
Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function

 , , and
, , and
Abstract
1. Introduction
2. Results
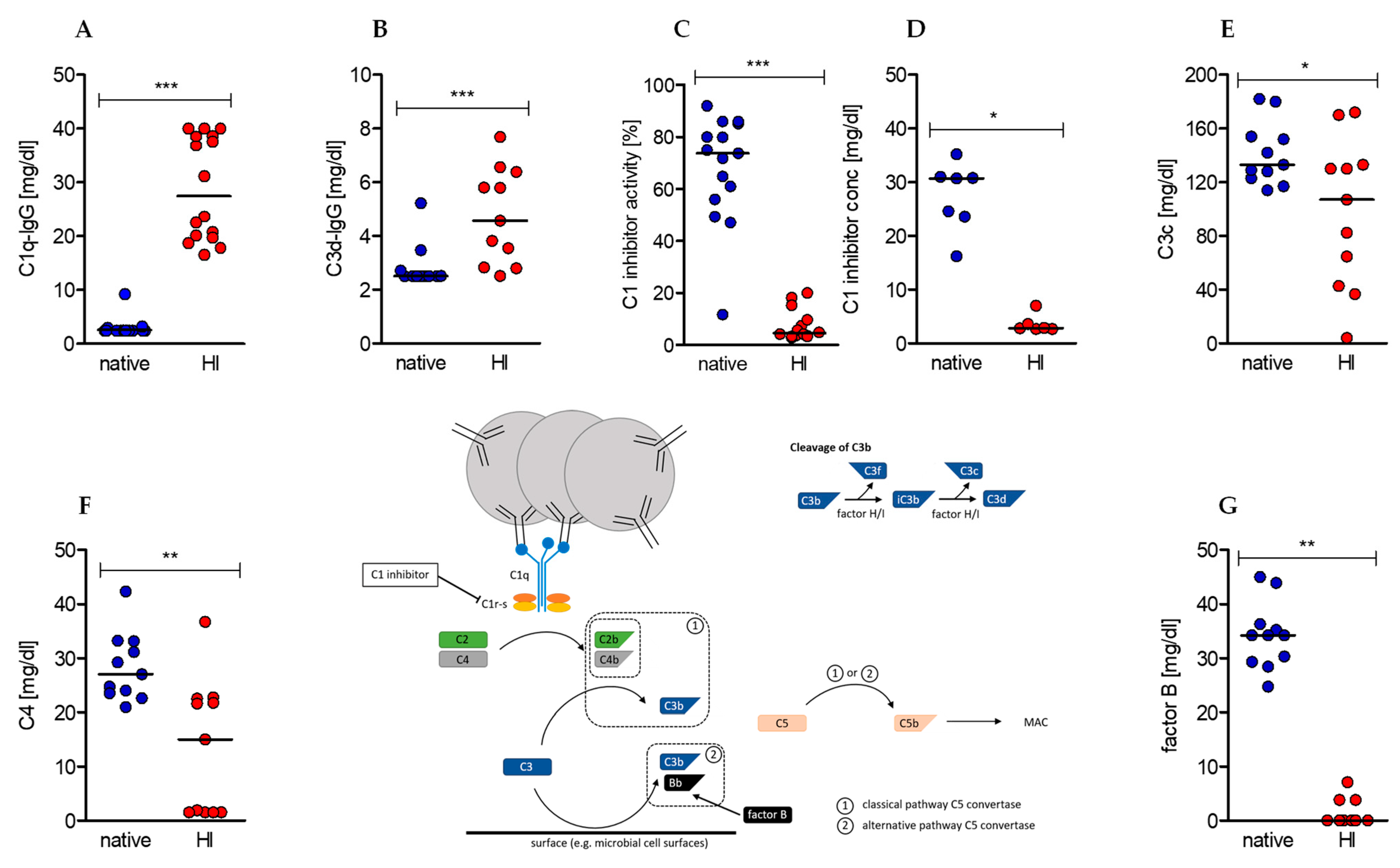
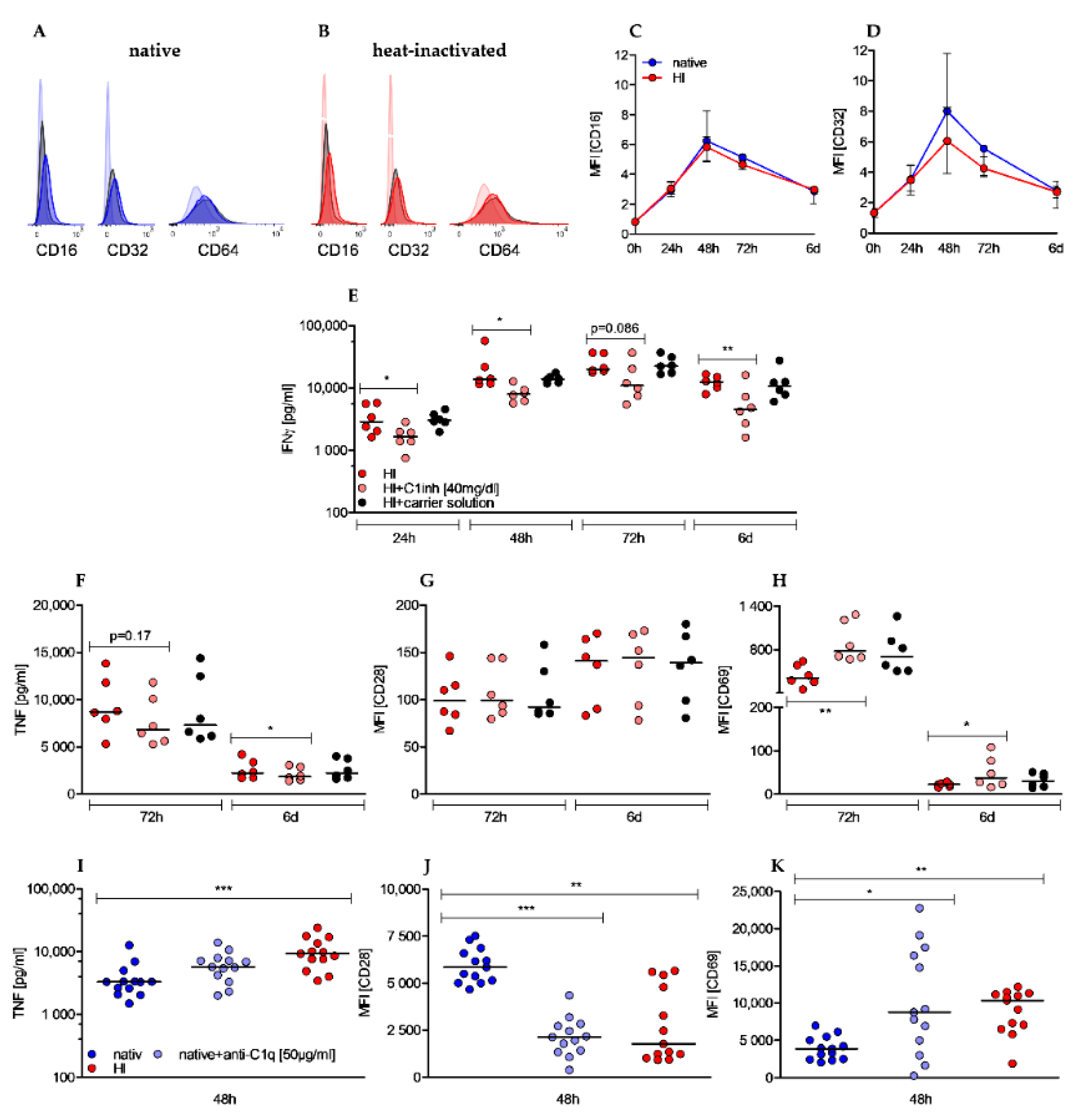
2.1. Heat-Inactivation of Human Serum Has Significant Impact on Complement Factors, Regulators, and Immune Complexes
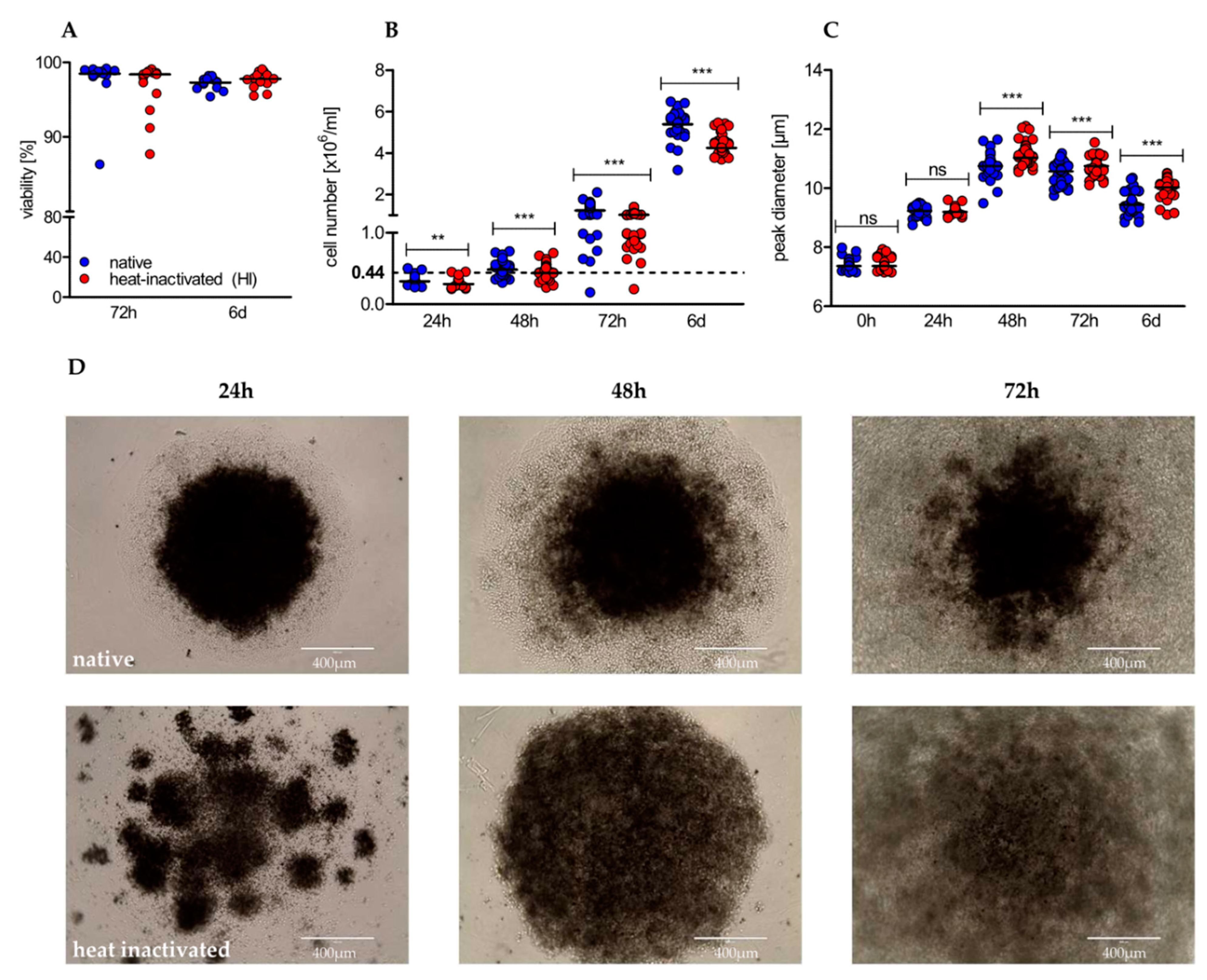
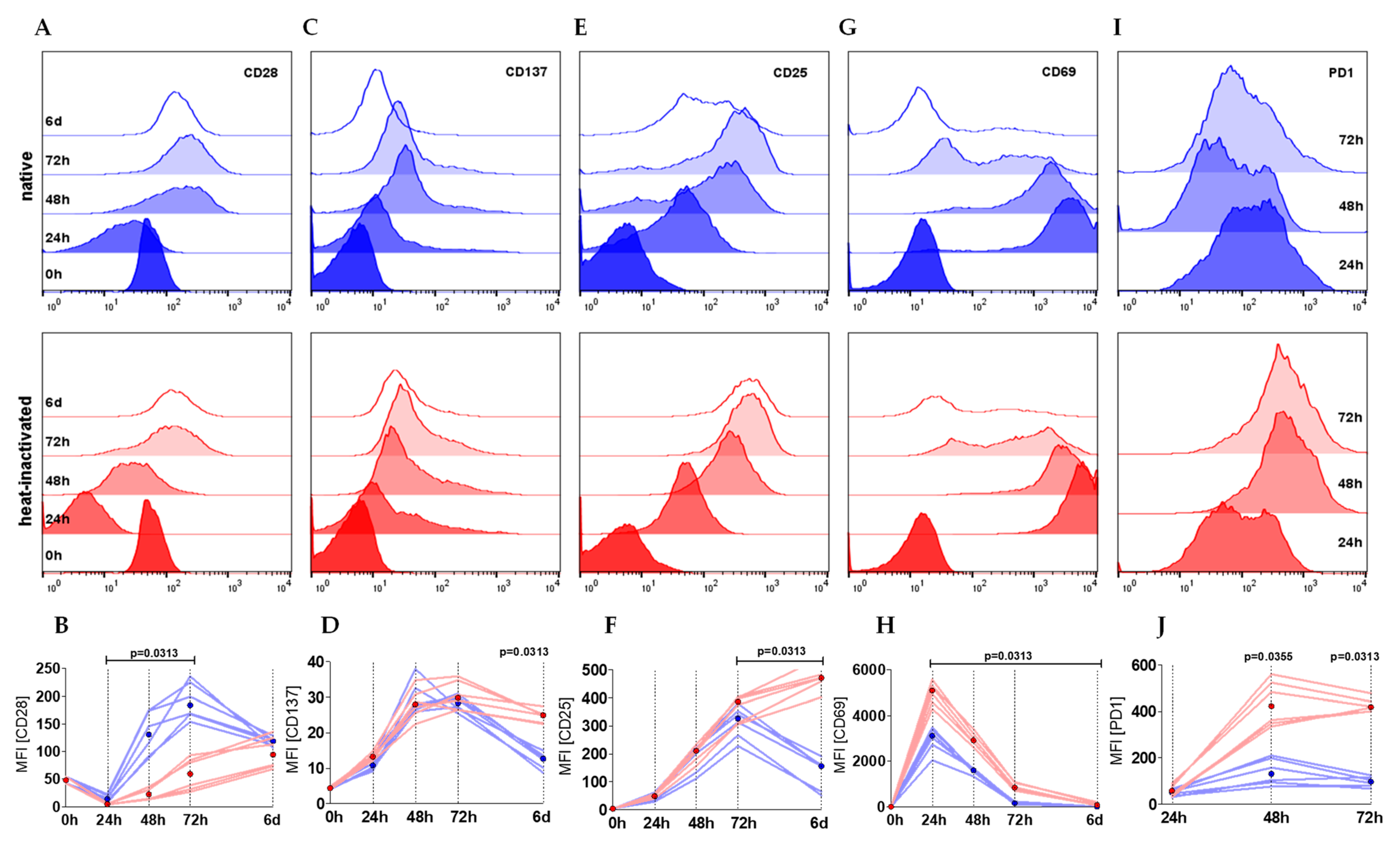
2.2. Heat-Inactivation of Human Serum Impairs Proliferation but Promotes on-Blast Formation of CD4+ T Cells
2.3. Heat-Inactivation of Human Serum Impairs Proliferation but Promotes on-Blast Formation of CD4+ T Cells
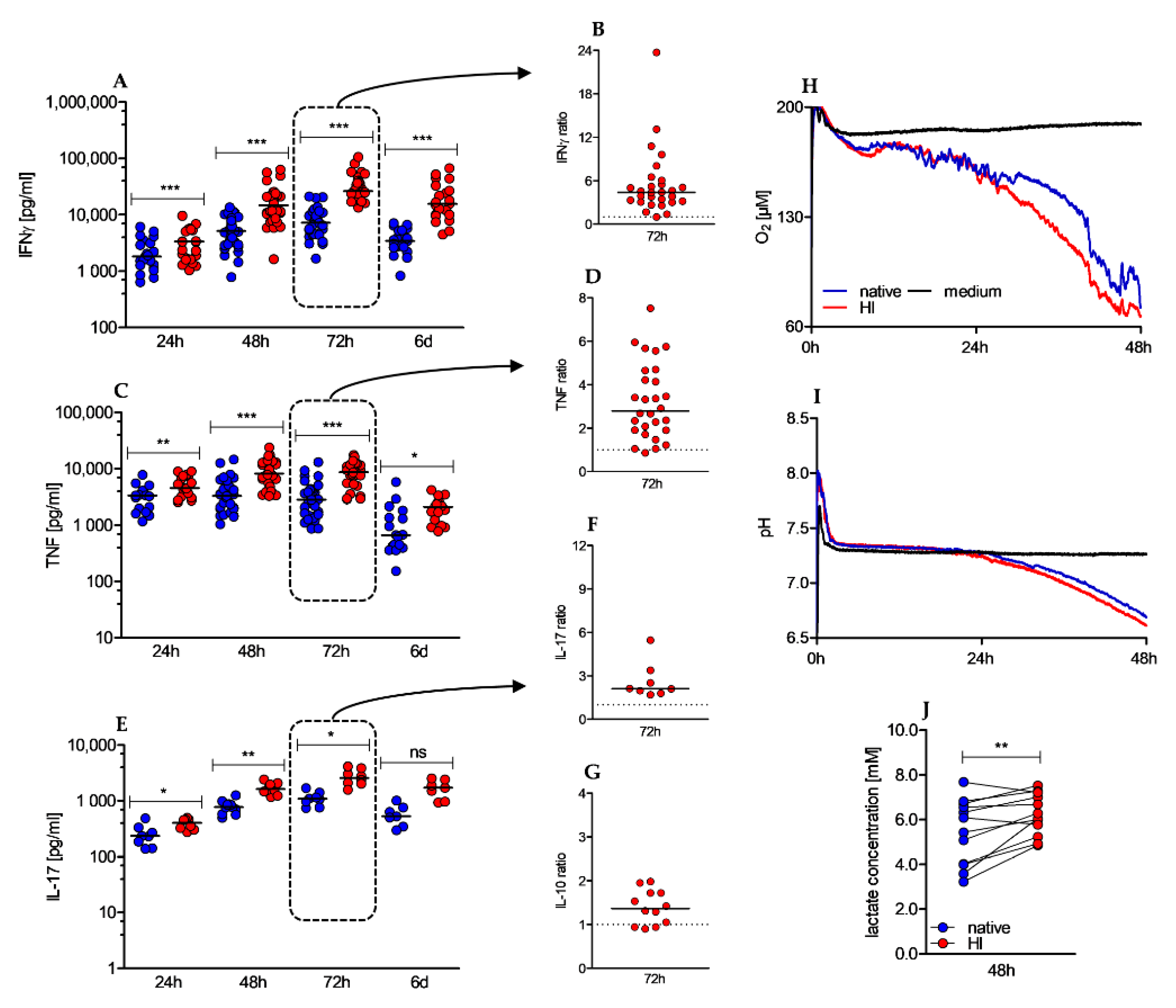
2.4. Heat-Inactivation of Human Serum Promotes Cytokine Secretion, and Increases Metabolic Activity of CD4+ T Cells
2.5. Effects of Heat-Inactivation Are Partially Reverted by c1 Inhibitor and Mimicked by Anti-C1q Antibody Supplementation
3. Discussion
4. Material and Methods
4.1. Heat-Inactivation of Human Sera
4.2. Determination of Heat-Induced Changes in Serum Composition
4.3. Human T Cell Isolation and Culture
4.4. Flow Cytometry
4.5. Determination of Cytokines
4.6. Monitoring of Oxygen Consumption and pH Development and Determination of Lactate Secretion
4.7. Supplementation of C1 Inhibitor to Heat Inactivated Sera
4.8. Modification of Native Sera by Anti-human Anti-C1q Antibody
4.9. Statistics and Design
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kohl, J. The Role of Complement in Danger Sensing and Transmission. IR 2006, 34, 157–176. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Webb, J.; Whaley, K. Complement and immune complex diseases. Aust. N. Z. J. Med. 1986, 16, 268–278. [Google Scholar] [CrossRef]
- van Lookeren Campagne, M.; Wiesmann, C.; Brown, E.J. Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 2007, 9, 2095–2102. [Google Scholar] [CrossRef]
- Müller-Eberhard, H.J. The Killer Molecule of Complement. J. Investig. Dermatol. 1985, 85, S47–S52. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Carroll, M.C. The complement system in B cell regulation. Mol. Immunol. 2004, 41, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B. Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J. Exp. Med. 1974, 140, 126–145. [Google Scholar] [CrossRef]
- Carroll, M.C. CD21/CD35 in B cell activation. Semin. Immunol. 1998, 10, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, R.; Carroll, M.C. Complement receptors CD21 and CD35 in humoral immunity. Immunol. Rev. 2007, 219, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Molina, H.; Holers, V.M.; Li, B.; Fung, Y.; Mariathasan, S.; Goellner, J.; Strauss-Schoenberger, J.; Karr, R.W.; Chaplin, D.D. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA 1996, 93. [Google Scholar] [CrossRef]
- Dempsey, P.W.; Allison, M.E.; Akkaraju, S.; Goodnow, C.C.; Fearon, D.T. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science 1996, 271, 348–350. [Google Scholar] [CrossRef]
- Dempsey, P.W.; Fearon, D.T. Complement: Instructing the acquired immune system through the CD21/CD19 complex. Res. Immunol. 1996, 147, 71–75. [Google Scholar] [CrossRef]
- Fischer, M.B.; Goerg, S.; Shen, L.; Prodeus, A.P.; Goodnow, C.C.; Kelsoe, G.; Carroll, M.C. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science 1998, 280, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Abel, B.; Gallimore, A.; Carroll, M.; Bachmann, M.F. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 2002, 8, 373–378. [Google Scholar] [CrossRef]
- Ghannam, A.; Fauquert, J.-L.; Thomas, C.; Kemper, C.; Drouet, C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol. Immunol. 2014, 58, 98–107. [Google Scholar] [CrossRef]
- Hawlisch, H.; Köhl, J. Complement and Toll-like receptors: Key regulators of adaptive immune responses. Mol. Immunol. 2006, 43, 13–21. [Google Scholar] [CrossRef]
- Li, K.; Fazekasova, H.; Wang, N.; Peng, Q.; Sacks, S.H.; Lombardi, G.; Zhou, W. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology 2012, 217. [Google Scholar] [CrossRef]
- Zhou, W.; Peng, Q.; Li, K.; Sacks, S.H. Role of dendritic cell synthesis of complement in the allospecific T cell response. Mol. Immunol. 2007, 44. [Google Scholar] [CrossRef]
- Kurita-Taniguchi, M.; Fukui, A.; Hazeki, K.; Hirano, A.; Tsuji, S.; Matsumoto, M.; Watanabe, M.; Ueda, S.; Seya, T. Functional modulation of human macrophages through CD46 (measles virus receptor): Production of IL-12 p40 and nitric oxide in association with recruitment of protein-tyrosine phosphatase SHP-1 to CD46. J. Immunol. 2000, 165, 5143–5152. [Google Scholar] [CrossRef]
- Csomor, E.; Bajtay, Z.; Sándor, N.; Kristóf, K.; Arlaud, G.J.; Thiel, S.; Erdei, A. Complement protein C1q induces maturation of human dendritic cells. Mol. Immunol. 2007, 44. [Google Scholar] [CrossRef]
- Clarke, E.V.; Weist, B.M.; Walsh, C.M.; Tenner, A.J. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J. Leukoc. Biol. 2015, 97, 147–160. [Google Scholar] [CrossRef]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lalli, P.N.; Strainic, M.G.; Yang, M.; Lin, F.; Medof, M.E.; Heeger, P.S. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 2008, 112, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.R.; Basheer, S.A.; Sacks, S.H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 2002, 8. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of signaling into CD4⁺ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3⁺ regulatory T cells. Nat. Immunol. 2013, 14, 162–171. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Post, T.W.; Atkinson, J.P. Membrane cofactor protein (MCP or CD46): Newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 1991, 9. [Google Scholar] [CrossRef]
- Seya, T.; Ballard, L.L.; Bora, N.S.; Kumar, V.; Cui, W.; Atkinson, J.P. Distribution of membrane cofactor protein of complement on human peripheral blood cells. An altered form is found on granulocytes. Eur. J. Immunol. 1988, 18. [Google Scholar] [CrossRef]
- Cole, J.L.; Housley, G.A.; Dykman, T.R.; MacDermott, R.P.; Atkinson, J.P. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc. Natl. Acad. Sci. USA 1985, 82, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Le Friec, G.; Sheppard, D.; Whiteman, P.; Karsten, C.M.; Shamoun, S.A.-T.; Laing, A.; Bugeon, L.; Dallman, M.J.; Melchionna, T.; Chillakuri, C.; et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat. Immunol. 2012, 13, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.; Trescol-Biémont, M.C.; Azocar, O.; Lamouille, B.; Rabourdin-Combe, C. Cutting edge: CD46, a new costimulatory molecule for T cells, that induces p120CBL and LAT phosphorylation. J. Immunol. 2000, 164, 6091–6095. [Google Scholar] [CrossRef]
- Cardone, J.; Le Friec, G.; Vantourout, P.; Roberts, A.; Fuchs, A.; Jackson, I.; Suddason, T.; Lord, G.; Atkinson, J.P.; Cope, A.; et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 2010, 11, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, Y.; Destaing, O.; Roux, A.; Ory, S.; Nheu, T.; Jurdic, P.; Rabourdin-Combe, C.; Astier, A.L. CD46/CD3 costimulation induces morphological changes of human T cells and activation of Vav, Rac, and extracellular signal-regulated kinase mitogen-activated protein kinase. J. Immunol. 2001, 167. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Dimeloe, S.; Le, F.G.; Navarini, A.; Arbore, G.; Povoleri, G.A.; Fischer, M.; Belle, R.; Loeliger, J.; Develioglu, L.; et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015, 42. [Google Scholar] [CrossRef]
- Hess, C.; Kemper, C. Complement-Mediated Regulation of Metabolism and Basic Cellular Processes. Immunity 2016, 45. [Google Scholar] [CrossRef]
- Jiang, K.; Chen, Y.; Xu, C.-S.; Jarvis, J.N. T cell activation by soluble C1q-bearing immune complexes: Implications for the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2003, 131, 61–67. [Google Scholar] [CrossRef]
- Chen, A.; Gaddipati, S.; Hong, Y.; Volkman, D.J.; Peerschke, E.I.; Ghebrehiwet, B. Human T cells express specific binding sites for C1q. Role in T cell activation and proliferation. J. Immunol. 1994, 153, 1430–1440. [Google Scholar]
- Ghebrehiwet, B.; Habicht, G.S.; Beck, G. Interaction of C1q with its receptor on cultured cell lines induces an anti-proliferative response. Clin. Immunol. Immunopathol. 1990, 54, 148–160. [Google Scholar] [CrossRef]
- Jiang, K.; Chen, Y.; Jarvis, J.N. Cord blood and adult T cells show different responses to C1q-bearing immune complexes. Cell. Immunol. 2004, 229. [Google Scholar] [CrossRef]
- Triglia, R.P.; Linscott, W.D. Titers of nine complement components, conglutinin and C3b-inactivator in adult and fetal bovine sera. Mol. Immunol. 1980, 17, 741–748. [Google Scholar] [CrossRef]
- Soltis, R.D.; Hasz, D.; Morris, M.J.; Wilson, I.D. The effect of heat inactivation of serum on aggregation of immunoglobulins. Immunology 1979, 36, 37–45. [Google Scholar] [PubMed]
- Pinyopummintr, T.; Bavister, B.D. Development of bovine embryos in a cell-free culture medium: Effects of type of serum, timing of its inclusion and heat inactivation. Theriogenology 1994, 41, 1241–1249. [Google Scholar] [CrossRef]
- Leshem, B.; Yogev, D.; Fiorentini, D. Heat inactivation of fetal calf serum is not required for in vitro measurement of lymphocyte functions. J. Immunol. Methods 1999, 223. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Testi, R.; Phillips, J.H.; Lanier, L.L. Leu 23 induction as an early marker of functional CD3/T cell antigen receptor triggering. Requirement for receptor cross-linking, prolonged elevation of intracellular Ca++ and stimulation of protein kinase C. J. Immunol. 1989, 142, 1854–1860. [Google Scholar] [PubMed]
- Testi, R.; Phillips, J.H.; Lanier, L.L. T cell activation via Leu-23 (CD69). J. Immunol. 1989, 143, 1123–1128. [Google Scholar]
- Herbel, C.; Patsoukis, N.; Bardhan, K.; Seth, P.; Weaver, J.D.; Boussiotis, V.A. Clinical significance of T cell metabolic reprogramming in cancer. Clin. Transl. Med. 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Cham, C.M.; Gajewski, T.F. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 2005, 174, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Greisman, S.G.; Redecha, P.B.; Kimberly, R.P.; Christian, C.L. Differences among immune complexes: Association of C1q in SLE immune complexes with renal disease. J. Immunol. 1987, 138, 739–745. [Google Scholar] [PubMed]
- Levinsky, R.J.; Cameron, J.S.; Soothill, J.F. Serum immune complexes and disease activity in lupus nephritis. Lancet 1977, 1. [Google Scholar] [CrossRef]
- Shukla, A.; Gaur, P. Hereditary C1 inhibitor deficiency associated with systemic lupus erythematosus. Lupus 2020, 29. [Google Scholar] [CrossRef]
- Cacoub, P.; Frémeaux-Bacchi, V.; De, L.I.; Guillien, F.; Kahn, M.F.; Kazatchkine, M.D.; Godeau, P.; Piette, J.C. A new type of acquired C1 inhibitor deficiency associated with systemic lupus erythematosus. Arthritis Rheum. 2001, 44. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Campoamor, M.T.; Chamorro, A.; Salvador, G.; Segura, S.; Botero, J.C.; Yagüe, J.; Cervera, R.; Ingelmo, M.; Font, J. Hypocomplementemia in systemic lupus erythematosus and primary antiphospholipid syndrome: Prevalence and clinical significance in 667 patients. Lupus 2004, 13. [Google Scholar] [CrossRef]
- Sim, R.B.; Arlaud, G.J.; Colomb, M.G. C1 inhibitor-dependent dissociation of human complement component C1 bound to immune complexes. Biochem. J. 1979, 179, 449–457. [Google Scholar] [CrossRef]
- Ziccardi, R.J. Activation of the early components of the classical complement pathway under physiologic conditions. J. Immunol. 1981, 126, 1769–1773. [Google Scholar]
- Cooper, N.R. The classical complement pathway: Activation and regulation of the first complement component. Adv. Immunol. 1985, 37. [Google Scholar] [CrossRef]
- Chan, J.Y.; Burrowes, C.E.; Habal, F.M.; Movat, H.Z. The inhibition of activated factor XII (Hageman factor) by antithrombin III: The effect of other plasma proteinase inhibitors. Biochem. Biophys. Res. Commun. 1977, 74. [Google Scholar] [CrossRef]
- Pixley, R.A.; Schapira, M.; Colman, R.W. The regulation of human factor XIIa by plasma proteinase inhibitors. J. Biol. Chem. 1985, 260, 1723–1729. [Google Scholar] [CrossRef]
- van der Graaf, F.; Koedam, J.A.; Bouma, B.N. Inactivation of kallikrein in human plasma. J. Clin. Investig. 1983, 71, 149–158. [Google Scholar] [CrossRef]
- Schapira, M.; Scott, C.F.; Colman, R.W. Contribution of plasma protease inhibitors to the inactivation of kallikrein in plasma. J. Clin. Investig. 1982, 69, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Sjögren, H.O. Inhibition of activation of human T lymphocytes by the complement C1 esterase inhibitor. Immunology 1995, 86, 304–310. [Google Scholar]
- Nissen, M.H.; Bregenholt, S.; Nording, J.A.; Claesson, M.H. C1-esterase inhibitor blocks T lymphocyte proliferation and cytotoxic T lymphocyte generation in vitro. Int. Immunol. 1998, 10, 167–173. [Google Scholar] [CrossRef][Green Version]
- Jiang, H.; Wagner, E.; Zhang, H.; Frank, M.M. Complement 1 inhibitor is a regulator of the alternative complement pathway. J. Exp. Med. 2001, 194, 1609–1616. [Google Scholar] [CrossRef]
- Muhlemann, M.F.; Macrae, K.D.; Am Smith; Beck, P.; Hine, I.; Hegde, U.; Milford-Ward, A.; Carter, G.D.; Wise, P.H.; Cream, J.J. Hereditary angioedema and thyroid autoimmunity. J. Clin. Pathol. 1987, 40. [Google Scholar] [CrossRef] [PubMed]
- Truedsson, L.; Bengtsson, A.A.; Sturfelt, G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 2007, 40. [Google Scholar] [CrossRef]
- Levy, D.; Craig, T.; Keith, P.K.; Krishnarajah, G.; Beckerman, R.; Prusty, S. Co-occurrence between C1 esterase inhibitor deficiency and autoimmune disease: A systematic literature review. Allergy Asthma Clin. Immunol. 2020, 16, 1–8. [Google Scholar] [CrossRef]
- Fridman, W.H.; Galon, J.; Pagès, F.; Tartour, E.; Sautès-Fridman, C.; Kroemer, G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011, 71, 5601–5605. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.-C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.-H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef]
- Förnvik, K.; Maddahi, A.; Persson, O.; Osther, K.; Salford, L.G.; Nittby Redebrandt, H. C1-inactivator is upregulated in glioblastoma. PLoS ONE 2017, 12, e0183086. [Google Scholar] [CrossRef]
- Osther, K.; Förnvik, K.; Liljedahl, E.; Salford, L.G.; Redebrandt, H.N. Upregulation of C1-inhibitor in pancreatic cancer. Oncotarget 2019, 10, 5703–5712. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Native Sera Mean (±SEM) | Heat-Inactivated Sera Mean (±SEM) | Significance Level | |
|---|---|---|---|
| IgG | 1146 (58.14) | 1369 (66.43) | ** |
| IgE | 0.0764 (0.0472) | 0.0017 (0.0006) | ** |
| IgD | 2.622 (0.562) | 0.510 (0.164) | ** |
| Albumin | 4254 (112.6) | 3797 (135.2) | * |
| Total protein | 7266 (138.1) | 7282 (187.5) | ns |
| Glucose | 71.25 (5.170) | 71.25 (5.218) | ns |
| Cholesterol | 196.3 (9.107) | 196.2 (9.646) | ns |
| −HDL | 56.89 (3.553) | 49.22 (3.582) | * |
| −LDL | 109.3 (9.042) | 110.7 (9.006) | ns |
| −VLDL | 30.11 (3.839) | 36.33 (4.752) | ns |
| Triglyceride | 122.4 (10.29) | 120.9 (9.945) | ns |
| 48/72 h | +anti-C1q [50µg/ml] | native | HI | +C1inh [40 mg/dL] |
|---|---|---|---|---|
| cell number | ↔ | ↓ | ↔ | |
| peak diameter | ↔ | ↑ | ↔ | |
| IFNγ | ↔ | ↑ | ↓ | |
| TNF | (↑) | ↑ | ↓ | |
| CD28 | ↓ | ↓ | ↔ | |
| CD69 | ↑ | ↑ | ↑ | |
| CD137 | ↔ | ↔ | ↔ | |
| CD25 | ↓ | ↑ | ↔ |
| Parameter | Company | Device | Method |
|---|---|---|---|
| CIC C1q-IgG | Human GmbH (Wiesbaden, Germany) | 1 | |
| CIC C3d-IgG | Human GmbH | 1 | |
| C1 inhibitor activity | TECO medical (Sissach, Swiss) | 1 | |
| C3 activator | The Binding Site GmbH (Schwetzingen, Germany) | 2 | |
| IgE | Roche Diagnostics (Mannheim, Germany) | Cobas e411 | 3 |
| IgG | Siemens Healthcare Diagnostics (Erlangen, Germany) | Dimension Vista 1500 | 4 |
| IgD | The Binding Site GmbH | BN ProSpec | 4 |
| C1 inhibitor concentration | Siemens Healthcare | BN ProSpec | 4 |
| C3c | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 4 |
| C3c | Roche Diagnostics (since 01/2020) | Cobas pro | 5 |
| C4 | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 4 |
| C4 | Roche Diagnostics (since 01/2020) | Cobas pro | 5 |
| Albumin | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| Electrophoresis | Sebia | Capillaries | 7 |
| Total protein | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| Glucose | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| Cholesterol | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| Triglyceride | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| HDL-cholesterol | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| LDL-cholesterol | Siemens Healthcare Diagnostics | Dimension Vista 1500 | 6 |
| VLDL-cholesterol | Calculated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fante, M.A.; Decking, S.-M.; Bruss, C.; Schreml, S.; Siska, P.J.; Kreutz, M.; Renner, K. Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function. Int. J. Mol. Sci. 2021, 22, 2646. https://doi.org/10.3390/ijms22052646
Fante MA, Decking S-M, Bruss C, Schreml S, Siska PJ, Kreutz M, Renner K. Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function. International Journal of Molecular Sciences. 2021; 22(5):2646. https://doi.org/10.3390/ijms22052646
Chicago/Turabian StyleFante, Matthias A., Sonja-Maria Decking, Christina Bruss, Stephan Schreml, Peter J. Siska, Marina Kreutz, and Kathrin Renner. 2021. "Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function" International Journal of Molecular Sciences 22, no. 5: 2646. https://doi.org/10.3390/ijms22052646
APA StyleFante, M. A., Decking, S.-M., Bruss, C., Schreml, S., Siska, P. J., Kreutz, M., & Renner, K. (2021). Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function. International Journal of Molecular Sciences, 22(5), 2646. https://doi.org/10.3390/ijms22052646