. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef]
- Sukumaran, P.; Sun, Y.; Schaar, A.; Selvaraj, S.; Singh, B.B. TRPC Channels and Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 976, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Zhang, L.; Liu, W.-B.; Fei, Z. Inhibition of Store-Operated Calcium Entry Attenuates MPP(+)-Induced Oxidative Stress via Preservation of Mitochondrial Function in PC12 Cells: Involvement of Homer1a. PLoS ONE 2013, 8, e83638. [Google Scholar] [CrossRef]
- Lee, K.S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.K.; Kang, H.Y.; Lu, B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853, Erratum in 2018, 115, E9992. [Google Scholar] [CrossRef]
- Fedorenko, O.A.; Popugaeva, E.; Enomoto, M.; Stathopulos, P.B.; Ikura, M.; Bezprozvanny, I. Intracellular calcium channels: Inositol-1,4,5-trisphosphate receptors. Eur. J. Pharmacol. 2014, 739, 39–48. [Google Scholar] [CrossRef]
- Huang, L.; Xue, Y.; Feng, D.; Yang, R.; Nie, T.; Zhu, G.; Tao, K.; Gao, G.; Yang, Q. Blockade of RyRs in the ER Attenuates 6-OHDA-Induced Calcium Overload, Cellular Hypo-Excitability and Apoptosis in Dopaminergic Neurons. Front. Cell. Neurosci. 2017, 11, 52. [Google Scholar] [CrossRef]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ATPase of animal cells: Structure, function and regulation, Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. Calcium Pumps in Health and Disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Brendel, A.; Renziehausen, J.; Behl, C.; Hajieva, P. Downregulation of PMCA2 increases the vulnerability of midbrain neurons to mitochondrial complex I inhibition. NeuroToxicology 2014, 40, 43–51. [Google Scholar] [CrossRef]
- Yamauchi, T. Neuronal Ca2+/Calmodulin-Dependent Protein Kinase II-Discovery, Progress in a Quarter of a Century, and Perspective: Implication for Learning and Memory. Biol. Pharm. Bull. 2005, 28, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Sanhueza, M.; Lisman, J. The CaMKII/NMDAR complex as a molecular memory. Mol. Brain 2013, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; General, I.J.; Furbee, E.; Ayoob, J.C.; Castro, S.L.; Bahar, I.; Greenamyre, J.T.; Pullara, F. Disulfide bridge formation prevents CaMKII/Calmodulin interaction in Parkinson’s disease. BioRxiv 2020. [Google Scholar] [CrossRef]
- Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2018, 19, 20. [Google Scholar] [CrossRef]
- Wiemerslage, L.; Schultz, B.J.; Ganguly, A.; Lee, D. Selective degeneration of dopaminergic neurons by MPP(+) and its rescue by D2 autoreceptors in Drosophila primary culture. J. Neurochem. 2013, 126, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Yabuki, Y.; Fukunaga, K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J. Neurochem. 2012, 120, 541–551. [Google Scholar] [CrossRef]
- Kawaai, K.; Mizutani, A.; Shoji, H.; Ogawa, N.; Ebisui, E.; Kuroda, Y.; Wakana, S.; Miyakawa, T.; Hisatsune, C.; Mikoshiba, K. IRBIT regulates CaMKIIα activity and contributes to catecholamine homeostasis through tyrosine hydroxylase phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, 5515–5520. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, Z.; Ding, X.; Wang, X.; Cui, G.; Hua, F.; Xiang, J. CaMKII inhibition ameliorated levodopa-induced dyskinesia by downregulating tyrosine hydroxylase activity in an experimental model of Parkinson’s disease. Brain Res. 2018, 1687, 66–73. [Google Scholar] [CrossRef]
- Picconi, B.; Gardoni, F.; Centonze, D.; Mauceri, D.; Cenci, M.A.; Bernardi, G.; Calabresi, P.; Di Luca, M. Abnormal Ca2+-Calmodulin-Dependent Protein Kinase II Function Mediates Synaptic and Motor Deficits in Experimental Parkinsonism. J. Neurosci. 2004, 24, 5283–5291. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, C.; Wang, Q.; Liu, Z. Interactions of CaMKII with dopamine D2 receptors: Roles in levodopa-induced dyskinesia in 6-hydroxydopamine lesioned Parkinson’s rats. Sci. Rep. 2014, 4, 6811. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, H.; Hadjian, R.A.; Dwyer, J.; Feinstein, M.B. Regulation of acetylcholine receptor phosphorylation by calcium and calmodulin. Proc. Natl. Acad. Sci. USA 1981, 78, 4708–4712. [Google Scholar] [CrossRef]
- Slonimsky, J.D.; Mattaliano, M.D.; Moon, J.-I.; Griffith, L.C.; Birren, S.J. Role for calcium/calmodulin-dependent protein kinase II in the p75-mediated regulation of sympathetic cholinergic transmission. Proc. Natl. Acad. Sci. USA 2006, 103, 2915–2919. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Sakowski, S.A.; Geddes, T.J.; Wilkerson, C.; Haycock, J.W. Phosphorylation and activation of tryptophan hydroxylase 2: Identification of serine-19 as the substrate site for calcium, calmodulin-dependent protein kinase II. J. Neurochem. 2007, 103, 1567–1573. [Google Scholar] [CrossRef]
- Sawada, M.; Nagatsu, T.; Ito, K.; Iizuka, R.; Kondo, T.; Narabayashi, H. Tryptophan hydroxylase activity in the brains of controls and parkinsonian patients. J. Neural Transm. 1985, 62, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Anxiety disorders and depressive disorders preceding Parkinson’s disease: A case-control study. Mov. Disord. 2000, 15, 669–677. [Google Scholar] [CrossRef]
- Diederich, N.J.; Goetz, C.G.; Stebbins, G.T. Repeated visual hallucinations in Parkinson’s disease as disturbed external/internal perceptions: Focused review and a new integrative model. Mov. Disord. 2005, 20, 130–140. [Google Scholar] [CrossRef]
- Diederich, N.J.; Vaillant, M.; Mancuso, G.; Lyen, P.; Tiete, J. Progressive sleep ‘destructuring’ in Parkinson’s disease. A polysomnographic study in 46 patients. Sleep Med. 2005, 6, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Fénelon, G.; Mahieux, F.; Huon, R.; Ziégler, M. Hallucinations in Parkinson’s disease: Prevalence, phenomenology and risk factors. Brain 2000, 123, 733–745. [Google Scholar] [CrossRef]
- Lim, J.; Bang, Y.; Choi, J.H.; Han, A.; Kwon, M.S.; Liu, K.H.; Choi, H.J. LRRK2 G2019S Induces Anxiety/Depression-like Behavior before the Onset of Motor Dysfunction with 5-HT1A Receptor Upregulation in Mice. J. Neurosci. 2018, 38, 1611–1621. [Google Scholar] [CrossRef]
- Piirainen, H.; Hellman, M.; Tossavainen, H.; Permi, P.; Kursula, P.; Jaakola, V.P. Human adenosine A2A receptor binds calmodulin with high affinity in a calcium-dependent manner. Biophys. J. 2015, 108, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Cieślak, M.; Komoszyński, M.; Wojtczak, A. Adenosine A(2A) receptors in Parkinson’s disease treatment. Purinergic Signal. 2008, 4, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Yoshida, R.; Nose, K.; Hayashi, Y.; Mishima, T.; Fukae, J.; Kitano, K.; Kikuchi, H.; Tsuboi, Y. A new therapeutic strategy with istradefylline for postural deformities in Parkinson’s disease. Neurol. Neurochir. Polska 2019, 53, 291–295. [Google Scholar] [CrossRef]
- Zhang, P.; Shao, X.-Y.; Qi, G.-J.; Chen, Q.; Bu, L.-L.; Chen, L.-J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Huang, W.; Huang, Y.; Xu, M.; Song, P.; Huang, Y.; Xie, H.; Hu, Y. Cdk5 Inhibitory Peptide Prevents Loss of Dopaminergic Neurons and Alleviates Behavioral Changes in an MPTP Induced Parkinson’s Disease Mouse Model. Front. Aging Neurosci. 2018, 10, 162. [Google Scholar] [CrossRef]
- Hosokawa, T.; Saito, T.; Asada, A.; Ohshima, T.; Itakura, M.; Takahashi, M.; Fukunaga, K.; Hisanaga, S.-I. Enhanced activation of Ca2+/Calmodulin-dependent protein kinase II upon downregulation of cyclin-dependent kinase 5-p35. J. Neurosci. Res. 2006, 84, 747–754. [Google Scholar] [CrossRef]
- Kuno, T.; Mukai, H.; Ito, A.; Chang, C.D.; Kishima, K.; Saito, N.; Tanaka, C. Distinct cellular expression of calcineurin Aa and Ab in rat brain. J. Neurochem. 1992, 58, 1643–1651. [Google Scholar] [CrossRef]
- Creamer, T.P. Calcineurin. Cell Commun. Signal. 2020, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H., 3rd; et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef]
- Luo, J.; Sun, L.; Lin, X.; Liu, G.; Yu, J.; Parisiadou, L.; Xie, C.; Ding, J.; Cai, H. A calcineurin- and NFAT-dependent pathway is involved in α-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum. Mol. Genet. 2014, 23, 6567–6574. [Google Scholar] [CrossRef] [PubMed]
- Martin, Z.S.; Neugebauer, V.; Dineley, K.T.; Kayed, R.; Zhang, W.; Reese, L.C.; Taglialatela, G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: Relevance to human synucleopathic diseases. J. Neurochem. 2012, 120, 440–452. [Google Scholar] [CrossRef]
- Pleiss, M.M.; Sompol, P.; Kraner, S.D.; Abdul, H.M.; Furman, J.L.; Guttmann, R.P.; Wilcock, D.M.; Nelson, P.T.; Norris, C.M. Calcineurin proteolysis in astrocytes: Implications for impaired synaptic function. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Sompol, P.; Furman, J.L.; Pleiss, M.M.; Kraner, S.D.; Artiushin, I.A.; Batten, S.R.; Quintero, J.E.; Simmerman, L.A.; Beckett, T.L.; Lovell, M.A.; et al. Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in ab-bearing mice. J. Neurosci. 2017, 37, 6132–6148. [Google Scholar] [CrossRef] [PubMed]
- Rozkalne, A.; Hyman, B.T.; Spires-Jones, T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 2011, 41, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.-Q.; Chen, L.-M.; Tan, B.-H.; Guo, C.-Y.; Li, Y.-N.; Zhang, Y.-F.; Li, S.-L.; Zhao, H.; Li, Y.-C. The effects of calcineurin inhibitor FK506 on actin cytoskeleton, neuronal survival and glial reactions after pilocarpine-induced status epilepticus in mice. Epilepsy Res. 2018, 140, 138–147. [Google Scholar] [CrossRef]
- Rojanathammanee, L.; Floden, A.M.; Manocha, G.D.; Combs, C.K. Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflamm. 2015, 12, 42. [Google Scholar] [CrossRef]
- Manocha, G.D.; Floden, A.M.; Puig, K.L.; Nagamoto-Combs, K.; Scherzer, C.R.; Combs, C.K. Defining the contribution of neuroinflammation to Parkinson’s disease in humanized immune system mice. Mol. Neurodegener. 2017, 12, 1–18. [Google Scholar] [CrossRef]
- Kim, S.; Violette, C.J.; Ziff, E.B. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol. Aging 2015, 36, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, N. Calcineurin inhibitors improve memory loss and neuropathological changes in mouse model of dementia. Pharmacol. Biochem. Behav. 2017, 153, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351, Erratum in 2007, 54, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, Y.; Wang, P.; Gu, L.; Wang, L.; Yang, H.; Wei, Q.; Li, Z.; Luo, J. The interaction between calcineurin and α-synuclein is regulated by calcium and calmodulin. Biochem. Biophys. Res. Commun. 2018, 496, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to a-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef]
- Chen, X.; Xue, B.; Wang, J.; Liu, H.; Shi, L.; Xie, J. Potassium Channels: A Potential Therapeutic Target for Parkinson’s Disease. Neurosci. Bull. 2018, 34, 341–348. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Rivard, A.F.; Bächinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nat. Cell Biol. 2001, 410, 1120–1124. [Google Scholar] [CrossRef]
- Mourre, C.; Manrique, C.; Camon, J.; Aidi-Knani, S.; Deltheil, T.; Turle-Lorenzo, N.; Guiraudie-Capraz, G.; Amalric, M. Changes in SK channel expression in the basal ganglia after partial nigrostriatal dopamine lesions in rats: Functional consequences. Neuropharmacology 2017, 113, 519–532. [Google Scholar] [CrossRef]
- Hartmann, A.; Müllner, J.N.M.; Meier, N.; Hesekamp, H.; Van Meerbeeck, P.; Habert, M.-O.; Kas, A.; Tanguy, M.-L.; Mazmanian, M.; Oya, H.; et al. Bee Venom for the Treatment of Parkinson Disease—A Randomized Controlled Clinical Trial. PLoS ONE 2016, 11, e0158235. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 2007, 369, 2097–2105. [Google Scholar] [CrossRef]
- Jiang, X.; Mu, D.; Manabat, C.; Koshy, A.A.; Christen, S.; Täuber, M.G.; Vexler, Z.S.; Ferriero, D.M. Differential vulnerability of immature murine neurons to oxygen-glucose deprivation. Exp. Neurol. 2004, 190, 224–232. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of Nitric Oxide Synthases in Parkinson’s Disease: A Review on the Antioxidant and Anti-inflammatory Activity of Polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Guillemette, J.G.; Dieckmann, T. Dynamics of Nitric Oxide Synthase–Calmodulin Interactions at Physiological Calcium Concentrations. Biochemistry 2015, 54, 1989–2000. [Google Scholar] [CrossRef]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Matthews, R.T.; Beal, M.F.; Fallon, J.; Fedorchak, K.; Huang, P.L.; Fishman, M.C.; Hyman, B.T. MPP+Induced Substantia Nigra Degeneration Is Attenuated in nNOS Knockout Mice. Neurobiol. Dis. 1997, 4, 114–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kurosaki, R.; Muramatsu, Y.; Michimata, M.; Matsubara, M.; Kato, H.; Imai, Y.; Itoyama, Y.; Araki, T. Role of nitric oxide synthase against MPTP neurotoxicity in mice. Neurol. Res. 2002, 24, 655–662. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kato, H.; Araki, T. Protective action of neuronal nitric oxide synthase inhibitor in the MPTP mouse model of Parkinson’s disease. Metab. Brain Dis. 2008, 23, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Riobó, N.A.; Carreras, M.C.; Cherńavsky, A.; Rubio, A.; Satz, M.L.; Poderoso, J.J. Overexpression of neutrophil neuronal nitric oxide synthase in Parkinson’s disease. Nitric Oxide 2000, 4, 534–539. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CaMBP | Norm | PD Pathology | Effect of Modification of Protein Level/Activity |
|---|---|---|---|
| L-type channels | Ca2+ influx | Increased expression in substantia nigra neurons of deceased PD patients [19]. | CaV1.2 channel blocker protects dopaminergic neurons exposed to rotenone or MPTP [19,31]. CaV1.2 channel knock-down in microglia impairs dopaminergic neurons in MPTP-treated mice [34]. |
| TRP channels | Ca2+ influx | Reduced expression in the substantia nigra neurons of PD model [37]. | Overexpression of TRPC1 protects MPP+-treated PC12 cells against apoptosis and increases their survival [38]. |
| NMDARs | L-glutamate receptors, mediate Ca2+ influx, role in learning and memory. | Increase in NMDA-sensitive glutamate binding in the striatum of PD patients [36]. Redistribution and altered ratio of NMDAR subunits in striatal synapses of both animal model and PD patient [35]. | ND |
| IP3R/RyR | Ca2+ release from ER and Ca2+ transfer from ER to mitochondria. | Increased expression of IP3R in cerebellum and motor cortex of rat PD model [42]. | RyR blockade attenuates Ca2+ overload, preserves excitability and protects dopaminergic neurons from apoptosis in animal and cellular models of PD [43]. |
| Orai1/STIM1 | Replenishment of ER Ca2+ stores. | STIM1 expression is unaltered in the substantia nigra of PD patients [38]. | STIM1 silencing decreases viability of human neuroblastoma SH-SY5Y cells [38]. STIM1 silencing in MPP+-treated PC12 cells prevents mitochondrial dysfunction and improves cell viability [40]. |
| PMCA | Ca2+ efflux | Decreased PMCA2 expression in a cellular PD model [46]. | PMCA2 downregulation sensitizes cells to, and upregulation protects from, MPP+ toxicity [46]. |
| CaMKII | Maintaining long-term potentiation (LTP), memory formation and neuronal excitability. | CaMKII activity is higher in a rat model of PD [56]. | CaMKII inhibition reverses deficits in synaptic function and motor behavior in a rat model of PD [56]. Reduction of CaMKII activity is associated with cognitive deficit and learning disability in mouse model of PD [52,53]. |
| Involvement in dopamine synthesis [54]. | Increased interaction of CaMKII-D2 receptor in striatal neurons of a rat model of PD after chronic administration of L-DOPA [57]. | CaMKII inhibition reduces of tyrosine hydroxylase phosphorylation [55]. | |
| CaMKII mediates cholinergic system by regulation of acetylcholine receptor and neurotrophin receptor p75 [58,59]. | ND | Inhibition of CaMKII results in loss of BDNF-induced inhibitory cholinergic transmission [59]. | |
| Activation of tryptophan hydroxylase, a key enzyme involved in serotonin synthesis [60]. | Activity of tryptophan hydroxylase is reduced in serotonergic neurons of PD patients [61]. | ND | |
| Regulation of A2AR activity [67]. | ND | Inhibition of A2ARs reverses movement dysfunction and is neuroprotective in animal models of PD [68]. | |
| CaN | Maintaining neuronal plasticity, long-term potentiation (LTP), memory formation. | CaN is activated in brain at early stages of cognitive decline [77,78]. | Inhibition of CaN protects brain cells from neurotoxicity [80,81], reduces neuroinflammation [82,83], improves the function of synapses [84], inhibits cognitive loss [85], and could extend lifespan [86]. |
| CaN interacts with α-synuclein [87]. | Overexpression of α-synuclein activates the CaN-NFAT pathway in cell lines and dopaminergic neurons; inhibition of this pathway protects dopaminergic neurons against α-synuclein-mediated toxicity [76]. Inhibition of CaN moves the α-synuclein-induced CaM-CaN cascade to a protective mode. This may have therapeutic implications for the treatment of PD [88]. | ||
| (SK) channels | Neurotransmitter release [89]. | Lower expression in PD models [91]. | SK channel inhibition protects nigral dopaminergic neurons and improves motor performance in PD model [91]. SK channel activation has protective effects [89]. |
| Glutamate decarboxylase | Decarboxylation of L-glutamic acid to GABA [93]. | ND | Improvement in motor function in PD patients after adeno-associated virus (AAV)-glutamic acid decarboxylase (GAD)- gene therapy [93]. |
| NOS | Learning, memory, neurogenesis | High levels of nNOS and iNOS in the substantia nigra and striatum of PD patients and PD models [97]. nNOS expression is increased in basal ganglia and in the respiratory burst of circulating neutrophils of PD patients; a significant increase in NO production and protein tyrosine nitration is observed [101]. | nNOS inhibitor protects neuronal cells against MPTP-induced neurotoxicity in animal models [99,100]. nNOS knockout mice are more resistant to MPTP-induced neurotoxicity compared with wild-type animals [98]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohush, A.; Leśniak, W.; Weis, S.; Filipek, A. Calmodulin and Its Binding Proteins in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 3016. https://doi.org/10.3390/ijms22063016
Bohush A, Leśniak W, Weis S, Filipek A. Calmodulin and Its Binding Proteins in Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(6):3016. https://doi.org/10.3390/ijms22063016
Chicago/Turabian StyleBohush, Anastasiia, Wiesława Leśniak, Serge Weis, and Anna Filipek. 2021. "Calmodulin and Its Binding Proteins in Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 6: 3016. https://doi.org/10.3390/ijms22063016
APA StyleBohush, A., Leśniak, W., Weis, S., & Filipek, A. (2021). Calmodulin and Its Binding Proteins in Parkinson’s Disease. International Journal of Molecular Sciences, 22(6), 3016. https://doi.org/10.3390/ijms22063016