
Clinical Potential of Kinase Inhibitors in Combination with Immune Checkpoint Inhibitors for the Treatment of Solid Tumors


{kind=link}
Abstract
1. Introduction
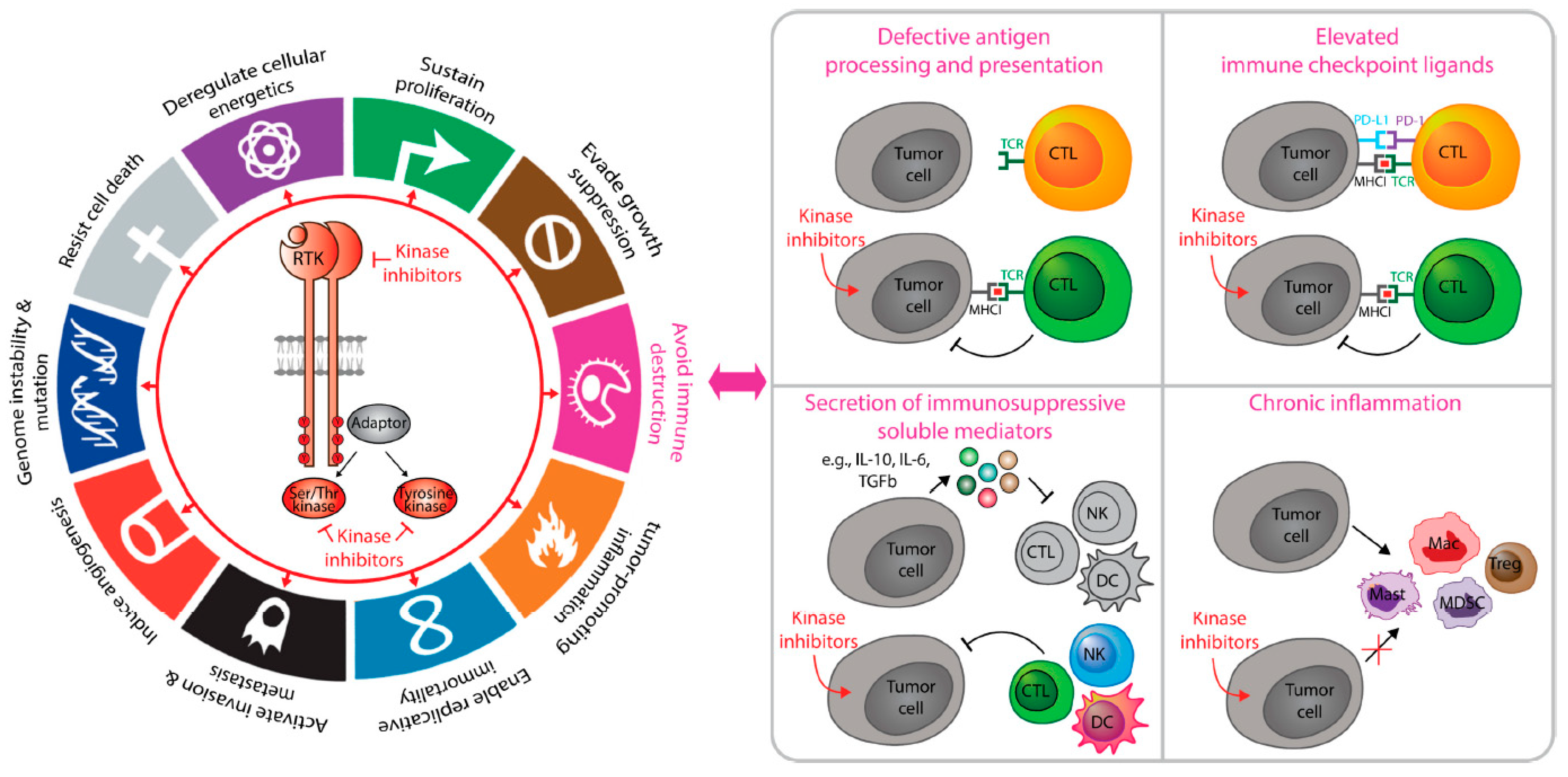
2. Cancer Immunosuppression and Anti-Tumor Immunity
3. Cancer Immunotherapy and Challenges
4. Tumor-Intrinsic Kinase Signaling That Coordinate Cancer Immunosuppression
5. Non-Receptor Kinases in Immunosuppression
6. Receptor Tyrosine Kinases in Immunosuppression
7. Adaptor Proteins in Immunosuppression
8. Kinase Inhibitors Potentiate the Tumoricidal Responses of Immunotherapy
9. Tyrosine Kinase Inhibitors
10. Serine/Threonine Kinase Inhibitors
10.1. CDK4/6 Inhibitors
10.2. RAF/MEK Inhibitors
10.3. PI3K/mTOR Inhibitors
11. Combination Strategies Targeting Kinase Inhibitors Improve the Efficacy of Immune Checkpoint Inhibitors in Breast Cancer
12. Kinase Inhibitors and Tumor-Intrinsic Antigen Processing and Presentation
13. Kinase Inhibitors Impact JAK/STAT-Mediated Tumor Immunity
14. Kinase Inhibitors Target Immune Cells in the Tumor Microenvironment
15. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shankaran, V.; Hiroaki, I.; Allen, T.; Bruce, J.M.; White, P.E.; Swanson, L.J.; Schreiber, R.D. IFN [Gamma] and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Dunn Gavin, P.; Allen, T.; Bruce, H.I.; Lloyd, J.; Robert, D.S. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cantor, H. The Path to Reactivation of Antitumor Immunity and Checkpoint Immunotherapy. Cancer Immunol. Res. 2014, 2, 926–936. [Google Scholar] [CrossRef][Green Version]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro-versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer Despite Immunosurveillance: Immunoselection and Immunosub-version. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and Granzymes: Function, Dysfunction and Human Pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Zachary, S.M.; Sondel, P.M. Nk Cell-Mediated Anti-Body-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. 2016, 76, 5597–5601. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.M.A.; Lee, A.H.S.; Paish, E.C.; Macmillan, R.D.; Ellis, I.O.; Green, A.R. The prognostic significance of B lymphocytes in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2011, 132, 545–553. [Google Scholar] [CrossRef]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef]
- Boero, S.A.; Morabito, B.; Banelli, B.; Cardinali, B.; Dozin, G.; Lunardi, P.; Piccioli, S.; Lastraioli, R.; Carosio, S.S.; Levaggi, F.A.; et al. Analysis of in Vitro Adcc and Clinical Response to Trastuzumab: Possible Relevance of Fcgammariiia/Fcgammariia Gene Polymorphisms and Her-2 Expression Levels on Breast Cancer Cell Lines. J. Transl. Med. 2015, 13, 324. [Google Scholar] [CrossRef]
- Lucarini, V.; Melaiu, O.; Tempora, P.; D’Amico, S.; Locatelli, F.; Fruci, D. Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment. Cancers 2021, 13, 433. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ott, P.A.; Adams, S. Small-Molecule Protein Kinase Inhibitors and Their Effects on the Immune System: Implications for Cancer Treatment. Immunotherapy 2011, 3, 213–227. [Google Scholar] [CrossRef]
- Kroemer, G.; Senovilla, L.; Galluzzi, L.; André, F.; Zitvogel, L. Natural and therapy-induced immunosurveillance in breast cancer. Nat. Med. 2015, 21, 1128–1138. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Hãc Ninger, E.; Krueger, T.E.G.; Lang, J.M.; Heninger, E. Augmenting Antitumor Immune Responses with Epigenetic Modifying Agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Joel, S.; Gupta, R.; Hou, L.; Kurc, T.; Singh, P.; Nguyen, V.; Samaras, D.; Shroyer, K.R.; Zhao, T.; Batiste, R.; et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep. 2018, 23, 23181. [Google Scholar]
- Li, X.; Gruosso, T.; Zuo, D.; Omeroglu, A.; Meterissian, S.; Guiot, M.-C.; Salazar, A.; Park, M.; Levine, H. Infiltration of CD8+ T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3678–3687. [Google Scholar] [CrossRef] [PubMed]
- König, L.; Mairinger, F.D.; Hoffmann, O.; Bittner, A.-K.; Schmid, K.W.; Kimmig, R.; Kasimir-Bauer, S.; Bankfalvi, A. Dissimilar patterns of tumor-infiltrating immune cells at the invasive tumor front and tumor center are associated with response to neoadjuvant chemotherapy in primary breast cancer. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Heindl, A.; Sestak, I.; Naidoo, K.; Cuzick, J.; Dowsett, M.; Yuan, Y. Relevance of Spatial Heterogeneity of Immune Infiltration for Predicting Risk of Recurrence After Endocrine Therapy of ER+ Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 166–175. [Google Scholar] [CrossRef]
- Nawaz, S.; Heindl, A.; Koelble, K.; Yuan, Y. Beyond immune density: Critical role of spatial heterogeneity in estrogen receptor-negative breast cancer. Mod. Pathol. 2015, 28, 766–777. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; De Souza, C.P.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769. [Google Scholar] [CrossRef]
- Corredor, G.; Wang, X.; Zhou, Y.; Lu, C.; Fu, P.; Syrigos, K.N.; Rimm, D.L.; Yang, M.; Romero, E.; Schalper, K.A.; et al. Spatial Architecture and Arrangement of Tumor-Infiltrating Lymphocytes for Predicting Likelihood of Recurrence in Early-Stage Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1526–1534. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Xia, Y.; Medeiros, L.J.; Young, K.H. Immune checkpoint blockade: Releasing the brake towards hematological malignancies. Blood Rev. 2016, 30, 189–200. [Google Scholar] [CrossRef]
- Kong, T.; Ahn, R.; Yang, K.; Zhu, X.; Fu, Z.; Morin, G.; Bramley, R.; Cliffe, N.C.; Xue, Y.; Kuasne, H.; et al. CD44 Promotes PD-L1 Expression and Its Tumor-Intrinsic Function in Breast and Lung Cancers. Cancer Res. 2019, 80, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Saibil, S.D.; Saltman, A.P. Immune checkpoint inhibitors in cancer immunotherapy. Can. Med. Assoc. J. 2020, 192, e651. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; De Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast Cancer Immunotherapy: An Update. Breast Cancer Basic Clin. Res. 2018, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.C.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2016, 47, 52–63. [Google Scholar] [CrossRef]
- Loi, S.N.; Sirtaine, F.; Piette, R.; Salgado, G.; Viale, F.; Van Eenoo, G.; Rouas, P.; Francis, J.P.; Crown, E.; Hitre, E.; et al. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes in a Phase Iii Randomized Adjuvant Breast Cancer Trial in Node-Positive Breast Cancer Com-paring the Addition of Docetaxel to Doxorubicin with Doxorubicin-Based Chemotherapy: Big 02-98. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Savas, P.P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.P.; Smyth, M.J.M.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef]
- Ali, H.R.; Provenzano, E.; Dawson, S.-J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12 439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Emens, A.L.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Duc, A.N.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 379, 2108–2121. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab Plus Chemotherapy Versus Placebo Plus Chemotherapy for Previously Untreated Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer (Key-note-355): A Randomised, Placebo-Controlled, Double-Blind, Phase 3 Clinical Trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Thomas, A.; Routh, E.D.; Pullikuth, A.; Jin, G.; Chou, J.W.; Hoadley, K.A.; Print, C.; Knowlton, N.; Black, M.A.; Demaria, S.; et al. Tumor Mutational Burden is a Determinant of Immune-Mediated Survival in Breast Cancer. Oncoimmunology 2018, 7, e1490854. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Zou, Y.; Zou, X.; Zheng, S.; Tang, H.; Zhang, L.; Liu, P.; Xie, X. Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: A systematic review and meta-analysis. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marté, J.L.; Lepone, L.M.; et al. Avelumab for Meta-static or Locally Advanced Previously Treated Solid Tumours (Javelin Solid Tumor): A Phase 1a, Multicohort, Dose-Escalation Trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Schmid, P.; Cruz, C.; Braiteh, F.S.; Eder, J.P.; Tolaney, S.; Kuter, I.; Nanda, R.; Chung, C.; Cassier, P.; Delord, J.P.; et al. Abstract 2986: Atezolizumab in Metastatic Tnbc (Mtnbc): Long-Term Clinical Outcomes and Biomarker Analyses. Cancer Res. 2017, 77, 2986. [Google Scholar]
- Adams, S.; Schmid, P.; Rugo, H.; Winer, E.; Loirat, D.; Awada, A.; Cescon, D.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Hudson, J.D.; Shoaibi, M.A.; Maestro, R.; Carnero, A.; Hannon, G.J.; Beach, D.H. A Proinflammatory Cytokine Inhibits P53 Tumor Suppressor Activity. J. Exp. Med. 1999, 190, 1375–1382. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; Prives, C. At the Crossroads of Inflammation and Tumorigenesis. J. Exp. Med. 1999, 190, 1367–1370. [Google Scholar] [CrossRef]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. Mmp-9 Supplied by Bone Mar-row-Derived Cells Contributes to Skin Carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; De Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The Braf-Mapk Signaling Pathway is Essential for Cancer-Immune Evasion in Human Melanoma Cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Natali, P.G.; Nicotra, M.R.; Nuti, M.; Bigotti, G.; Calabrò, A.; Schlom, J.; Giacomini, P. Molecular profile, tissue distribution and prognostic evaluation of a human melanoma-carcinoma antigen recognized by the murine monoclonal antibody B1.1. Int. J. Biol. Mark. 1988, 3, 211–220. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; Brin, J.H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.O.; Metzger-Filho, J.; et al. Cdk4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Chen, R.-H.; Xiao, Z.-W.; Yan, X.-Q.; Han, P.; Liang, F.-Y.; Wang, J.-Y.; Yu, S.-T.; Zhang, T.-Z.; Chen, S.-Q.; Zhong, Q.; et al. Tumor Cell-Secreted ISG15 Promotes Tumor Cell Migration and Immune Suppression by Inducing the Macrophage M2-Like Phenotype. Front. Immunol. 2020, 11, 64. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; Von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 1–18. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and Mertk (Tam Receptors): Implications for Macrophages in the Tumor Microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Xun, Q.; Wang, Z.; Hu, X.; Ding, K.; Lu, X. Small-Molecule CSF1R Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 3944–3966. [Google Scholar] [CrossRef]
- Hato, T.; Zhu, A.X.; Duda, D.G. Rationally Combining Anti-Vegf Therapy with Checkpoint Inhibitors in Hepatocellular Carcinoma. Immunotherapy 2016, 8, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Faham, N.; Welm, A.L. RON Signaling is a Key Mediator of Tumor Progression in Many Human Cancers. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.J.; Sung, J.H.; Kim, J.W.; Han, S.-H.; Lee, H.S.; Min, A.; Kang, M.H.; Kim, J.E.; Kim, S.H.; Lee, J.-O.; et al. EGFR or HER2 inhibition modulates the tumor microenvironment by suppression of PD-L1 and cytokines release. Oncotarget 2017, 8, 63901–63910. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Yu, S.; Sha, H.; Qin, X.; Chen, Y.; Li, X.; Shi, M.; Feng, J. Egfr E746-A750 Deletion in Lung Cancer Represses Anti-Tumor Immunity through the Exosome-Mediated Inhibition of Dendritic Cells. Oncogene 2020, 39, 2643–2657. [Google Scholar] [CrossRef]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajesh Kumar, N.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef]
- Ishihara, H.; Sasaoka, T.; Ishiki, M.; Takata, Y.; Imamura, T.; Usui, I.; Langlois, W.J.; Sawa, T.; Kobayashi, M. Functional Importance of Shc Tyrosine 317 on Insulin Signaling in Rat1 Fibroblasts Expressing Insulin Receptors. J. Biol. Chem. 1997, 272, 9581–9586. [Google Scholar] [CrossRef]
- Sasaoka, T.; Kobayashi, M. The Functional Significance of Shc in Insulin Signaling as a Substrate of the Insulin Receptor. Endocr. J. 2000, 47, 373–381. [Google Scholar] [CrossRef]
- Galvagni, F.; Pennacchini, S.; Salameh, A.; Rocchigiani, M.; Neri, F.; Orlandini, M.; Petraglia, F.; Gotta, S.; Sardone, G.L.; Matteucci, G.; et al. Endothelial Cell Adhesion to the Extracellular Matrix Induces c-Src–Dependent VEGFR-3 Phosphorylation Without the Activation of the Receptor Intrinsic Kinase Activity. Circ. Res. 2010, 106, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Hardy, W.R.; Murphy, J.M.; Jones, N.; Pawson, T. Screening for PTB Domain Binding Partners and LigandSpecificity Using Proteome-Derived NPXY Peptide Arrays. Mol. Cell. Biol. 2006, 26, 8461–8474. [Google Scholar] [CrossRef]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.R.; Siegel, P.M.; Ursini-Siegel, J. The Tyrosine Kinome Dictates Breast Cancer Heterogeneity and Therapeutic Responsiveness. J. Cell. Biochem. 2016, 117, 1971–1990. [Google Scholar] [CrossRef]
- Mishra, J.; Kumar, N. Adapter Protein Shc Regulates Janus Kinase 3 Phosphorylation. J. Biol. Chem. 2014, 289, 15951–15956. [Google Scholar] [CrossRef] [PubMed]
- Klint, P.; Kanda, S.; Claesson-Welsh, L. Shc and a Novel 89-kDa Component Couple to the Grb2-Sos Complex in Fibroblast Growth Factor-2-stimulated Cells. J. Biol. Chem. 1995, 270, 23337–23344. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Muller, W.J. The ShcA Adaptor Protein is a Critical Regulator of Breast Cancer Progression. Cell Cycle 2008, 7, 1936–1943. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Cory, S.; Zuo, D.; Hardy, W.R.; Rexhepaj, E.; Lam, S.; Schade, B.; Jirstrom, K.; Bjur, E.; Piccirillo, C.A.; et al. Receptor Tyrosine Kinase Signaling Favors a Protumorigenic State in Breast Cancer Cells by Inhibiting the Adaptive Immune Response. Cancer Res. 2010, 70, 7776–7787. [Google Scholar] [CrossRef]
- Ahn, R.; Sabourin, V.; Bolt, A.M.; Hébert, S.; Totten, S.; De Jay, N.; Festa, M.C.; Young, Y.K.; Im, Y.K.; Pawson, T.; et al. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. OncoImmunology 2017, 7, e1376155. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Rev. Cancer 2021, 1–17. [Google Scholar] [CrossRef]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib Inhibition of Stat3 Induces Renal Cell Carcinoma Tumor Cell Apoptosis and Reduces Immunosuppressive Cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef]
- Eyob, H.; Ekiz, H.A.; Derose, Y.S.; Waltz, S.E.; Williams, M.A.; Welm, A.L. Inhibition of Ron Kinase Blocks Conversion of Micrometastases to Overt Metastases by Boosting Antitumor Immunity. Cancer Discov. 2013, 3, 751–760. [Google Scholar] [CrossRef]
- Hannesdóttir, L.; Tymoszuk, P.; Parajuli, N.; Wasmer, M.-H.; Philipp, S.; Daschil, N.; Datta, S.; Koller, J.-B.; Tripp, C.H.; Stoitzner, P.; et al. Lapatinib and doxorubicin enhance the Stat1-dependent antitumor immune response. Eur. J. Immunol. 2013, 43, 2718–2729. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tam, K.Y. Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Paz-Ares, L.; Bennouna, J.; Felip, E.; Rodríguez Abreu, D.; Isla, D.; Barlesi, F.; Molinier, O.; Madelaine, J.; Audigier-Valette, C.; et al. Afatinib with Pembrolizumab for Treatment of Patients with Locally Advanced/Metastatic Squamous Cell Carcinoma of the Lung: The Lux-Lung Io/Keynote 497 Study Protocol. Clin. Lung Cancer 2019, 20, e407–e412. [Google Scholar] [CrossRef]
- Atkins, M.B.; Tannir, N.M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat. Rev. 2018, 70, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Blanc, J.F.; Okusaka, T.; Chau, I.; Cella, D.; Girvan, A.; Gable, J.; et al. 622pd-Ramucirumab as Second-Line Treatment in Patients with Advanced Hepatocellular Carcinoma (Hcc) and Elevated Alpha-Fetoprotein (Afp) Following First-Line Sorafenib: Patient Reported Outcome Results across Two Phase Iii Studies (Reach-2 and Reach). Ann. Oncol. 2018, 29, viii208. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Marino, D.; Zichi, C.; Audisio, M.; Sperti, E.; Di Maio, M. Second-line treatment options in hepatocellular carcinoma. Drugs Context 2019, 8, 1–13. [Google Scholar] [CrossRef]
- Ma, B.; Rudin, C.; Cervantes, A.; Dowlati, A.; Costa, D.; Schmid, P.; Heist, R.; Villaflor, V.; Sarkar, I.; Huseni, M.; et al. 441O Preliminary safety and clinical activity of erlotinib plus atezolizumab from a Phase Ib study in advanced NSCLC. Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Yang, J.; Yu, H.; Saka, H.; Ramalingam, S.; Goto, K.; Kim, S.-W.; Yang, L.; Walding, A.; Oxnard, G. 136O: Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: Results from the TATTON phase Ib trial. J. Thorac. Oncol. 2016, 11, S115. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Cuomo, A.; Spadoni, I.; Magni, E.; Silvola, A.; Conte, A.; Sigismund, S.; Ravenda, P.S.; Bonaldi, T.; Zampino, M.G.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Cabo, M.; Servitja, S.; Tusquets, I.; Martínez-García, M.; Rovira, A.; Rojo, F.; Albanell, J.; López-Botet, M. Interplay between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front. Immunol. 2017, 8, 1544. [Google Scholar] [CrossRef] [PubMed]
- Concha-Benavente, F.; Ferris, R. Jak2 Inhibition Prevents Nk-Released Ifnγ-Mediated Pd-L1 Upreg-ulation and Enhances Cetuximab Mediated Adcc of Hnc Cells (Tum2p.1014). J. Immunol. 2015, 194, 69. [Google Scholar]
- Lizotte, P.H.; Hong, R.L.; Luster, T.A.; Cavanaugh, M.E.; Taus, L.J.; Wang, S.; Dhaneshwar, A.; Mayman, N.; Yang, A.; Kulkarni, M.L.; et al. A High-Throughput Immune-Oncology Screen Identifies Egfr Inhibitors as Potent Enhancers of Antigen-Specific Cytotoxic T-Lymphocyte Tumor Cell Killing. Cancer Immunol. Res. 2018, 6, 1511–1523. [Google Scholar] [CrossRef]
- Liang, H.; Liu, X.; Wang, M. Immunotherapy combined with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer treatment. Oncol. Targets Ther. 2018, 11, 6189–6196. [Google Scholar] [CrossRef]
- Venugopalan, A.; Lee, M.-L.; Niu, G.; Medina-Echeverz, J.; Tomita, Y.; Lizak, M.J.; Cultraro, C.M.; Simpson, R.M.; Chen, X.; Trepel, J.B.; et al. Egfr-Targeted Therapy Results in Dramatic Early Lung Tumor Regression Accompanied by Imaging Response and Immune Infiltration in Egfr Mutant Transgenic Mouse Models. Oncotarget 2016, 7, 54137. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib as Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Press, M.F.; Chan, D.; Fernandez-Abad, M.; Petru, E.; Rostorfer, R.; Guarneri, V.; Huang, C.-S.; Barriga, S.; et al. Potent Cell-Cycle Inhibition and Upregulation of Immune Response with Abemaciclib and Anastrozole in Neomonarch, Phase Ii Neoadjuvant Study in Hr+/Her2− Breast Cancer. Clin. Cancer Res. 2020, 26, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The Cdk4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of Pd-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]
- Dowless, M.S.; Lowery, C.D.; Shackleford, T.J.; Renschler, M.; Stephens, J.R.; Flack, R.; Blosser, W.; Gupta, S.; Stewart, J.; Webster, Y.; et al. Abemaciclib is Active in Preclinical Models of Ewing Sarcoma via Multipronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin. Cancer Res. 2018, 24, 6028–6039. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nat. Cell Biol. 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Ebert, P.J.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab Plus Cobimetinib and Vemurafenib in Braf-Mutated Melanoma Patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Dummer, R.; Lebbé, C.; Atkinson, V.; Mandalà, M.; Nathan, P.D.; Arance, A.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. Combined PD-1, BRAF and MEK Inhibition in Advanced Braf-Mutant Melanoma: Safety Run-in and Biomarker Cohorts of COMBI-I. Nat. Med. 2020, 26, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved Antitumor Activity of Immunotherapy with Braf and Mek Inhibitors in Braf (V600e) Melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; See-staller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The Braf and Mek Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immuno-modulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef]
- Reinhard, D.; Ramelyte, E.; Schindler, S.; Thürigen, O.; Levesque, M.P.; Koelblinger, P. Mek Inhibition and Immune Responses in Advanced Melanoma. Oncoimmunology 2017, 6, e1335843. [Google Scholar]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. Braf Inhibition is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; Van Gool, M.; Kroon, P.; Pineda, C.; Foppen, M.H.G.; Scolyer, R.; Song, J.-Y.; et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 2016, 5, e1238557. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting Adenosine in BRAF-Mutant Melanoma Reduces Tumor Growth and Metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Chu, J.; Cargnello, M.; Topisirovic, I.; Pelletier, J. Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol. 2016, 26, 918–933. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. Pi3k-Akt-mTOR Inhibition in Cancer Immunotherapy, Re-dux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of Pd-L1 Expression by Oncogenic Activation of the Akt-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.-Y.; Chen, S.; Zhao, X.; Lei, C.-Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Erratum: Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 822. [Google Scholar] [CrossRef] [PubMed]
- Villegas, S.N.; Gombos, R.; García-López, L.; Gutiérrez-Pérez, I.; García-Castillo, J.; Vallejo, D.M.; Da Ros, V.G.; Balles-ta-Illán, E.; Mihály, J.; Dominguez, M. Pi3k/Akt Cooperates with Oncogenic Notch by Inducing Nitric Oxide-Dependent Inflammation. Cell Rep. 2018, 22, 2541–2549. [Google Scholar] [CrossRef]
- Xu, Y.; Poggio, M.; Jin, H.Y.; Shi, Z.; Forester, C.M.; Wang, Y.; Stumpf, C.R.; Xue, L.; Devericks, E.; So, L.; et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Guemiri, R.; Druillennec, S.; Girault, I.; Malka-Mahieu, H.; Shen, S.; Allard, D.; Martineau, S.; Welsch, C.; Agoussi, S.; et al. Translational Control of Tumor Immune Escape Via the Eif4f-Stat1-Pd-L1 Axis in Mela-noma. Nat. Med. 2018, 24, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Galvin, K.C.; Corcoran, A.-M.B.; Boon, L.; Higgs, R.; Mills, K.H. Immunotherapy with PI3K Inhibitor and Toll-Like Receptor Agonist Induces IFN-γ+IL-17+ Polyfunctional T Cells That Mediate Rejection of Murine Tumors. Cancer Res. 2012, 72, 581–591. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef]
- Prabhu, S.A.; Moussa, O.; Miller, W.H., Jr.; Del Rincón, S.V. The Mnk1/2-Eif4e Axis as a Potential Therapeutic Target in Melanoma. Int. J. Mol. Sci. 2020, 21, 4055. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR+ Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.R.; Liu, K.X.; Pham, T.V.; Zulcic, M.; Skola, D.; Chun, H.B.; Glass, C.K.; Morales, G.A.; Garlich, J.R.; et al. SF2523: Dual PI3K/BRD4 Inhibitor Blocks Tumor Immunosuppression and Promotes Adaptive Immune Responses in Cancer. Mol. Cancer Ther. 2019, 18, 1036–1044. [Google Scholar] [CrossRef]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.Y.; Subjeck, J.R.; Shrikant, P.A.; Kim, H.L. Temsirolimus, an mTOR Inhibitor, Enhances An-ti-Tumour Effects of Heat Shock Protein Cancer Vaccines. Br. J. Cancer 2011, 104, 643–652. [Google Scholar] [CrossRef]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bärtschi, D.; Dröge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Phase 2 Trial of Single-Agent Everolimus in Chemotherapy-Naive Patients with Castration-Resistant Prostate Cancer (Sakk 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Amandine, P.; Papaserafeim, M.; Anke Rietveld, N.L.; Kaestel, C.; Gruaz, L.; Vonarburg, C.; Spirig, R.; Puga Yung, G.L.; Seebach, J.G. Small-Molecule Immunosuppressive Drugs and Therapeutic Immu-noglobulins Differentially Inhibit Nk Cell Effector Functions in Vitro. Front. Immunol. 2019, 10, 556. [Google Scholar]
- Pascual, J.; Berger, S.P.; Witzke, O.; Tedesco, H.; Mulgaonkar, S.; Qazi, Y.; Chadban, S.; Oppenheimer, F.; Sommerer, C.; Oberbauer, R.; et al. Everolimus with Reduced Calcineurin Inhibitor Exposure in Renal Transplantation. J. Am. Soc. Nephrol. 2018, 29, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, D.; Li, J.; Zhu, L.; Zhang, C.; Lei, K.; Xu, Q.; You, R. Efficacy and Safety of Everolimus for Maintenance Immunosuppression of Kidney Transplantation: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2017, 12, e0170246. [Google Scholar] [CrossRef]
- Huijts, C.M.; Lougheed, S.M.; Bodalal, Z.; Van Herpen, C.M.; Hamberg, P.; Tascilar, M.; Haanen, J.B.; Verheul, H.M.; De Gruijl, T.D. The effect of everolimus and low-dose cyclophosphamide on immune cell subsets in patients with metastatic renal cell carcinoma: Results from a phase I clinical trial. Cancer Immunol. Immunother. 2019, 68, 503–515. [Google Scholar] [CrossRef]
- Hamilton, E.; Blackwell, K.; Hobeika, A.C.; Clay, T.M.; Broadwater, G.; Ren, X.-R.; Chen, W.; Castro, H.; Lehmann, F.; Spector, N.; et al. Phase I clinical trial of HER2-specific immunotherapy with concomitant HER2 kinase inhibition. J. Transl. Med. 2012, 10, 28. [Google Scholar] [CrossRef]
- Chen, G.; Gupta, R.; Petrik, S.; Laiko, M.; Leatherman, J.M.; Asquith, J.M.; Daphtary, M.M.; Garrett-Mayer, E.; Davidson, N.E.; Hirt, K.; et al. A Feasibility Study of Cyclophosphamide, Trastuzumab, and an Allogeneic GM-CSF–Secreting Breast Tumor Vaccine for HER2+ Metastatic Breast Cancer. Cancer Immunol. Res. 2014, 2, 949–961. [Google Scholar] [CrossRef]
- Kwilas, A.R.; Ardiani, A.; Donahue, R.N.; Aftab, D.T.; Hodge, J.W. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J. Transl. Med. 2014, 12, 1–15. [Google Scholar] [CrossRef]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib Enhances Mhc Induction in Braf (V600e) Homozygous Melanoma Cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Mesfin, J.M.; Joughin, B.A.; Lauffenburger, D.A.; White, F.M. Multiplexed Relative and Absolute Quantitative Immunopeptidomics Reveals Mhc I Repertoire Alterations Induced by Cdk4/6 Inhibition. Nat. Commun. 2020, 11, 2760. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.H.; Melief, C.J.M. Identification of T-cell epitopes for cancer immunotherapy. Leukemia 2007, 21, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The Molecular Regulation of Janus Kinase (Jak) Activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Ivashkiv, L.B. Role of Stat3 in Type I Interferon Responses. Negative Regulation of Stat1-Dependent Inflammatory Gene Activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between Cancer and Immune Cells: Role of Stat3 in the Tumour Microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Madera, S.; Rapp, M.; Firth, M.A.; Beilke, J.N.; Lanier, L.L.; Sun, J.C. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 2016, 213, 225–233. [Google Scholar] [CrossRef]
- Meissl, K.; Simonović, N.; Amenitsch, L.; Witalisz-Siepracka, A.; Klein, K.; Lassnig, C.; Puga, A.; Vogl, C.; Poelzl, A.; Bosmann, M.; et al. STAT1 Isoforms Differentially Regulate NK Cell Maturation and Anti-tumor Activity. Front. Immunol. 2020, 11, 2189. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Biswas, A.; Field, M.; Snapper, S.B. STAT1 signaling shields T cells from NK cell-mediated cytotoxicity. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Consales, C.; Valentini, E.; Albas, A.; Mendonca, R.; Fuches, R.; Soares, M.; Pereira, C. The preparation of cultured rabies virus and the production of antiserum for human use. J. Biol. Stand. 1988, 16, 27–32. [Google Scholar] [CrossRef]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; De Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Zhan, X.; Guo, S.; Li, Y.; Ran, H.; Huang, H.; Mi, L.; Wu, J.; Wang, X.; Xiao, D.; Chen, L.; et al. Glioma Stem-Like Cells Evade Interferon Suppression through Mbd3/Nurd Complex-Mediated Stat1 Downregulation. J. Exp. Med. 2020, 4, e20191340. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Pereira, R.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Wang, T.; et al. Activation of the IFN Signaling Pathway is Associated with Resistance to Cdk4/6 Inhibitors and Immune Checkpoint Activation in Er-Positive Breast Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Huang, J.; Chen, P.; Liu, K.; Liu, J.; Zhou, B.; Wu, R.; Peng, Q.; Liu, Z.X.; Li, C.; Kroemer, G.; et al. Cdk1/2/5 Inhibition Overcomes Ifng-Mediated Adaptive Immune Resistance in Pancreatic Cancer. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Showalter, L.E.; Oechsle, C.; Ghimirey, N.; Steele, C.; Czerniecki, B.J.; Koski, G.K. Th1 cytokines sensitize HER-expressing breast cancer cells to lapatinib. PLoS ONE 2019, 14, e0210209. [Google Scholar] [CrossRef]
- Chaganty, B.K.R.; Qiu, S.; Gest, A.; Lu, Y.; Ivan, C.; Calin, G.A.; Weiner, L.M.; Fan, Z. Trastuzumab Upregulates Pd-L1 as a Potential Mechanism of Trastuzumab Resistance through Engagement of Immune Effector Cells and Stimulation of Ifnγ Secretion. Cancer Lett. 2018, 430, 47–56. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.-C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef]
- Yamamoto, N.; Honma, M.; Suzuki, H. Off-Target Serine/Threonine Kinase 10 Inhibition by Erlotinib Enhances Lymphocytic Activity Leading to Severe Skin Disorders. Mol. Pharm. Ther. 2011, 80, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Al-Aubodah, T.-A.; Thebault, P.; Lapointe, R.; Hudson, M.; Johnson, N.A.; Baran, D.; Bhulaiga, N.; Takano, T.; Cailhier, J.-F.; et al. Targeting the mTOR pathway uncouples the efficacy and toxicity of PD-1 blockade in renal transplantation. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Lu, Q.; Lemke, G. Homeostatic Regulation of the Immune System by Receptor Tyrosine Kinases of the Tyro 3 Family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nat. Cell Biol. 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; De Ryckere, D.; Davies, K.D.; Earp, H.S. The Tam Family: Phosphatidylserine-Sensing Receptor Tyrosine Kinases Gone Awry in Cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Burstyn-Cohen, T.; Maimon, A. TAM receptors, Phosphatidylserine, inflammation, and Cancer. Cell Commun. Signal. 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor Tyrosine Kinases and Tlr/Il1rs Unexpectedly Activate Myeloid Cell Pi3kγ, a Single Convergent Point Promoting Tumor Inflammation and Progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Bartish, M.; Tong, D.; Pan, Y.; Wallerius, M.; Liu, H.; Ristau, J.; Ferreira, S.D.S.; Wallmann, T.; Van Hoef, V.; Masvidal, L.; et al. MNK2 governs the macrophage antiinflammatory phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 27556–27565. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Jin, Z.; Lu, X. Differential Targeting of Gr-MDSCs, T Cells and Prostate Cancer Cells by Dactolisib and Dasatinib. Int. J. Mol. Sci. 2020, 21, 2337. [Google Scholar] [CrossRef] [PubMed]
- Lu, X. 2025 Targeting immunosuppressive myeloid cells to enhance cancer immunotherapy. J. Clin. Transl. Sci. 2018, 2, 29. [Google Scholar] [CrossRef][Green Version]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia Promote Glioblastoma Via mTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated T (Reg) Subsets to Protect Tissue Homeo-stasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, X.; Zang, A.; Li, T.; Hu, Y.; Ma, S.; Lü, M.; Yin, H.; Wang, H.; Zhang, X.; et al. TSC1/mTOR-controlled metabolic–epigenetic crosstalk underpins DC control of CD8+ T-cell homeostasis. PLoS Biol. 2019, 17, e3000420. [Google Scholar] [CrossRef] [PubMed]
- Polk, A.; Svane, I.-M.; Andersson, M.; Nielsen, D. Checkpoint inhibitors in breast cancer—Current status. Cancer Treat. Rev. 2018, 63, 122–134. [Google Scholar] [CrossRef]
- Wang, F.; Meng, M.; Mo, B.; Yang, Y.; Ji, Y.; Huang, P.; Lai, W.; Pan, X.; You, T.; Luo, H.; et al. Crosstalks between mTORC1 and mTORC2 variagate cytokine signaling to control NK maturation and effector function. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Katholnig, K.; Schütz, B.; Fritsch, S.D.; Schörghofer, D.; Linke, M.; Sukhbaatar, N.; Matschinger, J.M.; Unterleuthner, D.; Hirtl, M.; Lang, M.; et al. Inactivation of mTORC2 in macrophages is a signature of colorectal cancer that promotes tumorigenesis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, R.; Ursini-Siegel, J. Clinical Potential of Kinase Inhibitors in Combination with Immune Checkpoint Inhibitors for the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 2608. https://doi.org/10.3390/ijms22052608
Ahn R, Ursini-Siegel J. Clinical Potential of Kinase Inhibitors in Combination with Immune Checkpoint Inhibitors for the Treatment of Solid Tumors. International Journal of Molecular Sciences. 2021; 22(5):2608. https://doi.org/10.3390/ijms22052608
Chicago/Turabian StyleAhn, Ryuhjin, and Josie Ursini-Siegel. 2021. "Clinical Potential of Kinase Inhibitors in Combination with Immune Checkpoint Inhibitors for the Treatment of Solid Tumors" International Journal of Molecular Sciences 22, no. 5: 2608. https://doi.org/10.3390/ijms22052608
APA StyleAhn, R., & Ursini-Siegel, J. (2021). Clinical Potential of Kinase Inhibitors in Combination with Immune Checkpoint Inhibitors for the Treatment of Solid Tumors. International Journal of Molecular Sciences, 22(5), 2608. https://doi.org/10.3390/ijms22052608