
Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
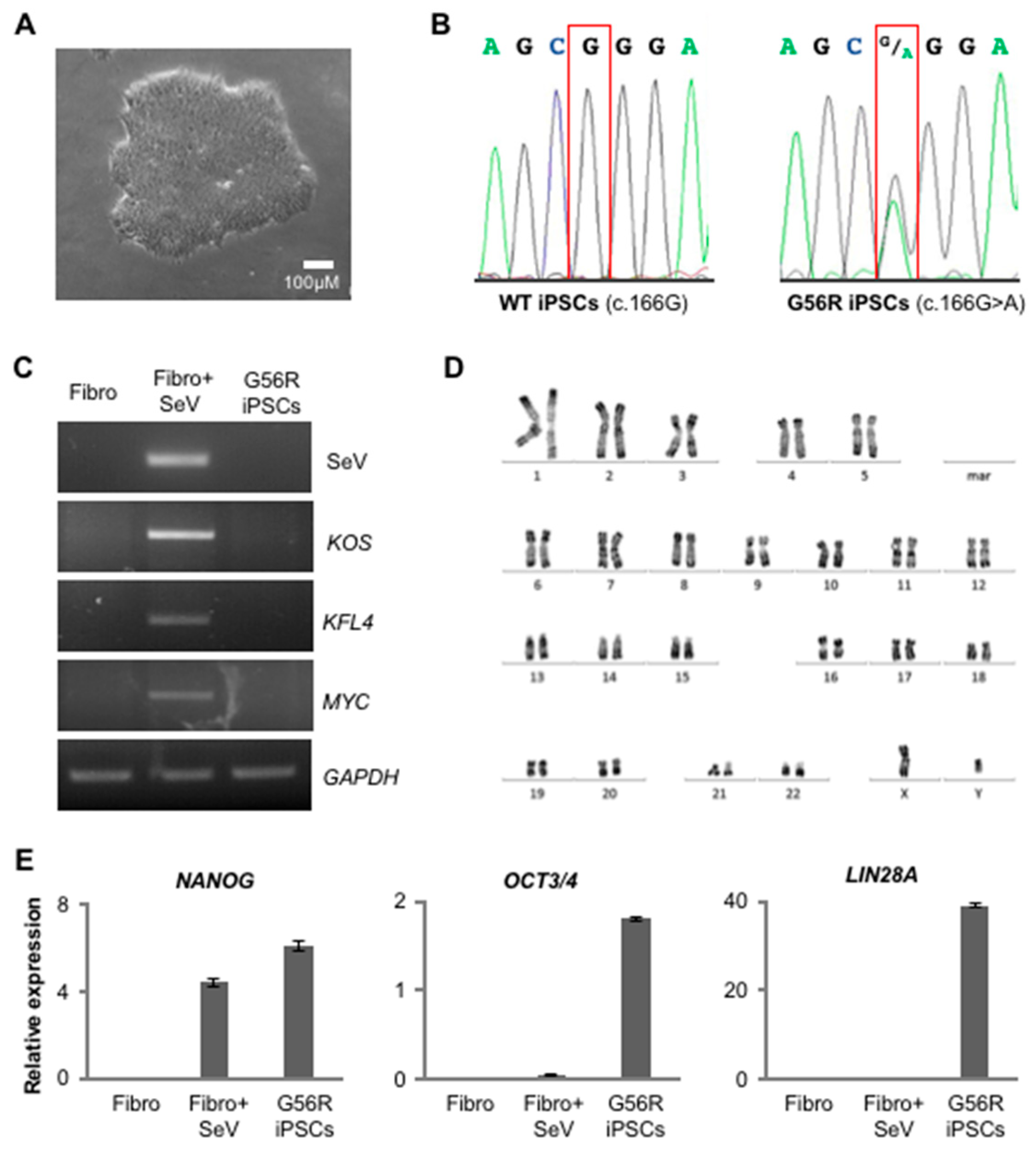
2.1. Reprogramming G56R Fibroblasts to iPSCs
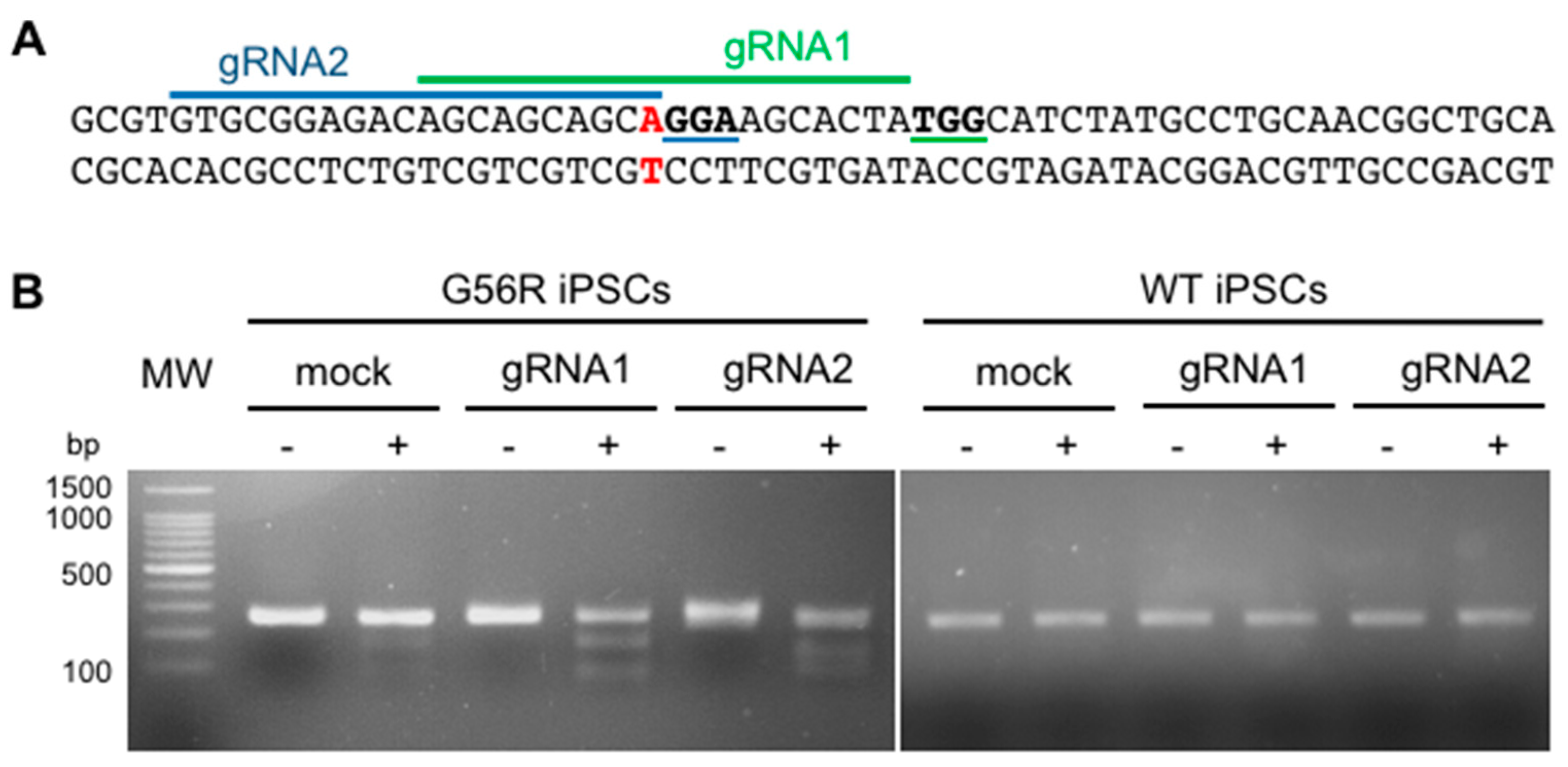
2.2. Designing a G56R-Specific Knockout Strategy
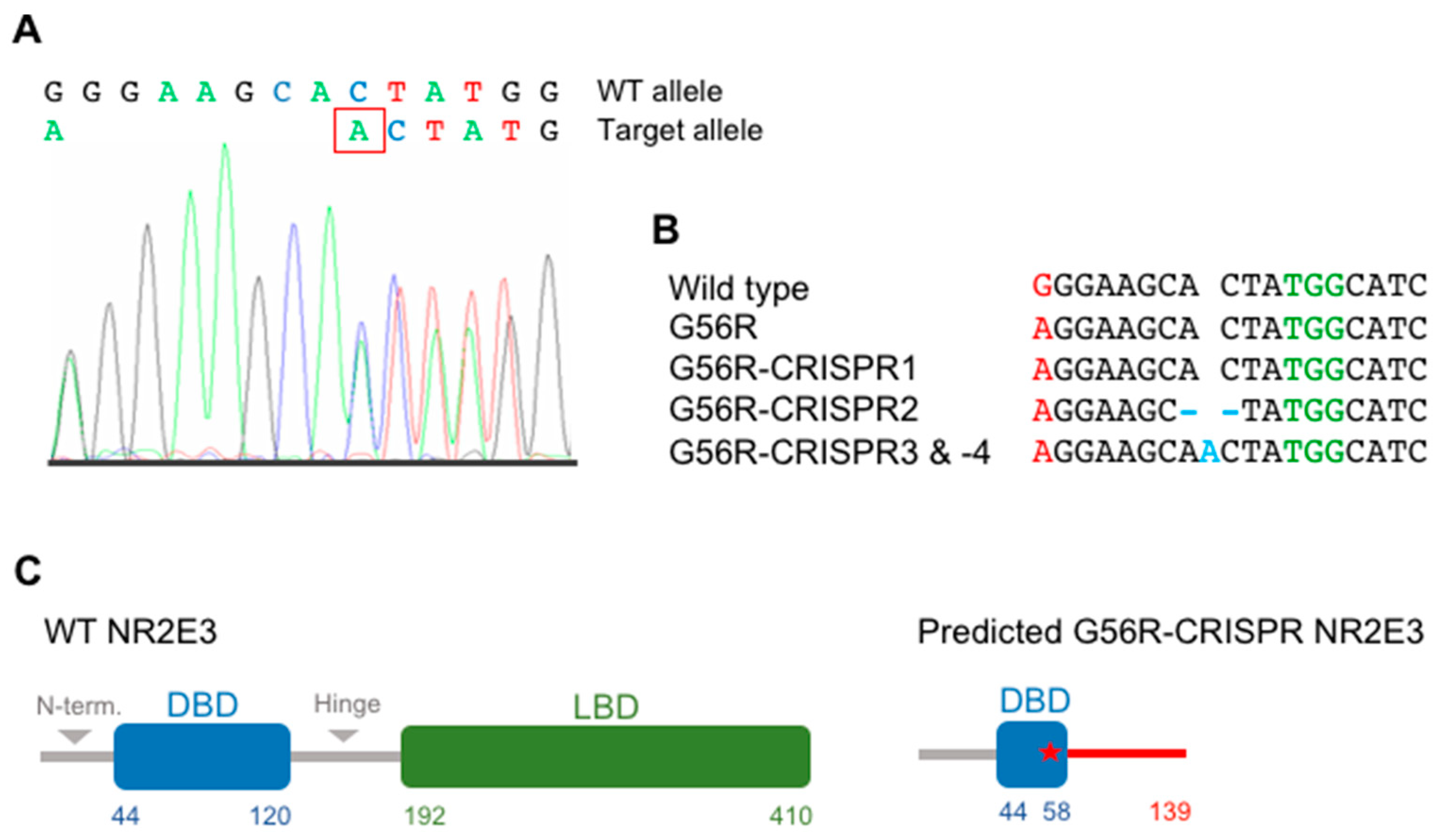
2.3. Knocking Out the G56R Allele in Patient iPSCs
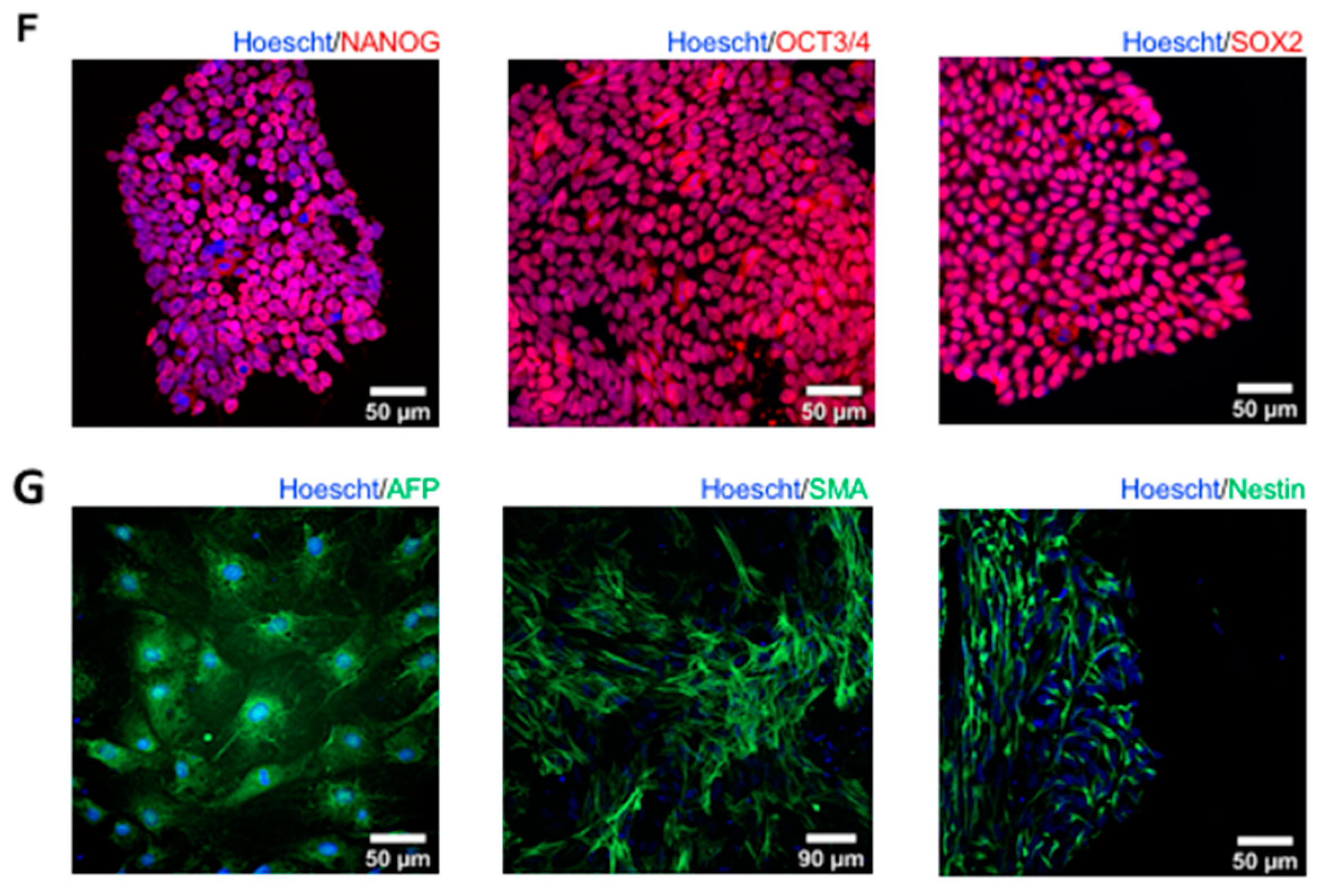
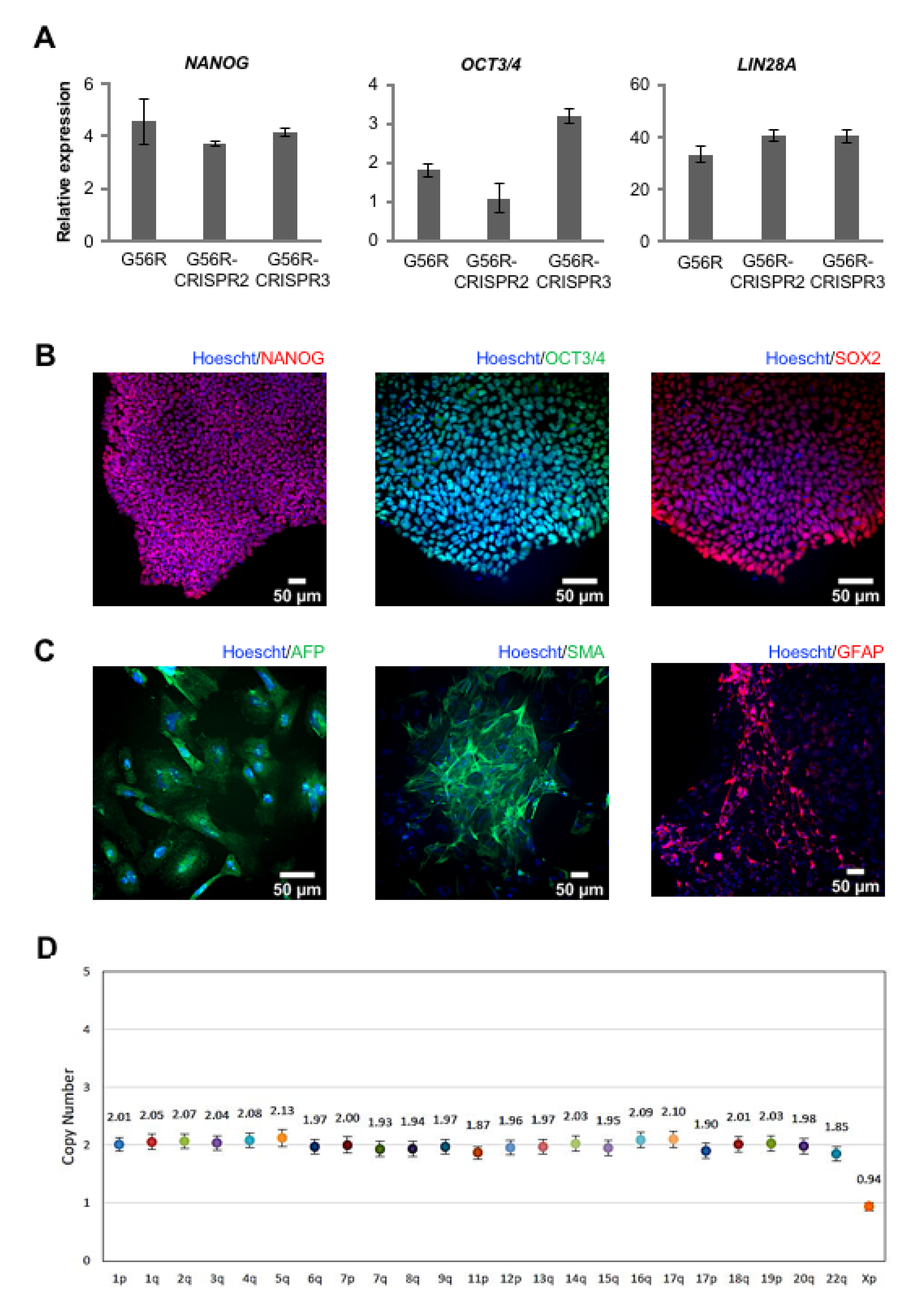
2.4. Characterizing the Pluripotency and Stability of G56R-CRISPR iPSC Lines
2.5. Validating Allele-Specific Knockout of Mutant G56R NR2E3 at the Protein Level
2.6. Assessing the Retinal Organoid Differentiation Potential of a G56R-CRISPR iPSC Line
3. Discussion
4. Materials and Methods
4.1. Skin Biopsy and Fibroblast Culture
4.2. iPSC Reprogramming and Culture
4.3. DNA Sequencing
4.4. RT-PCR and qPCR
4.5. Chromosome Integrity Analyses
4.6. Embryoid Body and Retinal Organoid Differentiation
4.7. Immunofluorescence Staining
4.8. Design of gRNAs and Plasmid Construction
4.9. iPSC Transfection and FACS
4.10. T7 Endonuclease I Assay
4.11. Off-Target Analysis
4.12. Mutagenesis and NR2E3 cDNA Plasmid Construction
4.13. HEK293 and COS7 Culture and Transfection
4.14. Western Blot Analyses
4.15. Differential Centrifugation
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino Acid(s) |
| ad | Autosomal Dominant |
| AFP | Alpha Fetal Protein |
| Cas | CRISPR-associated |
| COS7 | CV-1 Origin Sv40 |
| CNV | Copy Number Variant |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CRX | Cone-Rod Homeobox |
| DBD | DNA Binding Domain |
| DSB | Double-Strand Break |
| EGFP | Enhanced Green Fluorescent Protein |
| ESCS | Enhanced S Cone Syndrome |
| GFAP | Glial Fibrillary Acidic Protein |
| gRNA | Guide RNA |
| HDR | Homologous-Directed Repair |
| HEK293 | Human Embryonic Kidney |
| indels | Insertions/deletions |
| iPSC | induced Pluripotent Stem Cell |
| IRD | Inherited Retinal Dystrophies |
| KLF4 | Kruppel-Like Factor |
| LBD | Ligand Binding Domain |
| LIN28A | Lin-28 Homolog A |
| NANOG | Nanog Homeobox |
| NES | Nuclear Export Signal |
| NHEJ | Non-Homologous End Joining |
| NLS | Nuclear Localization Signal |
| NMD | Nonsense-Mediated Decay |
| NR1D1 | Nuclear Receptor Subfamily 1 Group D Member 1 |
| NR2E3 | Nuclear Receptor Subfamily 2 Group E Member |
| NRL | Neural Retina Leucine Zipper |
| OCT3/4 | Octamer Binding Transcription Factor |
| OTX2 | Orthodenticle Homeobox 2 |
| PAM | Protospacer Adjacent Motif |
| PTC | Premature Termination Codon |
| RP | Retinitis Pigmentosa |
| SeV | Sendai Virus |
| SMA | Smooth Muscle Actin |
| SOX2 | SRY-Box Transcription Factor 2 |
| Sp | Streptococcus Pyogenes |
| T7E1 | T7 Endonuclease 1 |
| WT | Wild type |
References
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome editing as a treatment for the most prevalent causative genes of autosomal dominant retinitis pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Gire, A.I.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1-2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar] [PubMed]
- Blanco-Kelly, F.; García Hoyos, M.; Lopez Martinez, M.A.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Fernandez-San Jose, P.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; García Sandoval, B.; et al. Dominant retinitis pigmentosa, p.Gly56Arg mutation in NR2E3: Phenotype in a large cohort of 24 cases. PLoS ONE 2016, 11, e0149473. [Google Scholar] [CrossRef]
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2e3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Khanna, H.; Oh, E.C.T.; Hicks, D.; Mitton, K.P.; Swaroop, A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 2004, 13, 1563–1575. [Google Scholar] [CrossRef]
- Peng, G.H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Mollema, N.J.; Yuan, Y.; Jelcick, A.S.; Sachs, A.J.; von Alpen, D.; Schorderet, D.; Escher, P.; Haider, N.B. Nuclear receptor Rev-erb alpha (Nr1d1) functions in concert with Nr2e3 to regulate transcriptional networks in the retina. PLoS ONE 2011, 6, e17494. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takezawa, S.; Hara, K.; Yu, R.T.; Umesono, Y.; Agata, K.; Taniwaki, M.; Yasuda, K.; Umesono, K. Identification of a photoreceptor cell-specific nuclear receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 4814–4819. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Zhou, X.E.; Soon, F.F.; Li, X.; Li, J.; Yong, E.L.; Melcher, K.; Xu, H.E. The Crystal Structure of the Orphan Nuclear Receptor NR2E3/PNR Ligand Binding Domain Reveals a Dimeric Auto-Repressed Conformation. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aísa-Marín, I.; López-Iniesta, M.J.; Milla, S.; Lillo, J.; Navarro, G.; de la Villa, P.; Marfany, G. Nr2e3 functional domain ablation by CRISPR-Cas9D10Aidentifies a new isoform and generates retinitis pigmentosa and enhanced S-cone syndrome models. Neurobiol. Dis. 2020, 146, 105122. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef]
- Roduit, R.; Escher, P.; Schorderet, D.F. Mutations in the DNA-binding domain of NR2E3 affect in vivo dimerization and interaction with CRX. PLoS ONE 2009, 4, e7379. [Google Scholar] [CrossRef]
- Kanda, A.; Swaroop, A. A comprehensive analysis of sequence variants and putative disease-causing mutations in photoreceptor-specific nuclear receptor NR2E3. Mol. Vis. 2009, 15, 2174–2184. [Google Scholar]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex Genome Engineering Using CRISPR/VCas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, N.B.; Piriev, N.I.; Chang, B.; Rapoport, A.L.; Hawes, N.L.; Nishina, P.M.; Nusinowitz, S.; Heckenlively, J.R.; Roderick, T.H.; Kozak, C.A.; et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 5551–5556. [Google Scholar] [CrossRef]
- Webber, A.L.; Hodor, P.; Thut, C.J.; Vogt, T.F.; Zhang, T.; Holder, D.J.; Petrukhin, K. Dual role of Nr2e3 in photoreceptor development and maintenance. Exp. Eye Res. 2008, 87, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Erkilic, N.; Sanjurjo-Soriano, C.; Diakatou, M.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi003-A, with a missense mutation in CRX associated with autosomal dominant cone-rod dystrophy. Stem Cell Res. 2019, 38, 101478. [Google Scholar] [CrossRef]
- Erkilic, N.; Sanjurjo-Soriano, C.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi004-A, with a point mutation in CRX associated with autosomal dominant Leber congenital amaurosis. Stem Cell Res. 2019, 38, 101476. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Baux, D.; Mamaeva, D.; Hamel, C.P.; Meunier, I.; Roux, A.F.; Kalatzis, V. Genome Editing in Patient iPSCs Corrects the Most Prevalent USH2A Mutations and Reveals Intriguing Mutant mRNA Expression Profiles. Mol. Ther. Methods Clin. Dev. 2020, 17, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Assou, S.; Girault, N.; Plinet, M.; Bouckenheimer, J.; Sansac, C.; Combe, M.; Mianné, J.; Bourguignon, C.; Fieldes, M.; Ahmed, E.; et al. Recurrent Genetic Abnormalities in Human Pluripotent Stem Cells: Definition and Routine Detection in Culture Supernatant by Targeted Droplet Digital PCR. Stem Cell Rep. 2020, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Slembrouck-Brec, A.; Rodrigues, A.; Rabesandratana, O.; Gagliardi, G.; Nanteau, C.; Fouquet, S.; Thuret, G.; Reichman, S.; Orieux, G.; Goureau, O. Reprogramming of adult retinal müller glial cells into human-induced pluripotent stem cells as an efficient source of retinal cells. Stem Cells Int. 2019, 2019, 18–21. [Google Scholar] [CrossRef]
- Chan, C.S.Y.; Lonfat, N.; Zhao, R.; Davis, A.E.; Li, L.; Wu, M.R.; Lin, C.H.; Ji, Z.; Cepko, C.L.; Wang, S. Cell type- And stage-specific expression of Otx2 is regulated by multiple transcription factors and cis-regulatory modules in the retina. Development 2020, 147, dev187922. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing for Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 2018, 5056279. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Popp, M.W.L.; Maquat, L.E. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol. Cells 2014, 37, 1–8. [Google Scholar] [CrossRef]
- Tuladhar, R.; Yeu, Y.; Tyler Piazza, J.; Tan, Z.; Rene Clemenceau, J.; Wu, X.; Barrett, Q.; Herbert, J.; Mathews, D.H.; Kim, J.; et al. CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Reber, S.; Mechtersheimer, J.; Nasif, S.; Benitez, J.A.; Colombo, M.; Domanski, M.; Jutzi, D.; Hedlund, E.; Ruepp, M.D. CRISPR-Trap: A clean approach for the generation of gene knockouts and gene replacements in human cells. Mol. Biol. Cell 2018, 29, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, D.; Sato, H.; Maquat, L.E. Chapter 9 Studying Nonsense-Mediated mRNA Decay in Mammalian Cells; Elsevier Masson SAS: Amsterdam, The Netherlands, 2008; Volume 449, pp. 177–201. [Google Scholar]
- Hegde, R.S.; Zavodszky, E. Recognition and degradation of mislocalized proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a033902. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six classes of nuclear localization signals specific to different binding grooves of importinα. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef]
- Robinson-Rechavi, M.; Garcia, H.E.; Laudet, V. The nuclear receptor superfamily. J. Cell Sci. 2003, 116, 585–586. [Google Scholar] [CrossRef]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and function of the nuclear receptor superfamily and current targeted therapies of prostate cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C. Molecular pathways involved in the transport of nuclear receptors from the nucleus to cytoplasm. J. Steroid Biochem. Mol. Biol. 2018, 178, 36–44. [Google Scholar] [CrossRef]
- Xu, L.; Marquis, K.; Pei, J.; Fu, S.C.; Cagatay, T.; Grishin, N.V.; Chook, Y.M. LocNES: A computational tool for locating classical NESs in CRM1 cargo proteins. Bioinformatics 2015, 31, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Burnight, E.R.; DeLuca, A.P.; Anffinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Penticoff, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci. Rep. 2016, 6, 30742. [Google Scholar] [CrossRef]
- Latella, M.C.; Di Salvo, M.T.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5, e389. [Google Scholar] [CrossRef]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Demontis, G.C.; Leocani, L.; Andreazzoli, M.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xie, B.; He, L.; Zhou, T.; Gao, G.; Liu, S.; Pan, G.; Ge, J.; Peng, F.; Zhong, X. Generation of Retinal Organoids with Mature Rods and Cones from Urine-Derived Human Induced Pluripotent Stem Cells. Stem Cells Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.J.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef]
- Ruan, G.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Collin, R.W.; den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common leber congenital amaurosis CEP290 mutation. Mol. Ther. Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Naessens, S.; Ruysschaert, L.; Lefever, S.; Coppieters, F.; De Baere, E. Antisense oligonucleotide-based downregulation of the G56R pathogenic variant causing NR2E3-associated autosomal dominant retinitis pigmentosa. Genes 2019, 10, 363. [Google Scholar] [CrossRef]
- Torriano, S.; Erkilic, N.; Faugère, V.; Damodar, K.; Hamel, C.P.; Roux, A.-F.; Kalatzis, V. Pathogenicity of a novel missense variant associated with choroideremia and its impact on gene replacement therapy. Hum. Mol. Genet. 2017, 26, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, N.; Cao, L.-H.; Peters, A.; Soon Park, T.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diakatou, M.; Dubois, G.; Erkilic, N.; Sanjurjo-Soriano, C.; Meunier, I.; Kalatzis, V. Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. Int. J. Mol. Sci. 2021, 22, 2607. https://doi.org/10.3390/ijms22052607
Diakatou M, Dubois G, Erkilic N, Sanjurjo-Soriano C, Meunier I, Kalatzis V. Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. International Journal of Molecular Sciences. 2021; 22(5):2607. https://doi.org/10.3390/ijms22052607
Chicago/Turabian StyleDiakatou, Michalitsa, Gregor Dubois, Nejla Erkilic, Carla Sanjurjo-Soriano, Isabelle Meunier, and Vasiliki Kalatzis. 2021. "Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3" International Journal of Molecular Sciences 22, no. 5: 2607. https://doi.org/10.3390/ijms22052607
APA StyleDiakatou, M., Dubois, G., Erkilic, N., Sanjurjo-Soriano, C., Meunier, I., & Kalatzis, V. (2021). Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. International Journal of Molecular Sciences, 22(5), 2607. https://doi.org/10.3390/ijms22052607